

Abstract

In an effort to develop potent, orally bioavailable compounds for the treatment of neoplastic diseases, we developed a class of dual VEGFR-2 kinase and tubulin inhibitors. Targeting the VEGFR receptor kinase and tubulin structure allows for inhibition of both tumor cells and tumor vasculature. Previously, a combination of two compounds, a VEGF receptor tyrosine kinase inhibitor and tubulin agent, was demonstrated to produce an enhanced antitumor response in animal studies. We have reaffirmed their results, with the added benefit that both activities are found in one compound.
Keywords: Tubulin, angiogenesis, VEGFR, tumor vasculature, antimitotic, vascular disrupting agent
A complex disease such as cancer requires an arsenal of therapies to help combat and stabilize its advancement. One approach is to target the tumor directly, employing chemotherapeutic agents and microtubule inhibitors to kill tumor cells that are actively dividing. Taxanes and Vinca drugs, which have been used successfully in the clinic as antimitotic agents because of their action on microtubules, are representative of this approach. It is well documented that microtubules are crucial for the maintenance of cell shape, vesicle and protein trafficking, for cell signaling, and in forming the mitotic spindle used in cell division.1 The predominant mode of action of microtubule inhibitors is disruption of mitotic spindle formation during cell division in rapidly dividing tumor cells, leading to mitotic arrest and subsequent apoptosis.2
An alternative treatment in cancer is the use of antiangiogenic drugs such as bevacizumab. Angiogenesis occurs in response to cues from the tumor, inducing the formation of blood vessels from the surrounding vasculature to deliver nutrients to the tumor. Vascular endothelial growth factor (VEGF) is considered the predominant growth factor required for angiogenesis by invading endothelial cells. VEGF binding to the receptor tyrosine kinase VEGFR-2, located on the surface of endothelial cells, induces trans-phosphorylation and dimerization of the receptor.3 This results in increased migration and proliferation of endothelial cells as well as enhanced permeability of the surrounding vasculature. Inhibiting the kinase activity of VEGFR-2 or blocking VEGF binding to its receptor has become an effective approach in treating cancer.
Combining a drug that inhibits tubulin with another agent that has antiangiogenic properties results in a synergistic effect on tumor growth inhibition.4,5 We attempted to combine these two activities into one compound by modifying oxadiazole and triazole derivatives that we have previously shown potently inhibit tubulin polymerization, with anti-VEGFR2 activity6,7 (Figure 1). Herein, we report a proof-of-concept study using an oxadiazole inhibitor of both tubulin and VEGFR-2 that is well tolerated and has antitumor efficacy in mice.
Figure 1.

Structures of lead compounds 1 and 17.
In addition to optimizing a 3-amino-1,2,4-triazole/3-1,2,4-aminooxadiazole series for tubulin inhibition,6,7 we submitted a patent application with examples of bis-aryl-substituted thiazoles/oxadiazoles of type 1 (Figure 1) as VEGFR-2 inhibitors.8 This work demonstrated that a trifluoromethyl group in the meta position in compound 1 or benzo[1,3]dioxol-5-yl functionality in triazole 4 results in an inhibitory activity in the VEGFR-2 enzymatic assay (Table 1). Moreover, triazole 1 along with several other analogues (not shown) exhibited VEGFR-2 activity around 1 μM in the cellular phosphorylation assay. We have previously established that the presence of a hydrogen bond acceptor in proximity to pyridyl nitrogen of compound 1 is required for kinase activity.9,10 Conversely, substitution of an alternative heterocyclic group for the pyridin-4-yl binding element (2) leads to a complete loss of VEGFR-2 activity. Finally, we demonstrated that the triazole core could be substituted with an oxadiazole group (3) without loss of function.
Table 1. Cross-Reactivity Profile of the Initial VEGFR-2 Lead Class.
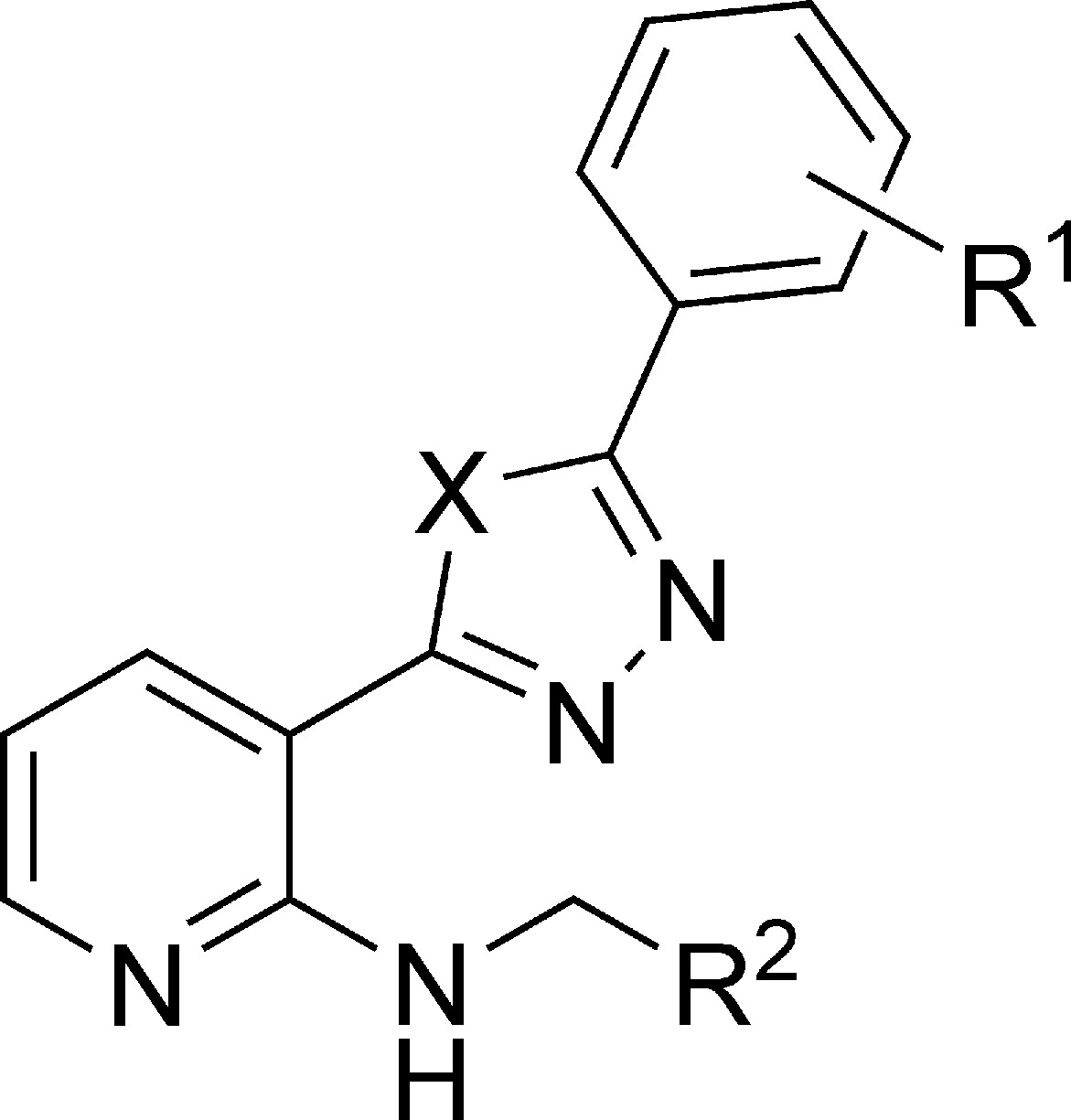
| compd | R1 | R2 | X | ITPa IC50 (μM) | G2/M blockb EC50 (μM) | VEFGR-2 enzyme IC50 (μM) | VEGFR-2 cell IC50 (μM) |
|---|---|---|---|---|---|---|---|
| 1 | 3-trifluoromethyl | pyridin-4-yl | NH | >10 | >10 | 1.1 ± 0.14 | 1.0 ± 0.19 |
| 2 | 3-trifluoromethyl | pyrazol-4-yl | NH | >10 | >10 | >10 | >10 |
| 3 | 3-trifluoromethyl | pyridin-4-yl | O | >10 | >10 | 2.5 ± 0.14 | >10 |
| 4 | benzo[1,3]dioxol-5-yl | pyridin-4-yl | NH | >10 | >10 | 2.3 ± 0.06 | >10 |
| 5 | 4-methylsulfonyl | 1,4-dioxan-2-yl | NH | 6.6 ± 0.07 | >10 | 4.5 ± 0.14 | >10 |
Compound concentration required for 50% inhibition of maximum tubulin assembly.
Compound concentration required for 50% of A431 cells to accumulate at the G2/M phase of the cell cycle.
Noting some similarity between the tubulin inhibitors6,7 and the VEGFR-2 inhibitors,8 all triazole and oxadiazole analogues that were produced as angiogenesis inhibitors were screened in a tubulin polymerization assay. The effort resulted in the identification of compound 5 with dual activity in tubulin polymerization and VEGFR-2 enzymatic assays. However, this hit lacked cellular potency in both VEGFR-2 cellular and tubulin G2M block assays. Nevertheless, this observation and the structural similarity of triazoles 1−5 with tubulin active scaffolds that we had identified6,7 led us to believe that we could incorporate both activities into the triazole/oxadiazole motif.
A variety of chemical entities that contained an oxadiazole group attached to benzene or pyridyl rings with an adjacent substituted amino functionality were prepared and tested for kinase and tubulin inhibitory activity (Table 2). The analogues demonstrated nanomolar inhibitory potency toward VEGFR-2 in both enzymatic and cellular phosphorylation assays. The same analogues exhibited nanomolar activity in tubulin cellular G2M block assay. The trifluoromethyl group on the phenyl moiety that had previously provided us with VEGFR-2 activity (Table 1) resulted in a loss of tubulin potency in both tubulin polymerization and G2M block assays. Therefore, we focused on the benzo[1,3]dioxol-5-yl (piperonyl) group of analogue 4 since we had described the tubulin activity of piperonyl-containing compounds in our earlier work on tubulin active scaffolds.6,7 We reasoned that the addition of a 4-pyridyl group, a common element of multiple VEGFR-2 active classes,8−10 would lend VEGFR-2 activity to our compounds. This was supported by the VEGFR-2 enzymatic activity of piperonyl analogues 6−8, our first attempt at incorporating both VEGFR-2 and tubulin elements into a single molecule. The minor difference in VEGFR-2 and tubulin potencies between central pyridyl ring compound 6 and its correspondent benzene ring analogue 7 allowed us to focus on more synthetically accessible oxadiazoles with a 2-amino-pyridyl core. As it was demonstrated in multiple publications on VEGFR-2 kinase inhibitors,8−10 the repositioning of pyridyl nitrogen into the meta or ortho positions leads to the complete or partial loss of activity (9 and 10). The piperonyl group has a hydrogen bond-accepting moiety, which can often serve as a substituent for 4-pyridyl group.9,11−13 However, the presence of groups without a para-oriented nitrogen atom proved to be insufficient in achieving VEGFR-2 inhibitory activity (11). An attempt to extend the basic nitrogen atom chain length (12 and 13) also did not improve VEGFR-2 or tubulin activities.
Table 2. Cross-Reactivity Profile of Oxadiazole Series.

| compd | R1 | R2 | Y | ITPa IC50 (μM) | G2/M blockb EC50 (μM) | VEFGR-2 enzyme IC50 (μM) | VEGFR-2 cell IC50 (μM) |
|---|---|---|---|---|---|---|---|
| 6 | benzo[1,3]dioxol-5-yl | pyridin-4-yl | N | 1.0 ± 0.15 | 0.020 ± 0.003 | 1.89 ± 0.13 | >10 |
| 7 | benzo[1,3]dioxol-5-yl | pyridin-4-yl | CH | 0.69 ± 0.01 | 0.62 ± 0.03 | 0.83 ± 0.07 | >10 |
| 8 | 2,2-difluoro-benzo[1,3]dioxol-5-yl | pyridin-4-yl | N | 3.9 ± 0.28 | >10 | 3.16 ± 0.22 | >10 |
| 9 | benzo[1,3]dioxol-5-yl | pyridin-3-yl | N | 8.8 ± 0.7 | 0.031 ± 0.001 | >10 | >10 |
| 10 | benzo[1,3]dioxol-5-yl | pyridin-2-yl | N | 1.7 ± 0.20 | 0.049 ± 0.01 | >10 | >10 |
| 11 | benzo[1,3]dioxol-5-yl | benzo[1,3]dioxol-5-yl | N | 2.5 ± 0.4 | 0.52 ± 0.09 | >10 | >10 |
| 12 | benzo[1,3]dioxol-5-yl | 2-(pyridin-3-yl) ethyl | N | 1.7 ± 0.02 | >10 | 3.41 ± 0.09 | >10 |
| 13 | benzo[1,3]dioxol-5-yl | 2-(pyridin-4-yl) ethyl | N | >10 | >10 | >10 | >10 |
| 14 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | pyridin-4-yl | N | 0.5 ± 0.01 | 0.018 ± 0.0024 | 0.17 ± 0.03 | 1.0 ± 0.05 |
| 15 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | pyridin-3-yl | N | 0.70 ± 0.03 | 0.013 ± 0.002 | >10 | >10 |
| 16 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | pyridin-2-yl | N | 1.9 ± 0.11 | 0.016 ± 0.001 | >10 | >10 |
| 17 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | pyridin-4-yl | CH | 1.09 ± 0.13 | 0.009 ± 0.002 | 0.21 ± 0.001 | 0.11 ± 0.02 |
| 18 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | benzo[1,3]dioxol-5-yl | N | 0.71 ± 0.11 | 0.018 ± 0.002 | 2.6 ± 0.1 | >10 |
| 19 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | 2-(pyridin-3-yl) ethyl | N | >10 | >10 | >10 | >10 |
| 20 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | 2-(pyridin-4-yl) ethyl | N | >10 | >10 | >10 | >10 |
| 21 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | imidazol-5-yl | N | 1.5 ± 0.15 | 0.01 ± 0.017 | 0.16 ± 0.02 | 0.83 ± 0.04 |
| 22 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | indazol-5-yl | N | 1.25 ± 0.07 | 0.034 ± 0.001 | 1.4 ± 0.3 | >10 |
| 23 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | quinoline-6-yl | N | 1.45 ± 0.11 | 0.018 ± 0.002 | 0.30 ± 0.02 | 0.22 ± 0.04 |
Compound concentration required for 50% inhibition of maximum tubulin assembly.
Compound concentration required for 50% of A431 cells to accumulate at the G2M phase of the cell cycle.
A breakthrough in our effort to introduce both VEGFR-2 kinase and tubulin activities simultaneously came from a substitution of the benzo[1,3]dioxol-5-yl functionality with the closely related 2,3-dihydro-benzo[1,4]dioxin-6-yl group (Table 2). This group was shown to improve tubulin properties.6,7 We incorporated a VEGFR-2-specific 4-pyridyl functionality into this new oxadiazole 2,3-dihydro-benzo[1,4]dioxin-6-yl scaffold by preparing analogues 14 and 17. Analogue 14 was the first compound that combined appreciable tubulin and kinase cellular activities. VEGFR-2 cellular potency was further improved to submicromolar levels by substituting a pyridine central core for the benzene ring. Structure−activity relationship trends similar to the benzo[1,3]dioxol-5-yl subseries were also observed within the 2,3-dihydro-benzo[1,4]dioxin-6-yl subclass (compare 9−13 to 14, 16, and 18−20, respectively). Heterocyclic groups other than the 4-pyridyl moiety likewise showed dual kinase/tubulin activity within the 2,3-dihydro-benzo[1,4]dioxin-6-yl subclass (imidazolyl 21 and quinolinyl 23).
As previously stated, our synthetic efforts to combine kinase and tubulin activities started with several triazole leads in Table 1. However, our attempt to reproduce a dual profile of oxadiazole 17 with the corresponding triazole analogue 24 (Table 3) failed to display VEGFR-2 cellular potency. Additionally, triazole analogues in Table 3 showed no VEGFR-2 cellular activity including the quinolinyl compound 28.
Table 3. Cross-Reactivity Profile of Triazole Series.
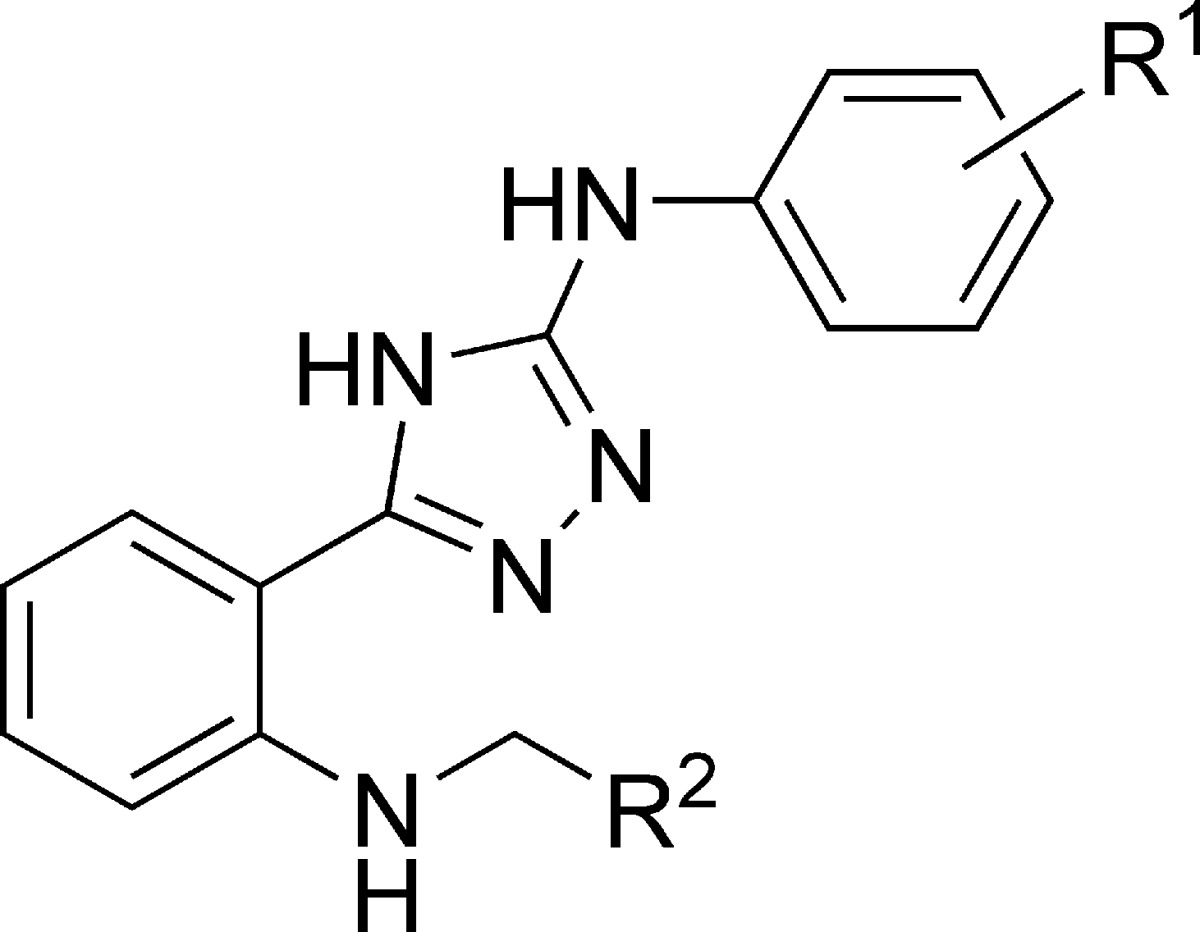
| compd | R1 | R2 | ITPa IC50 (μM) | G2/M blockb EC50 (μM) | VEFGR-2 enzyme IC50 (μM) | VEGFR-2 cell IC50 (μM) |
|---|---|---|---|---|---|---|
| 24 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | pyridin-4-yl | 8.1 ± 0.07 | 0.049 ± 0.028 | 5.1 ± 0.17 | >10 |
| 25 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | pyridin-3-yl | >10 | >10 | 3.2 ± 0.05 | 2.8 ± 0.25 |
| 26 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | imidazol-5-yl | >10 | >10 | 4.6 ± 0.13 | >10 |
| 27 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | indazol-5-yl | >10 | >10 | 3.1 ± 0.06 | 7.3 ± 0.21 |
| 28 | 2,3-dihydro-benzo[1,4]dioxin-6-yl | quinoline-6-yl | 0.07 | >10 | 0.15 | >10 |
Compound concentration required for 50% inhibition of maximum tubulin assembly.
Compound concentration required for 50% of A431 cells to accumulate at the G2M phase of the cell cycle.
Lead compounds 14, 17, and 23 were demonstrated to have little activity, other than against VEGFR-2, in a panel of tyrosine and serine/threonine kinases (see the Supporting Information). Even the close family member VEGFR-1 had an IC50 10-fold higher than VEGFR-2. In addition, compounds 17 and 23 were screened against several kinases known to affect G2M transition, to rule out that the cell cycle arrest seen was due to kinase-inhibiting effects rather than tubulin destabilization. There was limited inhibition of cell cycle dependent kinases CDK1, Aurora A and PLK1. Inhibition of kinase activity was in the micromolar range, far greater than the nanomolar quantities required in our G2M block assay. Therefore, cell cycle arrest is most probably due to compounds 17 and 23 inhibiting tubulin polymerization.
Drug plasma levels in mice were determined using rapid assessment of compound exposure (RACE). A dose of 60 mg/kg of either compound 17 or 23 was injected via interperitoneal route (ip) or administred orally (see the Supporting Information). Exposure of compound 23 in mouse sera was substantially lower than that seen with compound 17. Thus, we decided to focus on compound 17 because of its better exposure in mice and its potential use as an oral drug. Compound 17 was well tolerated up to 100 mg/kg in MTD studies when dosed once a week, similar to other tubulin drugs (data not shown).14 No neuropathy or white blood cell depletion was seen at 100 mg/kg, in marked contrast to Taxanes. Compound 17 was administered via mouth to animals implanted with the human breast cancer cells MDA-MB435LM2 to access its affect on tumor growth (Figure 2 and Table 4). As compared to Paclitaxel, compound 17 demonstrated excellent efficacy by keeping tumors stable throughout treatment. On the other hand, Paclitaxel showed little efficacy and high toxicity, with animals becoming moribund by day 26, thus requiring termination of the study. Even though animals in the Paclitaxel arm did not exhibit weight loss (Table 4), an indicator of health, they had severely distended stomachs and enlarged and inflamed intestines at the time of necropsy. Animals treated with compound 17 on the other hand had no visible signs of discomfort or abnormal pathology.
Figure 2.
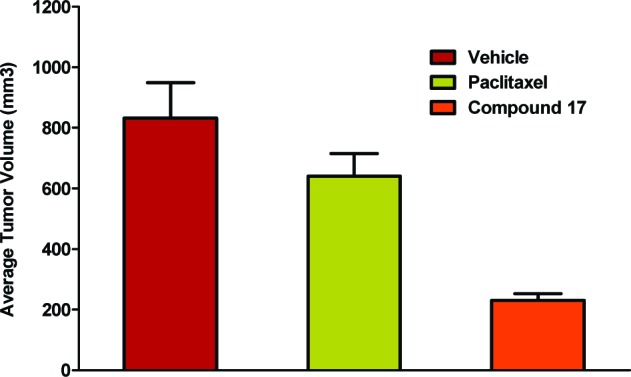
Effect of treatment with compound 17 on MDA-MB-435LM2 tumor volume. MDA-MB-435LM2 tumor size is an average of 10 animals per group on day 26 of the indicated treatment. The experiment was terminated because animals in the Paclitaxel arm were moribund.
Table 4. Effects of Compound 17 on Tumor Size and Body Weighta.
| treatment group | T/C% | RM ANOVA for tumor volume | average % body weight change |
|---|---|---|---|
| vehicle | 3.8 | ||
| compound 17 vs vehicle | 28 | P = <0.0001 | 3.1 |
| paclitaxel vs vehicle | 78 | P = 0.2750 | 2.8 |
| compound 17 vs paclitaxel | P = <0.0001 |
Treatment/control % (T/C%) and two-way ANOVA were calculated to determine statistical differences between the treatment groups. Weight change, as an indicator of health, was evaluated for each treatment group.
Compound 17 in this study has been shown to be a well-tolerated and efficacious molecule. In addition, it can be administered orally, an advantage over many drugs previously used as tubulin inhibitors. Therefore, dual inhibition of VEGFR-2 and tubulin represents a potential and improved new therapy for human cancer.
Acknowledgments
We thank E. Wu, C. Balagtas, R. Rolser, K. H. Yu, S. Patel, and Dr. D. Milligan for providing in vitro data on the compounds. We also acknowledge helpful discussions with Dr. M. Labelle, Dr. M. Duncton, Dr. W. Wong, L. Smith, Dr. K. Kim, and Dr. J. Kawakami.
Supporting Information Available
Experimental details and analytical data for listed compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Wyeth Research, 500 Arcola Rd, Collegeville, Pennsylvania 19406.
deCODE, 2501 Davey Road, Woodridge, Illinois 60517.
Stanford University Schools of Medicine, 269 Campus Drive, CCSR3150, Stanford, California 94305.
Supplementary Material
References
- Gardner M. K.; Hunt A. J.; Goodson H. V.; Odde D. J. Microtubule assembly dynamics: new insights at the nanoscale. Curr. Opin. Cell Biol. 2008, 20, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan M. A.; Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Schenone S.; Bondavalli F.; Botta M. Antiangiogenic agents: an update on small molecule VEGFR inhibitors. Curr. Med. Chem. 2007, 14, 2495–2516. [DOI] [PubMed] [Google Scholar]
- Wedge S. R.; Kendrew J.; Ogilvie D. J.; Hennequin L. F.; Brave S. R.; Ryan A. J; Ashton S. E.; Calvete J. A.; Blakey D. C.. Combination of the VEGF receptor tyrosine kinase inhibitor ZD6474 and tubulin agent ZD6126 produces an enhanced anti-tumor response. American Association for Cancer Research, Annual Meeting, 2002; Poster 5351.
- Varghese H. J.; Mackenzie L. T.; Groom A. C.; Ellis C. G.; Ryan A. J.; MacDonald I. C.; Chambers A. F.. In vivo videomicroscopy reveals differential effects of the vascular-targeting agent ZD6126 and the anti-angiogenic agent ZD6474 on vascular function in a liver metastasis model. Clin. Cancer Res. 2003, 9 (Suppl. 1). [DOI] [PubMed] [Google Scholar]
- Ouyang S.; Piatnitski E. L.; Pattaropong V.; Chen X.; He H.-Y.; Kiselyov A. S.; Velankar A.; Kawakami J.; Labelle M.; Smith L. II; Lohman J.; Lee S. P.; Malikzay A.; Fleming J.; Gerlak J.; Wang Y.; Rolser R. L.; Zhou K.; Mitelman S.; Camara M.; Surguladze D.; Doody J. F.; Tuma M. C. Oxadiazole derivatives as a novel class of antimitotic agents: Synthesis, inhibition of tubulin polymerization, and activity in tumor cell lines. Bioorg. Med. Chem. Lett. 2006, 16, 1191–1196. [DOI] [PubMed] [Google Scholar]
- Ouyang S.; Chen X..; Piatnitski E. L.; Kiselyov A. S.; He H.-Y.; Mao Y.; Pattaropong V.; Yu Y.; Ki K. H.; Kincaid J.; Smith L. II; Wong W. C.; Lee S. P.; Milligan D. A.; Malikzay A.; Fleming J.; Gerlak J.; Deevi D.; Doody J. F.; Chiang H.-H.; Patel S. N.; Wang Y.; Rolser R. L.; Kussie P.; Labelle M.; Tuma M. C. Synthesis and structure-activity relationships of 1,2,4-triazoles as a novel class of potent tubulin polymerization inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 5154–5159. [DOI] [PubMed] [Google Scholar]
- Piatnitski E. L.; Kiselyov A. S.; Doody J.; Hadari Y.; Ouyang X.. Preparation of triazole, oxadiazole, and imidazole derivatives as angiogenesis inhibitors for treatment of cancer. WO 052280, 2004.
- Kiselyov A. S.; Piatnitski E. L.; Milligan D.; Ouyang X. 1H-1,2,4-triazol-3-ylanilines: novel potent inhibitors of vascular endothelial growth factor receptors 1 and 2. Chem. Biol. Drug Des. 2007, 69, 331–337. [DOI] [PubMed] [Google Scholar]
- Piatnitski Chekler E. L.; Katoch-Rouse R.; Kiselyov A. S.; Sherman D.; Ouyang X.; Kim K.; Wang Y.; Hadari Y. R.; Doody J. F. Synthesis and evaluation of heteroaryl-ketone derivatives as a novel class of VEGFR-2 inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4344–4347. [DOI] [PubMed] [Google Scholar]
- Bold G.; Altmann K. H.; Frei J.; Lang M.; Manley P. W.; Traxler P.; Wietfeld B.; Bruggen J.; Buchdunger E.; Cozens R.; Ferrari S.; Fure P.; Hofmann F.; Martiny-Baron G.; Mestan J.; Rosel J.; Sills M.; Stover D.; Acemoglu F.; Boss E.; Emmenegger R.; Lasser L.; Masso E.; Roth R.; Schlachter C.; Vetterli W. New anilinophthalazines as potent and orally well absorbed inhibitors of the VEGF receptor tyrosine kinases useful as antagonists of tumor-driven angiogenesis. J. Med. Chem. 2000, 43122310–2323. [DOI] [PubMed] [Google Scholar]
- Kiselyov A. S.; Piatnitski E. L.; Samet A. V.; Kisliy V. P.; Semenov V. V. Ortho-substituted azoles as selective and dual inhibitors of VEGF receptors 1 and 2. Bioorg. Med. Chem. Lett. 2007, 1751369–1375. [DOI] [PubMed] [Google Scholar]
- Dominguez C.; Smith L.; Huang Q.; Yuan C.; Ouyang X.; Cai L.; Chen P.; Kim J.; Harvey T.; Syed R.; Kim T.-S.; Tasker A.; Wang L.; Zhang M.; Coxon A.; Bready J.; Starnes C.; Chen D.; Gan Y.; Neervannan S.; Kumar. G.; Polverino A.; Kendall E. Discovery of N-phenyl nicotinamides as potent inhibitors of Kdr. Bioorg. Med. Chem. Lett. 2007, 17216003–6008. [DOI] [PubMed] [Google Scholar]
- Tuma M. C.; Malikzay A.; Ouyang X.; Surguladze D.; Fleming J.; Mitelman S.; Camara M.; Kearney J.; Finnerty B.; Doody J.; Piatnitski Chekler E. L.; Kussie P.; Tonra J. R.. IMC-038525: an orally available inhibitor of tubulin polymerization efficacious against MDR and paclitaxel-resistant tumor cells. Transl. Oncol. 2010, accepted for publication. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.