

Abstract
Hematopoietic prostaglandin D synthase (HPGDS) is primarly expressed in mast cells, antigen-presenting cells, and Th-2 cells. HPGDS converts PGH2 into PGD2, a mediator thought to play a pivotal role in airway allergy and inflammatory processes. In this letter, we report the discovery of an orally potent and selective inhibitor of HPGDS that reduces the antigen-induced response in allergic sheep.
Keywords: Hematopoietic prostaglandin D synthase (HPGDS); PGH2; PGD2; airway allergy; inflammatory processes, cyclooxygenase (COX)
Asthma is a chronic inflammatory disorder of the airways that causes recurrent episodes of wheezing, breathlessness, chest tightness, and coughing in susceptible individuals.1,2 Prostaglandin D2 (PGD2), a mediator of allergy and inflammation, is produced by mast cells and Th-2 cells in a variety of human tissues. PGD2 is the most abundant de novo cyclooxygenase-derived mediator produced following IgE-mediated degranulation of mast cells.3 The mast cell, following allergen-provoked degranulation, is believed to be the major source of PGD2 found in the nasal exudates of patients with allergic rhinitis. PGD2 levels increase dramatically in bronchoalveolar lavage fluid following allergen challenge, and the observation that patients with asthma exhibit bronchoconstriction upon inhalation of PGD2 underscores the pathologic consequences of high levels of PGD2 in the lung.4 Treatment with PGD2 produces significant nasal congestion and fluid secretion in man and dogs, and PGD2 is 10 times more potent than histamine and 100 times more potent than bradykinin in producing nasal blockage in humans, demonstrating a role for PGD2 in allergic rhinitis.5,6 Mast cell-derived PGD2 exerts its effect by activating two distinct G-protein-coupled receptors (GPCRs): the DP-1 receptor, a member of the prostanoid receptor subfamily, and the recently discovered chemoattractant receptor-homologous molecule expressed on T-helper 2 cells (CRTH2 receptor). The DP-1 receptor is localized on the nasal vasculature and in mucin-secreting cells and is associated with tissue swelling and a concomitant increase in nasal airway resistance, while the CRTH2 receptor is expressed on a subset of infiltrating T cells in inflamed nasal mucosa and is associated with the induction of chemotactic migration and/or activation of Th-2, eosinophils, and basophils.7−9 Collectively, these data support a role for PGD2 in inflammatory diseases of the upper airways and suggest that blockade of PGD2 action at either or both receptors might be beneficial for the treatment of nasal allergies and other PGD2-mediated inflammatory conditions. This has created significant interest in both DP-1 and CRTH2 as targets, and several publications have appeared describing inhibitor design.10,11
PGD2 is synthesized from arachidonic acid via the oxidation by cyclooxygenase PGH2 (COX) and isomerization of PGH2 to PGD2 by prostaglandin D synthase (PGDS). Hematopoietic PGDS (HPGDS) is responsible for the synthesis of PGD2 by mast cells and Th-2 cells, whereas a lipocalin type PGDS (LPGDS) catalyzes the production of PGD2 in the central nervous system, male genital organs, and heart.12 The production of PGD2 by HPGDS is thought to play a pivotal role in airway allergic and inflammatory processes and induces vasodilatation, bronchoconstriction, pulmonary eosinophil and lymphocyte infiltration, and cytokine release in asthmatics.12 Inhibition of HPGDS may be therapeutically beneficial in the treatment of allergic disease and may be more effective than blocking either DP-1 or CRTH2 alone.13
HPGDS and LPGDS are genetically distinct synthases that have presumably arisen as a consequence of convergent evolution. These isoenzymes are named according to their differential requirement for GSH as a cofactor. The GSH-independent PGD2 synthase is a member of the lipocalin superfamily, while the GSH-dependent synthase is the only vertebrate member of the class Sigma family of glutathione S-transferases (GSTs) identified to date.14,15 HPGDS is a cytosolic homodimer of 26 kDa subunits, and several crystal structures have been published.16,17 Those structures reveal a well-defined active site, enabling a structure-based design of inhibitors. There have been several reports of potent enzyme inhibitors18−20 of HPGDS along with HQL-79 as an in vivo efficacious inhibitor21 (Figure 1). In this letter, we will describe our effort to identify an orally active inhibitor of HPGDS.22
Figure 1.

Examples of literature inhibitors.
A subset screen was performed on the Pfizer compound collection based on structural diversity. This resulted in numerous leads for further manipulation. Our lead generation group at the Research and Technology Center completed a triage and lead validation identifying the thiazole 1 as an advanced lead for further optimization (Figure 2). Thiazole 1 is a potent inhibitor of HPGDS enzyme (IC50, 10 nM) and cell (IC50, 300−1300 nM). In addition, the compound had a very acceptable pharmacokinetic (PK) profile (Cl, 14 mL/min/kg; T1/2, 2 h; and BA, 20%). In analysis of this compound, we identified several parameters that required optimization, most importantly, the poor cell activity of the compound. In addition, we desired a scaffold with improved novelty as judged by substructure searching in Scifinder.
Figure 2.

Advanced lead as an inhibitor of HPGDS.
The thiazole could easily be divided into three major areas for exploration utilizing high-throughput chemistry: the head, the body, and the tail. Our initial effort was focused on the body, and a series of heterocycles were explored that could retain a similar hydrogen-bonding network as 1 with a conserved water molecule in the active site.23 Those compounds were easily prepared from available acids by an amide coupling reaction with 2; selective examples are illustrated in Table 1. Most of those building blocks were commercially available with a few exceptions, which were available in our in-house chemical storeroom.
Table 1. Heterocyclic Variation of the Body.
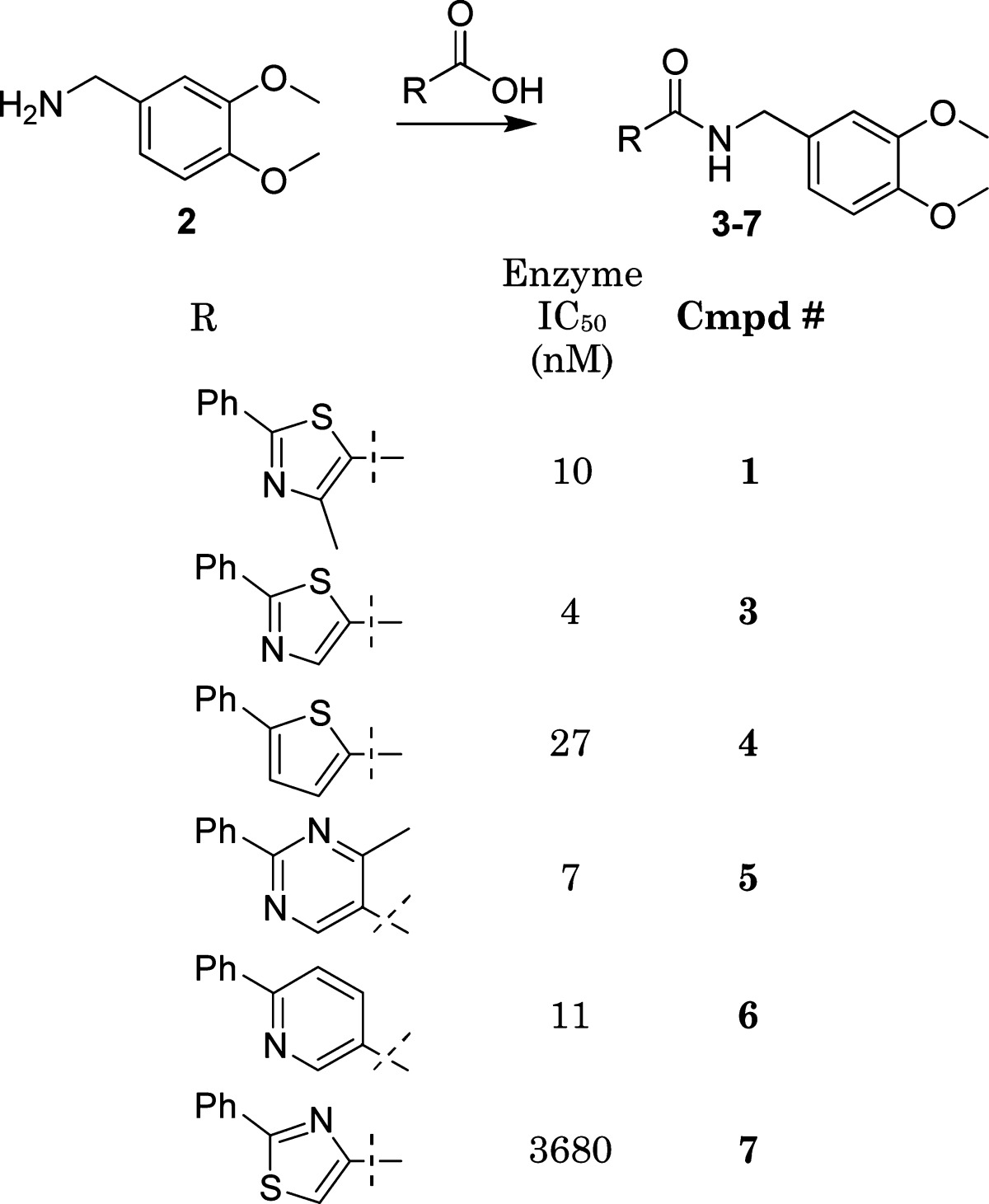 |
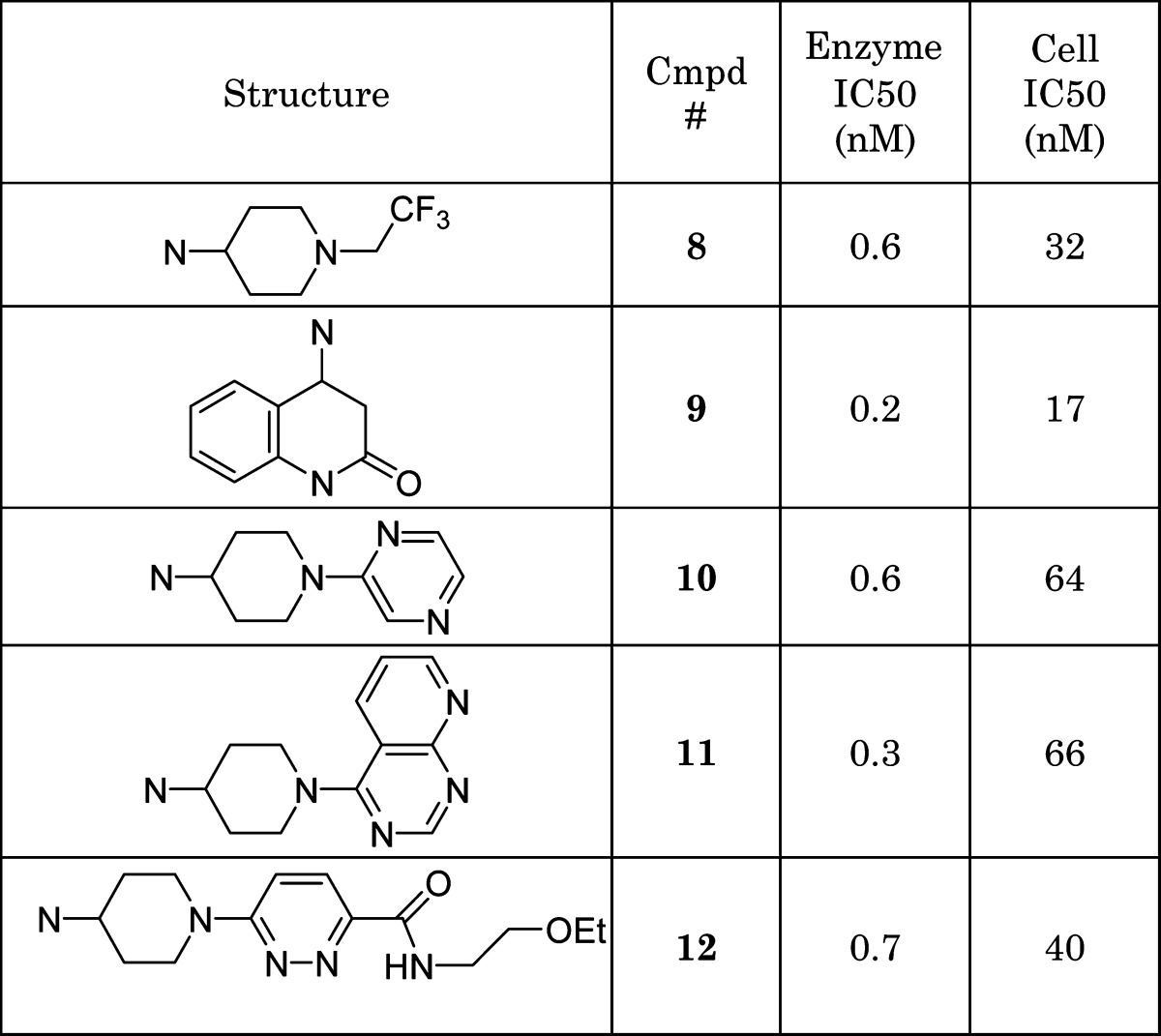
This initial iteration provided a wealth of information regarding the shape and the nature of the heteroatom equal to the nitrogen in the thiazole. Only heterocycles with a N location similar to the thiazole such as the potent compounds 5 and 6 were found to be active. The location of the nitrogen in relationship to the trajectory of the tail was found to be just as important, as 7 illustrated. The activity of 4 (27 nM), which lacks a central ring nitrogen atom, was therefore very surprising and confounded the structure−activity relationship interpretation until isothermal calorimetry illustrated that entropy was the major driving force for affinity.24 The series represented by scaffolds such as 5 and 6 were subsequently evaluated further by several cycles of high-throughput synthesis focused on the tail variation. This was done to address one of our major objectives, improved cell activity (Figure 3). It was clear that we consistently could prepare potent inhibitors of HPGDS, but only a handful of compounds illustrated potency in our cell assay. A key observation was made that certain substructures in the tail had a higher probability for good cell activity. The most noticeable fragments were benzylamines and 4-aminopiperdines. A focused effort in preparing analogues of 5 and 6 containing a tail with the 4-aminopiperdine substructure was therefore undertaken. Those compounds were designed utilizing a range of both structural and physical properties (MW, <500; cLogp, <4; and TPSA, <120) based criteria. Incorporating these design features resulted in significantly improved compounds with meaningful activity in the cell assay (4% to 34%, as judged by IC50 ∼100 nM) (Figure 4). This does not explain why potent compounds illustrated poor activity, and a range of hypothesis such as permeability and efflux were explored to explain that without success. Noticeable was the tolerance of a wide range of structural diversity in the piperidine scaffold, resulting in cell active compounds. The structures of several of those cell active analogues are depicted in Table 2.
Figure 3.
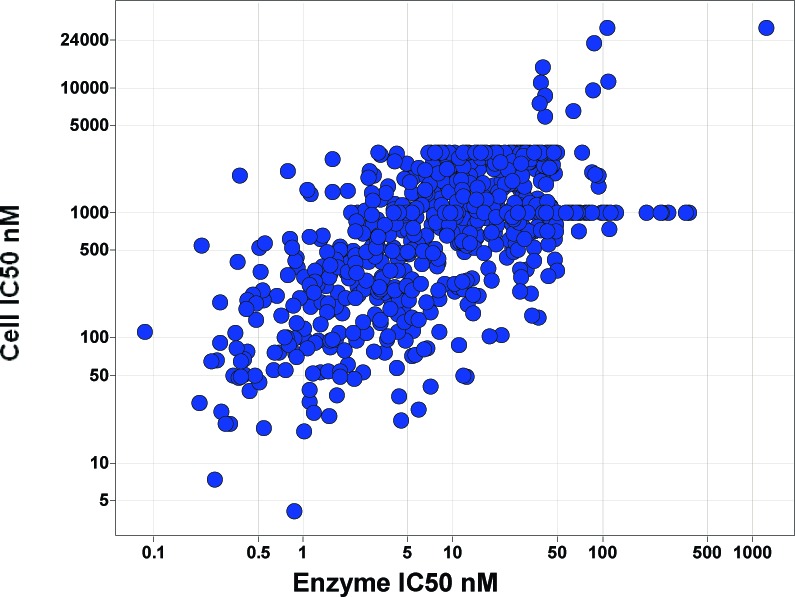
Correlation of enzyme potency with cell activity for 651 analogue prepared spanning a range of templates.
Figure 4.
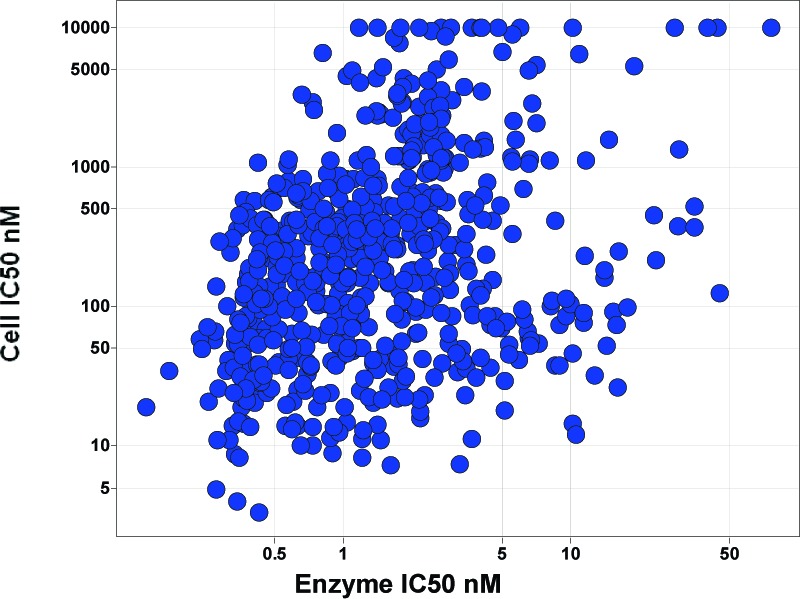
Focused effort on pyridine and pyrimidine cores containing a 4-aminopiperdine core, a total of 631 compounds.
Table 2. Selected Examples of Pyridines and Pyrimidine Analogues with a 4-Aminopiperdine Tail.
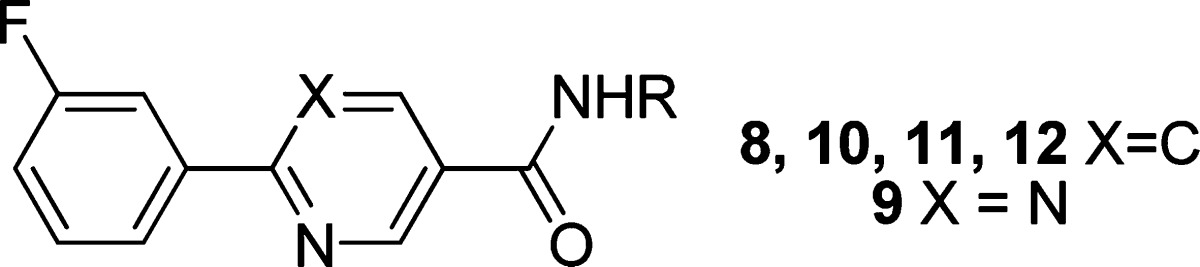
 |
The simplistic tail found in compound 8 made it ideal for a brief survey of the phenyl headgroup. A series of analogues guided by SBDD were prepared as depicted in Table 3. Despite the selection of small changes, it was found that only the 3-fluorine substituent (8) was tolerated as judged by the cell assay. The fluoro substituent (8) provided a significant improvement with regard to stability in an in vitro human microsome assay as compared to the unsubstituted analogue 13.
Table 3. Optimization of the Aryl Head Group with a Pyridine Body and a Fixed Piperidine Tail.
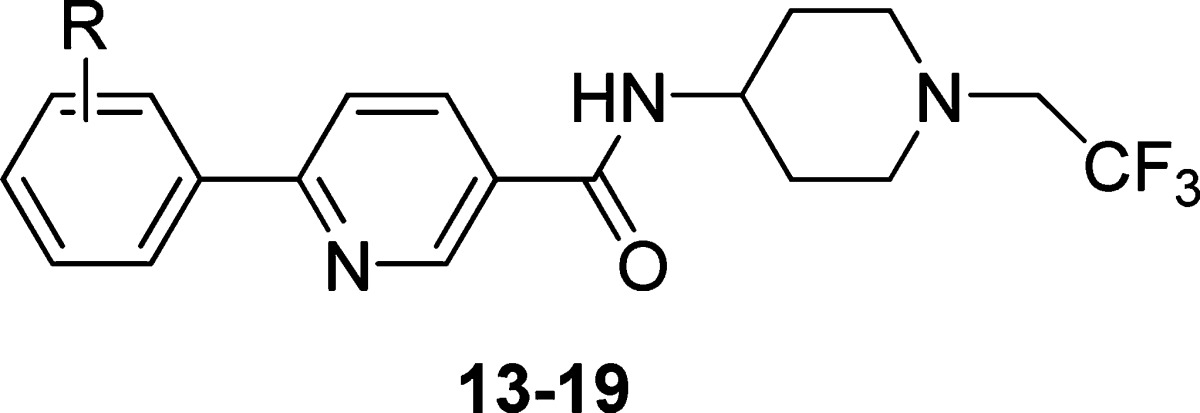
| IC50 (nM) |
|||
|---|---|---|---|
| parent ID | R | enzyme | cell |
| 13 | H | 0.2 | 30 |
| 14 | 4-F | 23 | 600 |
| 15 | 3-OMe | 4 | 344 |
| 16 | 3-Me | 0.8 | 70 |
| 8 | 3-F | 0.7 | 32 |
| 17 | 3,5-F | 1.8 | 155 |
| 18 | 3,6-F | 1 | 160 |
| 19 | 2-F | 0.7 | 153 |
Compound 8 was elected for further profiling based on its enzyme and cell potency. The compound illustrated equal potency against purified HPGDS from human, rat, dog, and sheep (IC50, 0.5−2.3 nM). Compound 8 was profiled in a panel of cellular assays to screen for activity against several relevant human enzyme targets. For example, those assays indicated that 8 does not inhibit human L-PGDS, mPGES, COX-1, COX-2, or 5 LOX (IC50 values > 10000 nM). These data illustrate that 8 has good selectivity against several relevant human targets.
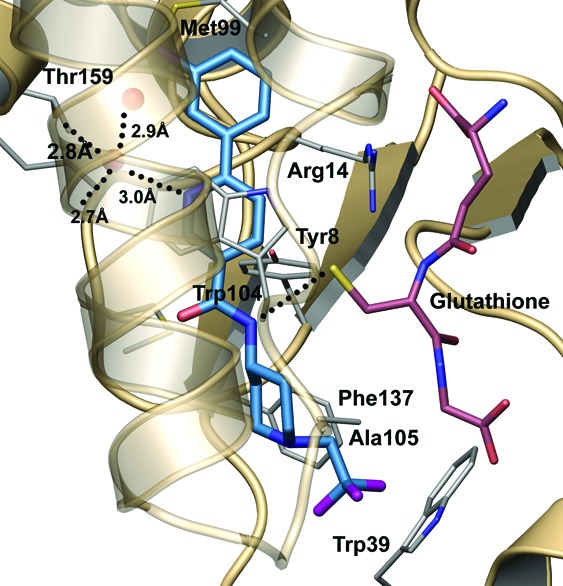
The crystal structure of 8 in complex with HPGDS was determined and refined at 2.1 Å resolution (Rfree = 22.2%, and RMSd bonds = 0.007 Å) by soaking the compound into preformed crystals (Figure 5). The majority of interactions made between the inhibitor and the protein are van der Waals type, including favorable π−π stacking interaction made between the central pyridyl ring of the compound and the Trp104 of the protein. Additionally, two key hydrogen bonds are made to the inhibitor, both of which likely have ionic character. The first hydrogen bond is made through a solvent molecule to the buried carboxyl terminus of the protein. Elimination of this interaction by replacement of the pyridine with a phenyl ring significantly alters the binding position and potency (>100-fold difference) of the compound. The second polar interaction is made between the amide nitrogen of the inhibitor and the thiolate anion of the glutathione. Hydrogen bonds between amide nitrogen atoms and thiolate anions remain strong with interatomic distances of 3.4−4.0 Å.25 Similar distances are routinely observed between the glutathione and other HPGDS inhibitors in this series.
Figure 5.
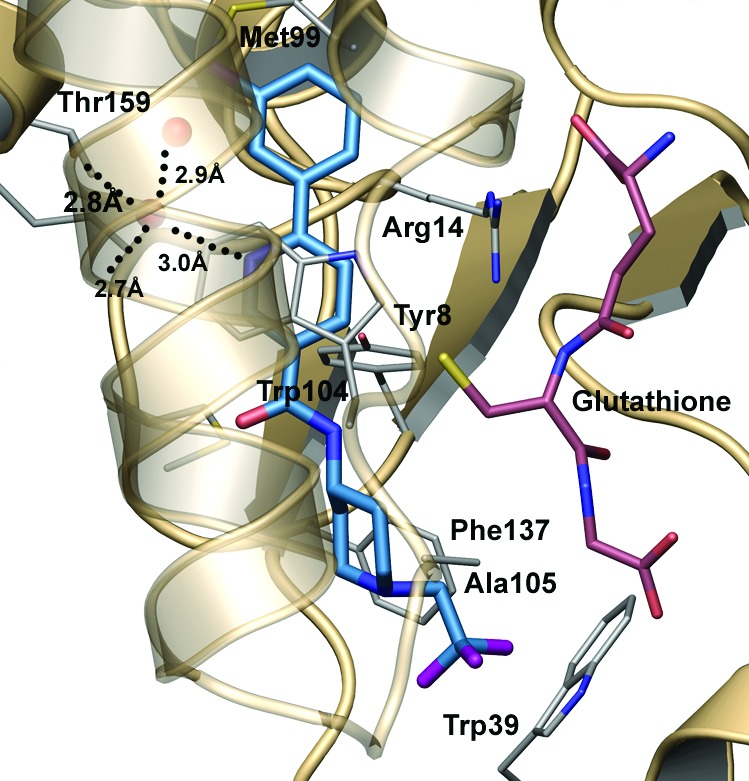
Crystal structure of 8 bound at the HPGDS active site. The inhibitor makes two key hydrogen bonds (dotted lines) in addition to substantial van der Waals contacts within the pocket.
The physical properties of the compound were evaluated, and it was found that 8 had a solubility of 1.5 μg/mL (3.9 μM) at pH 6.50. The compound had excellent PK characteristics when dosed in rats at 1 mpk with 76% bioavailability (Table 4).26
Table 4. PK Properties of 8 in Rata.
| property | entry |
|---|---|
| AUC (μg h/mL) | 1.9 |
| CL (mL/min/kg) | 8.7 |
| Vss (L/kg) | 2.1 |
| T1/2 (h) | 4.1 |
| F (%) | 76 |
Unmilled crystalline powder at 1mpk, 0.5% HPMC/0.1% Tween 80 in dH2O.
Rats dosed orally with 1 and 10 mpk 8 were sacrificed at various times, and plasma concentrations of 8 and spleen PGD2 concentrations were measured. Oral administration of 8 blocked PGD2 production in the rat spleen; inhibition of PGD2 was inversely correlated with the plasma concentration of 8 in a time- and dose-dependent manner (Figure 6). Spleen PGD2 levels fall as 8 plasma levels increase over time; PGD2 levels return to baseline levels as 8 plasma levels decline.
Figure 6.
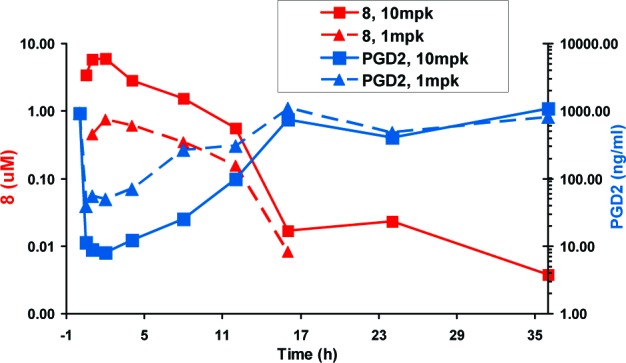
Rat H-PGDS target modulation assay. Rats dosed with 1 and 10 mpk 8 were sacrificed at various times, and the plasma concentration of 8 and spleen PGD2 concentration was measured. The concentration time curve for 1 and 10 mpk doses of 8 with corresponding spleen PGD2 levels.
The effect of 8 in a model of antigen-induced airway response in allergic sheep was explored. Representative responses to allergen challenge and the response to carbachol provocation for the two trials are shown in Figures 7 and 8. The same animals were used for the drug and vehicle trials. In the vehicle-treated animals, mean lung resistance (RL) increased 4−5-fold over baseline value immediately after airway challenge with Ascaris sum antigen. RL then slowly returned to baseline values at 4 h (EAR). RL then increased again 4−8 h postchallenge (LAR). Airway hyper-responsiveness (AHR) was determined at 24 h postchallenge by measuring the change in RL in response to administration of carbachol. In the vehicle trial, AHR was evident as shown by the decrease in the PC400 after antigen challenge.27 In contrast, treatment of sheep with 8 almost completely prevented the LAR (maximum was 86% inhibition of LAR) and blocked the AHR (no change in post antigen PC400), although 8 had no effect on the EAR.
Figure 7.
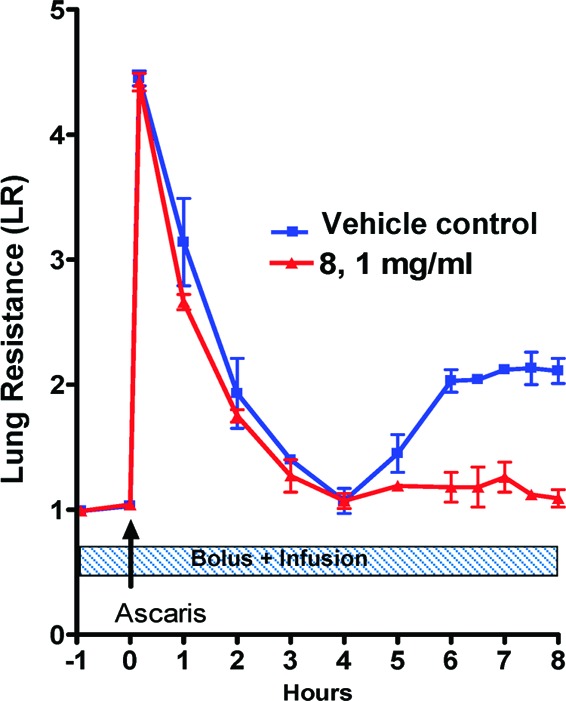
Time course of antigen-induced changes in lung resistance of sheep treated with vehicle or vehicle containing 8. Values are means ± SDs; 2 sheep/group.
Figure 8.
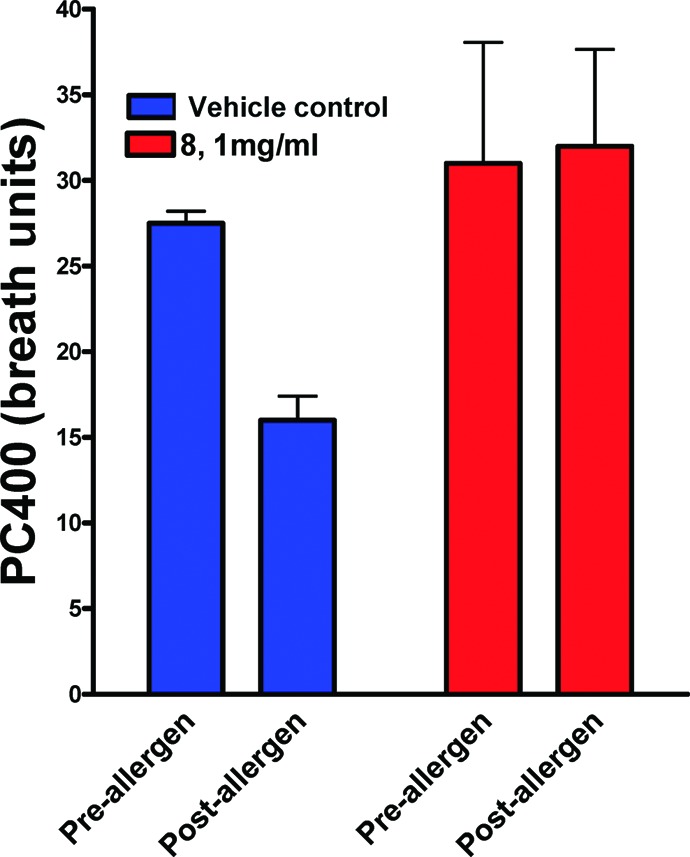
The sheep used in these experiments develop AHR 24 h post Ascaris challenge. Vehicle did not inhibit allergen-induced AHR (significant fall in postantigen PC 400), whereas administration of 8 completely protected against allergen-induced AHR to carbachol provocation; the PC400% pre- and postchallenges were not significantly different (p = 0.89).
In summary, we have described the discovery of a series of orally active inhibitors of HPGDS. Compound 8 is a selective inhibitor for HPGDS, and the compound has excellent PK properties that allowed us to evaluate it in several animal models. Following oral administration, the compound inhibits PGD2 production in a dose-dependent manner in an in vivo rat model. Furthermore, 8 illustrates efficacy in an in vivo sheep model of asthma.
Acknowledgments
We thank the lead generation group at the RTC lead by Patricia Soulard and Cyrille F. Kuhn for the delivery of compound 1, without which none of the work described here would have been possible.
Supporting Information Available
Procedures and characterization for all newly prepared compounds, coordinates for the cocrystal structure of 8 with HPGDS (3KXO), experimental procedures for the enzyme, and the cellular assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Barnes P. Nat. Rev. Drug Discovery 2004, 3, 831. [DOI] [PubMed] [Google Scholar]
- Bloemen K.; Verstraelen S.; Van Den Heuvel R.; Witters H.; Nelissen I.; Schoeters G. Immunol. Lett. 2007, 113, 6. [DOI] [PubMed] [Google Scholar]
- Lewis R. A.; Soter N. A.; Diamond P. T.; Austen K. F.; Oates J. A.; Roberts L. J. 2nd J. Immunol. 1982, 129, 1627. [PubMed] [Google Scholar]
- Murray J. J.; Tonnel A. B.; Brash A. R.; Roberts L. J. 2nd; Gosset P.; Workman R.; Capron A.; Oates J. A. N. Engl. J. Med. 1986, 315, 800. [DOI] [PubMed] [Google Scholar]
- Hardy C. C.; Robinson C.; Tattersfield A. E.; Holgate S. T. N. Engl. J. Med. 1984, 311, 209. [DOI] [PubMed] [Google Scholar]
- Howarth P. H. Allergy 1997, 52, 12. [DOI] [PubMed] [Google Scholar]
- Hata A. N.; Breyer R. M. Pharmacol. Ther. 2004, 103, 147. [DOI] [PubMed] [Google Scholar]
- Matsuoka T.; Hirata M.; Tanaka H.; Takahashi Y.; Murata T.; Kabashima K.; Sugimoto Y.; Kobayashi T.; Ushikubi F.; Aze Y.; Eguchi N.; Urade Y.; Yoshida N.; Kimura K.; Mizoguchi A.; Honsda Y.; Nagai H.; Narumiya S. Science 2000, 287, 2013. [DOI] [PubMed] [Google Scholar]
- Nantel F.; Fong C.; Lamontagne S.; Wright D. H.; Giaid A.; Desrosiers M.; Metters K. M.; O'Neill G. P.; Gervais F. G. Prostaglandins Other Lipid Mediators 2004, 73, 87. [DOI] [PubMed] [Google Scholar]
- Medina J. C.; Liu Annu. Rep. Med. Chem. 2006, 40, 221. [Google Scholar]
- Pettipher R.; Hansel T. T.; Armer R. Nat. Rev. Drug Discovery 2007, 6, 313. [DOI] [PubMed] [Google Scholar]
- Kanaoka Y.; Urade Y. Prostaglandins, Leukotrienes Essent. Fatty Acids 2003, 69, 163. [DOI] [PubMed] [Google Scholar]
- Trivedi S. G.; Newson J.; Rajakariar R.; Jacques T. S.; Hannon R.; Kanaoka Y.; Eguchi N.; Colville-Nash P.; Gilroy D. W. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowsey I. R.; Thomson A. M.; Flanagan J. U.; Murdock P. R.; Moore G. B. T.; Meyer D. J.; Murphy G. J.; Smith S. A.; Hayes J. D. Biochem. J. 2001, 359, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urade Y.; Eguchi N. Prostaglandins Other Lipid Mediators 2002, 68−69, 375. [DOI] [PubMed] [Google Scholar]
- Kanaoka Y.; Ago H.; Inagaki E.; Nanayama T.; Miyano M.; Kikuno R.; Fujii Y.; Eguchi N.; Toh H.; Urade Y.; Hayaishi O. Cell 1997, 90, 1085. [DOI] [PubMed] [Google Scholar]
- Inoue T.; Irikura D.; Okazaki N.; Kinugasa S.; Matsumura H.; Uodome N.; Yamamoto M.; Kumasaka T.; Miyano M.; Kai Y.; Urade Y. Nat. Struct. Biol. 2003, 10, 291. [DOI] [PubMed] [Google Scholar]
- A very similar lead has been disclosed by AstraZeneca, and Evotec, see refs 18 and 19: ; Hohwy M.; Spadola L.; Lundquist B.; Hawtin P.; Dahmen J.; Groth-Clausen I.; Nilsson E.; Persdotter S.; Wachenfeldt K. V.; Folmer R. H. A.; Edman K. J. Med. Chem. 2008, 51, 2178. [DOI] [PubMed] [Google Scholar]
- Hesterkamp T.; Barker J.; Davenport A.; Whittaker M. Curr. Top. Med. Chem. 2007, 7, 1582. [DOI] [PubMed] [Google Scholar]
- Aldous S. C.; Jiang J. Z.; Lu J.; Ma L.; Mu L.; Munson H. R.; Sabol J. S.; Thurairatnam S.; Vandeusen C. L. PCT Int. Appl. WO 2007041634, 2007.
- Aritake K.; Kado Y.; Inoue T.; Miyano M.; Urade Y. J. Biol. Chem. 2006, 281, 15277. [DOI] [PubMed] [Google Scholar]
- Blake T. D.; Hamper B. C.; Huang W.; Kiefer J. R.; Moon J. B.; Neal B. E.; Olson K. L.; Pelc M. J.; Schweitzer B. A.; Thorarensen A.; Trujillo J. I.; Turner S. R. U.S. Pat. Appl. US 2008146569, 2008.
- The importance of a hydrogen bond to a water molecule has been previously described; see refs (18) and (19).
- The detailed analysis of this will be published in due course.
- Naor M. M.; Jensen J. H. PROTEINS: Struct., Funct., Bioinf. 2004, 57, 799. [DOI] [PubMed] [Google Scholar]
- The Pfizer Institutional Animal Care and Use Committee reviewed and approved the animal use in all of these studies. The animal care and use program is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International.
- PC400 is the provocative concentration of carbachol that increases pulmonary resistance (rL) by 400%. Kasaian M. T.; Donaldson D. D.; Tchistiakova L.; Marquette K.; Tan X.-Y.; Ahmed A.; Jacobson B. A.; Widom A.; Cook T. A.; Xu X; Barry A. B.; Goldman S. J.; Abraham W. M. Am. J. Respir. Cell Mol. Biol. 2007, 36, 368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.