

Abstract
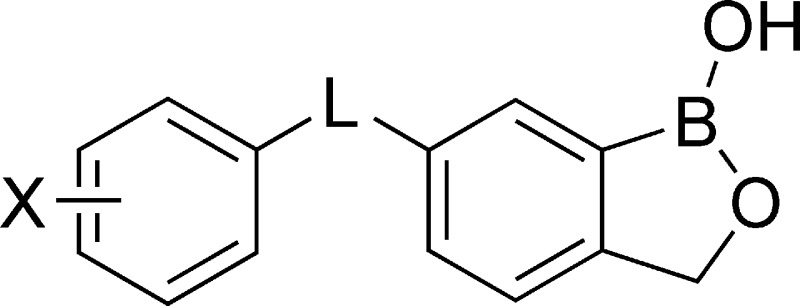
We report the discovery of benzoxaborole antitrypanosomal agents and their structure−activity relationships on central linkage groups and different substitution patterns in the sulfur-linked series. The compounds showed in vitro growth inhibition IC50 values as low as 0.02 μg/mL and in vivo efficacy in acute murine infection models against Tryapnosoma brucei.
Keywords: Tryapnosoma brucei, African trypanosomiasis, benzoxaborole
African sleeping sickness (African trypanosomiasis), a fatal disease, is caused by the protozoan parasite Trypanosoma brucei and is transmitted by the bite of the tsetse fly.1 Although it affects a large population in Africa, drug discovery has been largely neglected during the past half century.2 The currently available treatments for early stage infection, pentamidine and suramin, and melarsoprol and eflornithine for late stage infection, have the problems of high toxicity, high cost, or low efficacy.3 There is an urgent need to develop new therapies with low toxicity, improved efficacy, and affordable cost.4,5
We report here the discovery and structure−activity relationship (SAR) of novel benzoxaborole antitrypanosomal agents. During an initial screening of a focused library of antiinfective benzoxaboroles, compound 12 was found to inhibit in vitro T. brucei growth (IC50 = 0.12 μg/mL). There was no previous report on benzoxaboroles as effective antiprotozoals, although they had been studied as antifungal6 and antiinflammatory7 agents. In this study, we explored the effect of a variety of linkage groups at C(6) and different substitution patterns in the 6-sulfur linked series on T. brucei growth inhibition.
The synthesis of benzoxaboroles with thioether, sulfoxide, and sulfone linkage groups at C(6) is outlined in Scheme 1. Nucleophilic substitution of 2-bromo-4-fluorobenzaldehyde by phenylthiol gave thioether 1, where an ice bath was necessary in some cases to minimize side reactions due to the substitution of bromide. After the aldehyde was converted to MOM-protected hydroxyl, the oxaborole ring was installed by halogen−metal exchange with n-butyllithium followed by in situ trapping with triisopropylborate and deprotection with HCl to give benzoxaboroles 4−8. An alternative route utilized a palladium-mediated boronylation of 1 to provide aldehyde 19, followed by reduction with NaBH4 and acid-catalyzed cyclization to the benzoxaboroles 20−24. Thioethers were oxidized to their sulfoxide analogues either by heating with NaIO4 at 60 °C for an hour or by treatment with an equivalent of m-CPBA at −20 °C. Sulfones were obtained by treatment of thioethers with NaIO4 at 60 °C for 12 h or by treatment with 2 equiv of m-CPBA at −60 °C. Analogously, ether 28 was obtained from 2-bromo-4-fluorobenzaldehyde and phenol as depicted in Scheme 2.
Scheme 1. Synthesis of Benzoxaboroles with Thioether, Sulfoxide, and Sulfone Linkage Groups.

Reagents and conditions: (a) K2CO3, DMF, 0 or 100 °C. (b) NaBH4, CH3OH. (c) MOMCl, DIPEA. (d) n-BuLi, B(i-PrO)3, −78 °C to room temperature. (e) HCl. (f) NaIO4, MeOH−H2O, 60 °C, 1 h or 1 equiv of m-CPBA, DCM−THF, −20 °C. (g) NaIO4, 60 °C, 12 h or 2 equiv of m-CPBA, −60 °C to room temperature. (h) Bis(pinacol-diboron), PdCl2(dppf)2, KOAc, dioxane. (i) NaBH4, EtOH.
Scheme 2. Synthesis of Benzoxaboroles with Ether Linkage Group.

Reagents and conditions: (a) K2CO3, DMF, 100 °C. (b) NaBH4, MeOH. (c) MOMCl, DIPEA, DCM. (d) (i-PrO)3B, n-BuLi, THF, −78 °C to room temperature. (e) HCl.
Benzoxaboroles with carbonyl and carbinol linkage groups were synthesized as shown in Scheme 3. Diphenyl ketone 29 was prepared by Friedel−Crafts reaction and was subsequently brominated with NBS in the presence of Bz2O2 and converted to hydroxymethyl group by treatment with sodium acetate followed by basic hydrolysis to give compound 30. After oxidation with PCC, both carbonyl groups were protected as acetals (32). Introduction of the boronic acid functionality via lithiation and trapping with triisopropyl borate afforded boronic acid 33 after hydrolysis of the acetal groups. Reduction of compound 33 led to the formation of carbinol 34. Oxidation of compound 34 with PCC resulted in ketone 35.
Scheme 3. Synthesis of Benzoxaboroles with Carbonyl and Carbinol Linkage Groups.

Reagents and conditions: (a) SOCl2. (b) Benzene, AlCl3, 50 °C. (c) NBS, Bz2O2, CCl4, reflux. (d) NaOAc, DMF, 60 °C. (e) NaOH, MeOH−H2O, reflux. (f) PCC, DCM. (g) Eethylene glycol, p-TsOH, toluene, reflux, 96 h. (h) B(i-PrO)3, n-BuLi, −78 °C to room temperature. (i) HCl. (j) NaBH4, THF−H2O. (k) PCC, DCM.
Benzoxaboroles with linkage groups derived from 6-OH and 6-NH2 were also synthesized (Scheme 4). First, compound 37 was prepared from acetal 36 by treatment with benzyl alcohol and NaH. Benzoxaborole 39 was obtained after boronylation and reduction as described above. Hydrogenation of compound 39 in the presence of Pd/C resulted in the 6-OH benzoxaborole 40, which was coupled with phenyl isocyanate to give carbamate 41. Coupling of amine 42(8) with benzoic acid, benzyl bromide, or phenylsulfonyl chloride led to the formation of benzoxaboroles with amide (43), aminomethylene (44), and sulfonamide (45) linkage groups, respectively. The N-methylbenzyl amine 48 was prepared in three steps from 2-bromo-4-fluorobenzaldehyde. Hydrogenolysis of compound 48 afforded the N-methyl benzoxaborole 49, which was treated with benzoyl chloride to provide the N-methylbenzamide 50. The reversed amide 53 was synthesized by coupling of aniline with carboxylic acid 52 that was prepared from 6-formyl benzoxaborole 51.9
Scheme 4. Synthesis of Benzoxaboroles with 6-OH and 6-NH2 Derived and Reversed Amide Linkage Groups.

Reagents and conditions: (a) Ethylene glycol, TsOH, toluene, reflux. (b) BnOH, NaH, DMF, 0−65 °C. (c) n-BuLi, B(iPrO)3, −78 °C to room temperature. (d) HCl. (e) NaBH4, THF, 0 °C. (f) Pd/C, H2, MeOH. (g) PhNCO, Et3N, DMF, 0 °C to room temperature. (h) PhCOCl, NaHCO3, CH3CN. (i) BnBr, NaHCO3, DMF, 100 °C. (j) PhSO2Cl, K2CO3, CH3CN. (k) K2CO3, DMF. (l) Bis(pinacol-diboron), PdCl2(dppf)2, KOAc, dioxane. (m) NaBH4, MeOH. (n) Aqueous HCl. (o) Pd/C, HCO2NH4. (p) PhCOCl, Et3N, CH2Cl2. (q) AgNO3, NaOH, H2O, 0 °C. (r) Analine, EDCI, DCM, room temperature, 60 h.
As shown in Scheme 5, benzoxaboroles with NH and methylene linkage groups were also synthesized. Diphenylamine 54 was obtained by the coupling of iodobenzene and 3-bromo-4-methylanaline in the presence of CuI and l-proline.10 After Boc protection, compound 55 was converted to alcohol 56 as described above. Subsequent THP protection and boronylation gave boronic acid 58. Attempts to remove the Boc and THP protecting groups simultaneously with HCl resulted in a complex mixture. Thus, THP was first removed with pyridinium p-toluenesulfonate to give compound 59, which was treated with trifluoroacetic acid at 0 °C to give amine 60. Compound 65 with a methylene linkage group was synthesized from benzyl boronic acid and 2-methoxy-4-bromobenzaldehyde. Suzuki coupling gave biarylmethane 61, which was converted to triflate 63 by demethylation using CeCl3/NaI followed by treatment with Tf2O. Boronylation followed by reduction and acidic treatment gave benzoxaborole 65.
Scheme 5. Synthesis of Benzoxaboroles with NH and CH2 Linkage Groups.

Reagents and conditions: (a) CuI, l-proline, t-BuONa, DMSO, 50 °C. (b) LiHMDS, Boc2O, THF, −80 °C to room temperature. (c) NBS, Bz2O2, CCl4, reflux. (d) NaOAc, DMF, 70 °C. (e) NaOH, MeOH−H2O, reflux. (f) DHP, pyridine, p-TsOH, CH2Cl2. (g) n-BuLi, B(iPrO)3, −78 °C to room temperature. (h) p-TsOH, pyridine, EtOH, 50 °C. (i) TFA, CH2Cl2, 0 °C to room temperature. (j) Pd(dppf)Cl2, CsF, K2CO3, dioxane, 80 °C. (k) CeCl3, NaI, CH3CN, reflux. (l) Tf2O, Et3N, CH2Cl2, −78 °C. (m) Bis(pinacol-diboron), Pd(dppf)Cl2, KOAc, dioxane, 80 °C. (n) NaBH4, MeOH−THF, 0 °C. (o) HCl.
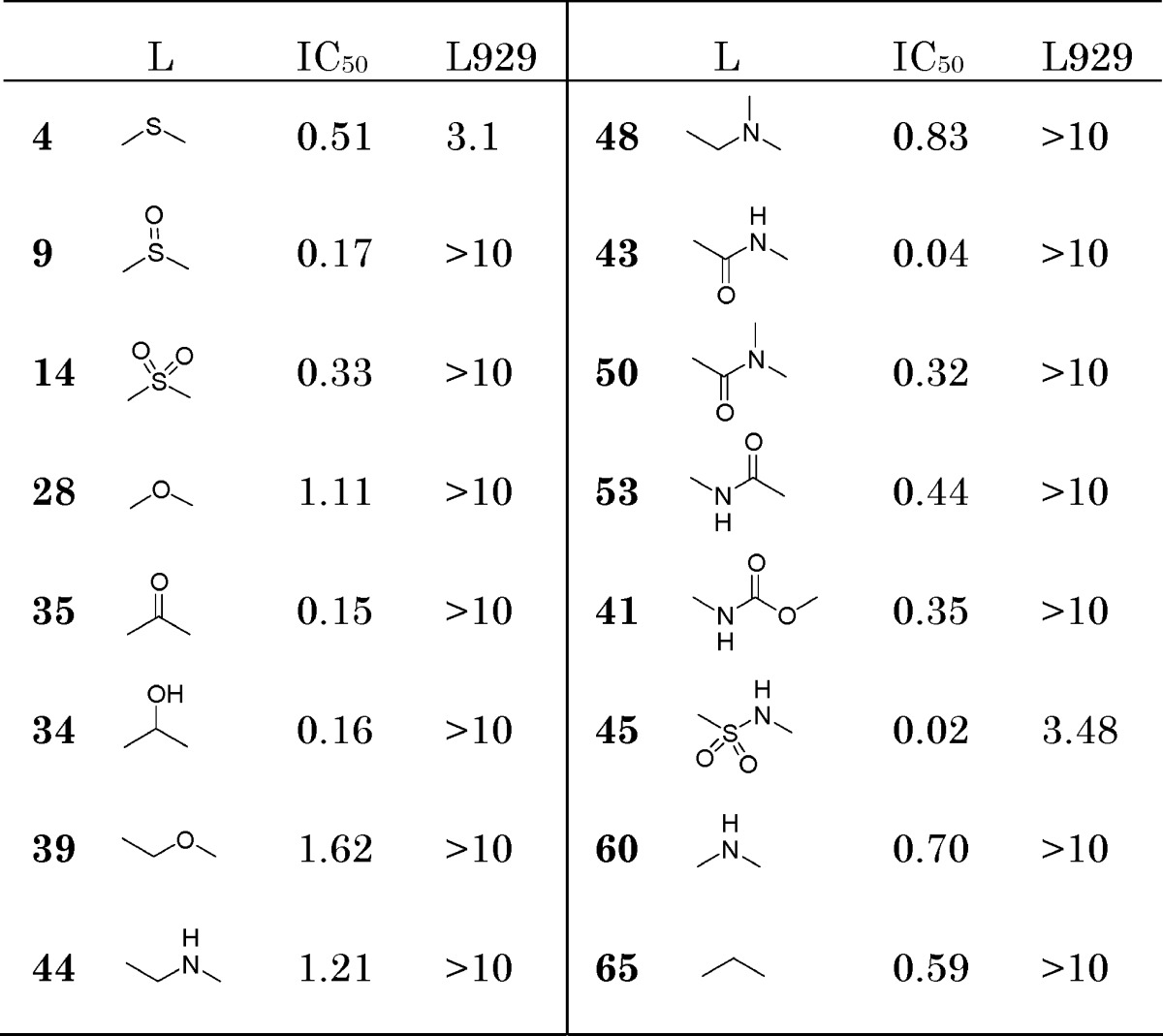
To probe the effect of different C(6) linker groups on antitrypanosomal activity, the above benzoxaboroles were tested for their ability to inhibit growth of T. brucei. They showed good inhibitory effect with IC50 values ranging from 1.62 to 0.02 μg/mL. They also showed a satisfactory (>10 μg/mL) cytotoxicity profile against mouse lung fibroblast cells (L929) in a 72 h in vitro assay. First, the oxaborole functionality was essential for the observed antitrypanosomal activity as demonstrated by the loss of activity (IC50 > 10 μg/mL) upon removal of the oxaborole ring from compounds 4 and 35. Second, the length and hydrogen-bonding properties of the linkage group “L” at C(6) had a significant effect on the antitrypanosomal activity (Table 1). The mechanism of action of these benzoxaboroles is unclear, so the possibility of interaction with multiple biomolecular targets remains, and their membrane permeability and serum-binding properties may also have contributed to cellular activity.
Table 1. Effect of Linkage Group “L” on T. brucei Growth Inhibition and Cytotoxicitya.
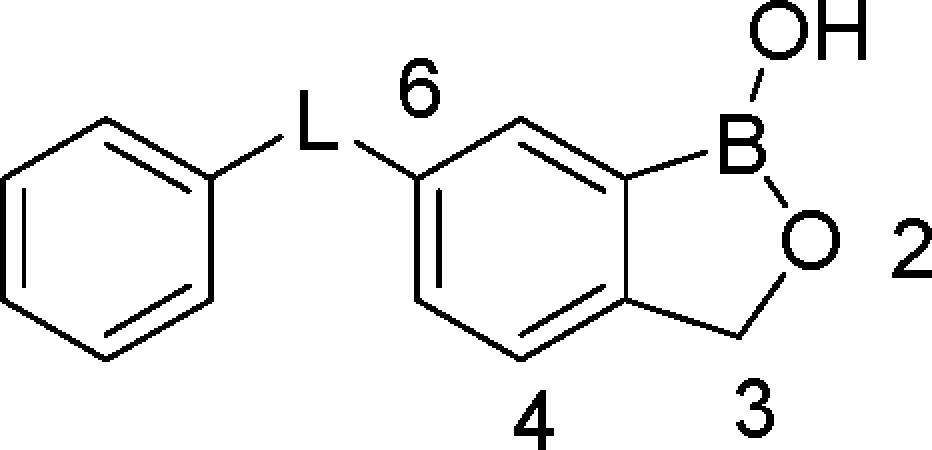
 |
IC50: growth inhibition of T. b. brucei 427 strain (μg/mL). L929: IC50 against L929 cells (μg/mL). References: suramin and pentamidine.
Compounds with thioether (4), ether (28), methylene (65), and amino (60) linkage groups showed IC50 values in the range of 0.51−1.11 μg/mL. The methylene linker is the most flexible with a wide range of allowed conformations, while amino is the most rigid and prefers a near planar conformation.11
The benzoxaboroles bridged with sulfoxide (9), sulfone (14), carbonyl (35), and carbinol (34) represent the category of linkage groups with a hydrogen bond acceptor that is two covalent bonds away from C(6) and showed improved potency (0.15−0.33 μg/mL). It is known that benzophenones such as 35 are a rather rigid chemical motif with sp2 geometry and closely clustered conformations,11,12 while sulfoxide 9, sulfone 14, and carbinol 34 have sp3 geometry and more conformational flexibility. Carbinol 34 showed comparable activity, suggesting that the hydroxyl group may serve as a hydrogen bond acceptor as well.
Benzoxaboroles with amide (43) and sulfonamide (45) linkers showed further improvement of antitrypanosomal activity and represent the most potent compounds among the series (IC50: 0.04 and 0.02 μg/mL). The N-methylbenzamide 50 is 8-fold less potent than amide 43. It is interesting that benzylamine 44 and N-methylbenzyl amine 48 are significantly less active (IC50: 1.21 and 0.83 μg/mL), indicating that carbonyl is essential for high potency and may contribute as a strong hydrogen bond acceptor. Benzyl ether 39 lacking a carbonyl also showed low potency. The reversed amide 53 has a 10-fold lower activity than amide 43, suggesting that the distance between the hydrogen bond acceptor O and the benzoxaborole C(6) has a significant effect. Compounds 9, 14, 34, and 35 have an O−C(6) distance in the range of 2.38−2.70 Å and IC50 values of 0.15−0.24 μg/mL. Amide 43 and sulfonamide 45 have an O−C(6) distance of 2.96 and 3.52 Å and improved IC50 of 0.04 and 0.02 μg/mL. With the exception of thioether 4 and sulfonamide 45, the benzoxaboroles described in Table 1 exhibited very little cytotoxicity in a L929 cell line.
We next explored the SAR of the 6-sulfur-linked benzoxaboroles (Table 2). In the thioether oxidation state, the 3-chloro (6) and 4-chloro (7) analogues improved the antiparasite potency approximately 3-fold, but cytotoxicity was also increased relative to the unsubstituted thioether (4). Cytotoxicity was further increased in the 3,4-dichloro (8) analogue, but the antiparasite potency was diminished. By contrast, the 2-chloro analogue (5) was approximately equipotent with the unsubstituted phenyl but did not exhibit cytotoxicity. In the sulfoxide oxidation state, the antiparasitic potency was not significantly increased by chloro substitution (10−13), but cytotoxicity was observed for the 4-chloro (12) and 3,4-dichloro (13) analogues. In the sulfone oxidation state, the antiparasite potency was similar for the three analogues (15−17) and slightly diminished for the 3,4-dichloro analogue (18). No cytotoxicity was observed at the sulfone oxidation state. Furthermore, a few more thioethers 4-nitro (20), 2-carbomethoxy (21), 4-acetamido (23), and 4-amino (24) analogues exhibited good antiparasite activity and low cytotoxicity, while the 3-methoxy (22) analogue was cytotoxic.
Table 2. SAR of S-Linked Seriesa.
| X | IC50 | L929 | X | IC50 | L929 | ||
|---|---|---|---|---|---|---|---|
| thioether | |||||||
| 5 | 2-Cl | 0.58 | >10 | 21 | 2-CO2Me | 0.14 | >10 |
| 6 | 3-Cl | 0.12 | 1.11 | 22 | 3-OMe | 0.12 | 2.64 |
| 7 | 4-Cl | 0.15 | 2.02 | 23 | 4-NHCOMe | 0.04 | >10 |
| 8 | 3,4-Cl2 | 0.49 | 0.56 | 24 | 4-NH2 | 0.03 | >10 |
| 20 | 4-NO2 | 0.32 | >10 | ||||
| sulfoxide | |||||||
| 10 | 2-Cl | 0.18 | >10 | 12 | 4-Cl | 0.12 | 8.87 |
| 11 | 3-Cl | 0.15 | >10 | 13 | 3,4-Cl2 | 0.30 | 2.67 |
| sulfone | |||||||
| 15 | 2-Cl | 0.10 | >10 | 17 | 4-Cl | 0.16 | >10 |
| 16 | 3-Cl | 0.22 | >10 | 18 | 3,4-Cl2 | 0.52 | >10 |
IC50: growth inhibition of T. b. brucei 427 strain (μg/mL). L929: IC50 against L929 cells (μg/mL). References: suramin and pentamidine.
The 4-chlorophenylsulfoxide 12 was evaluated in a murine model of blood stage T. brucei infection. Treatment of mice infected with 600 T. b. brucei (EATRO 221 strain) at 50 mg/kg, b.i.d., i.p. × 5 days resulted in 100% survival and no parasitemia 40 days after infection. Treatment of T. b. rhodesiense (strain IL1852, 104 parasites)-infected mice at the same dose also cleared parasites 60 days after infection (Figure 1).
Figure 1.
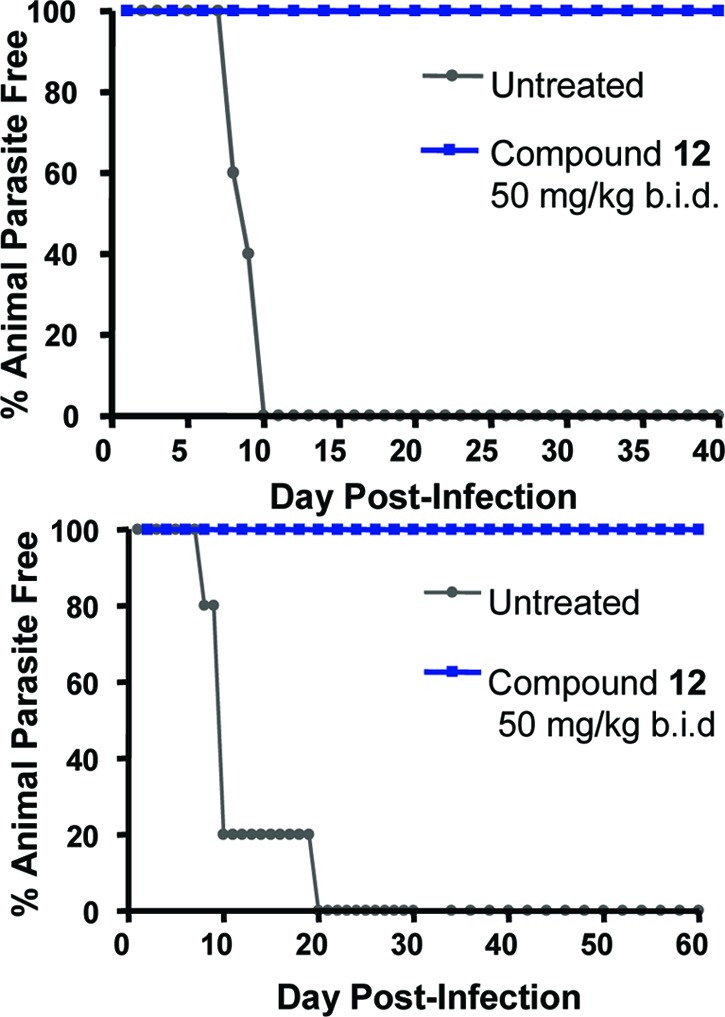
(a) Female BALB/c mice were inoculated IP with 600 T. b. brucei (EATRO 221) parasites. Treatment with compound 12 (50 mg/kg, b.i.d.) cured 100% of mice. (b) Treatment of T. b. rhodesiense (IL1852, 104/mouse)-infected mice with compound 12 (50 mg/kg, b.i.d.) also gave parasite-free survival of the mice. Reference compound: suramin.
We further explored the efficacy of several 6-S-linked benzoxaboroles in the murine model of blood stage T. b. brucei infection, using the EATRO 110 strain. The sulfoxide 12 was effective at 20 mg/kg, i.p., but failed to show complete cure of infection via an oral route. The sulfone 17 was more efficacious, with complete cure observed at 20 mg/kg, p.o., but with only limited efficacy at 10 mg/kg, p.o. None of the thioether analogues (7, 23, or 24) demonstrated meaningful reduction of parasitemia in this model.
In summary, we report novel benzoxaborole-based antitrypanosomal agents. Their in vitro antitrypanosomal IC50 values ranged from 0.02 to 1.62 μg/mL, and they also showed satisfactory cytotoxicity above 10 μg/mL against L929 cells. The SAR of the linkage groups suggested that while most linkers can provide compounds with acceptable antiparasite potency, those containing a hydrogen bond acceptor offer superior potency. The effects of substitution and sulfur oxidation state were investigated, and it was found that they had a significant effect on cytotoxicity. Finally, compounds 12 and 17 showed efficacy in the mice infected with T. brucei. Further optimization of the benzoxaboroles, in particular 6-carboxamides, exemplified by compound 43, is underway and will be the subject of future communications. The discovery of benzoxaboroles as a novel class of antiparasitic agents offers a new opportunity in the war against pathogenic parasites.
Supporting Information Available
Synthetic experimental details, analytical data of compounds, and biological assay protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
We thank The Sandler Foundation, Drugs for Neglected Diseases initiative, National Science Foundation of China (20702031), and Ministry of Science and Technology of China (2009CB918404) for financial support of this work.
Supplementary Material
References
- Legros D.; Ollivier G.; Gastellu-Etchegorry M.; Paquet C.; Burri C.; Jannin J.; Buscher P. Treatment of human African trypanosomiasis-present situation and needs for research and development. Lancet Infect. Dis. 2002, 2, 437–440. [DOI] [PubMed] [Google Scholar]
- Hotez P. J.; Molyneux D. H.; Fenwick A.; Kumaresan J.; Sachs S. E.; Sachs J. D.; Savioli L. Control of neglected tropical diseases. N. Engl. J. Med. 2007, 357, 1018–1027. [DOI] [PubMed] [Google Scholar]
- Croft S. L.; Barrett M. P.; Urbina J. A. Chemotherapy of trypanosomiases and leishmaniasis. Trends Parasitol. 2005, 21, 508–512. [DOI] [PubMed] [Google Scholar]
- Renslo A. R.; McKerrow J. H. Drug discovery and development for neglected parasitic diseases. Nat. Chem. Biol. 2006, 2, 701–710. [DOI] [PubMed] [Google Scholar]
- Pink R.; Hudson A.; Mouries M. A.; Bendig M. Challenges in antiparasitic drug discovery. Nat. Rev. Drug Discovery 2005, 4, 727–740. [DOI] [PubMed] [Google Scholar]
- Rock F. L.; Mao W.; Yaremchuk A.; Tukalo M.; Crepin T.; Zhou H.; Zhang Y. K.; Hernandez V.; Akama T.; Baker S. J.; Plattner J. J.; Shapiro L.; Martinis S. A.; Benkovic S. J.; Cusack S.; Alley M. R. K. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 2007, 316, 1759–1761. [DOI] [PubMed] [Google Scholar]
- Akama T.; Baker S. J.; Zhang Y. K.; Hernandez V.; Zhou H.; Sanders V.; Freund Y.; Kimura R.; Maples K. R.; Plattner J. J. Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis. Bioorg. Med. Chem. Lett. 2009, 19, 2129–2132. [DOI] [PubMed] [Google Scholar]
- Lennarz W. J.; Snyder H. R. Arylboronic acids. IV. Reactions of boronophthalide. J. Am. Chem. Soc. 1960, 82, 2172–2175. [Google Scholar]
- Ye L.; Ding D.; Feng Y.; Xie D.; Wu P.; Guo H.; Meng Q.; Zhou H. Convenient and versatile synthesis of formyl-substituted benzoxaboroles. Tetrahedron 2009, 65, 8738–8744. [Google Scholar]
- Ma D.; Cai Q. Copper/amino acid catalyzed cross-couplings of aryl and vinyl halides with nucleophiles. Acc. Chem. Res. 2008, 41, 1450–1460. [DOI] [PubMed] [Google Scholar]
- Hao M. H.; Haq O.; Muegge I. Torsion angle preference and energetics of small-molecule ligands bound to proteins. J. Chem. Inf. Model. 2007, 47, 2242–2252. [DOI] [PubMed] [Google Scholar]
- Gough K. M.; Wildman T. A. Hindered internal rotation in benzophenone. J. Am. Chem. Soc. 1990, 112, 9141–9144. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.