

Abstract

Novel oxazolobenzimidazoles are described as potent and selective positive allosteric modulators of the metabotropic glutamate receptor 2. The discovery of this class and optimization of its physical and pharmacokinetic properties led to the identification of potent and orally bioavailable compounds (20 and 21) as advanced leads. Compound 20 (TBPCOB) was shown to have robust activity in a PCP-induced hyperlocomotion model in rat, an assay responsive to clinical antipsychotic treatments for schizophrenia.
Keywords: Oxazolobenzimidazoles, allosteric modulators, metabotropic glutamate 2 receptor, hyperlocomotion model, schizophrenia, GPCR
Schizophrenia is a chronic, debilitating disorder affecting 1% of the world population.1−3 Current treatments target dopamine D2 and serotonin 5-HT2A receptors and are effective in treating the positive symptoms of schizophrenia (hallucinations and delusions) but offer limited efficacy for the negative symptoms (apathy, anhedonia, and social withdrawal) and do not improve cognitive deficit characteristic of this disease.4 In addition, patient use of some of the currently prescribed antipsychotic agents is accompanied by undesirable side effects including significant weight gain, hyperglycemia, diabetes, sedation, and various dyskinesias.5 Thus, there is a clear need for improved therapeutics with novel mechanisms of action.
l-Glutamate is the major excitatory neurotransmitter in the mammalian central nervouse system (CNS), and it is hypothesized that elevated glutamate transmission in the forebrain is associated with schizophrenia symptomatology. Therefore, a treatment that could reduce synaptic glutamate levels might be therapeutically beneficial for treating this disease.6,7 As glutamate levels are regulated presynaptically by group II mGlu receptors (mGluR2 and -3) as well as group III (mGluR4, -6, -7, and -8),8 these mGluR's have been identified as potential targets for novel treatments. Importantly, and in support of this “glutamate hypothesis”, (−)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY2140023, 1), a nonselective mGluR2/3 agonist prodrug (Figure 1), has demonstrated efficacy against both positive and negative symptoms in a 4 week phase IIb study in schizophrenia patients.9
Figure 1.

Dual mGluR2/3 agonist and prodrug.
Evidence from behavioral studies in mGluR2 and mGluR3 KO mice suggest that the antipsychotic activity of the mGluR2/3 agonist is derived from the activation of mGluR2;10,11 however, because of a high degree of homology in the orthosteric sites of group II mGlu receptors, selective mGluR2 agonists have not been discovered. An alternative approach takes advantage of the less homologous transmembrane domain of these receptors where positive allosteric modulators (PAMs) have been shown to bind selectively to mGluR2.12 In fact, a significant number of nonamino acid mGluR2-selective PAMs have been reported in the literature, by both Merck13 and others.15−17 Selective mGluR2 PAMs have several potential advantages over an orthosteric agonist: (1) avoidance of potential mGluR3-mediated effects, (2) reduced potential for tachyphylaxis, which has been observed with GPCR agonists upon chronic treatment,18 and (3) the selective activation of this receptor only in relevant tissues in the presence of endogenous agonist glutamate.
In this report, the discovery of a novel series of oxazolobenzimidazole mGluR2 PAMs is described. Following optimization of physical and pharmacokinetic properties, an advanced compound was demonstrated to be orally efficacious in a rat PCP-induced hyperlocomotion model predictive of antipsychotic potential.
In a previous publication,19 the HTS hit to lead optimization for an oxazolidinone series of mGluR2 PAMs was described and culminated in the identification of tert-butylphenoxymethyl oxazolidinone 2. Despite significant effort, further optimization of potency and pharmacokinetic properties proved elusive within this architecture. An important observation from the oxazolidinones structure−activity relationship (SAR) was that ortho substitution on the N-aryl ring was not tolerated, suggesting a preference for coplanarity between the oxazolidinone and the aryl ring. In this second generation approach, a central ring constraint was envisioned (Figure 2), leading to a proposed oxazolobenzimidazole core 3. It was postulated that the enforced coplanar disposition of the rings in this tricycle would mimic the bioactive conformation of 2 and further improve physical and pharmacokinetic properties in this series with its enhanced basicity and reduced rotatable bonds.20
Figure 2.

Proposed constraint of oxazolidinone lead to oxazolobenzimidazole.
A versatile and efficient chemical route to these oxazolobenzimidazoles was developed following literature precedent (Scheme 1). Alkylation of 4-tert-butyl phenol with (R)-epichlorohydrin provided the (S)-glycidyl phenyl ether 6 due to direct attack by the phenol on the epoxide carbon and not the one bearing the chloride, followed by subsequent ring closure to reform the terminal epoxide. Combination of 6 with 2-chloro-benzimidazole in the presence of base and heat directly afforded the desired oxazolobenzimidazole213 in moderate yield.
Scheme 1. Synthesis of 3.

Reagents and conditions: (a) NaOH, H2O, 64 °C, 47%. (b) Cs2CO3; EtOH, 23 °C, 54%.
Compounds were evaluated in a Chinese hamster ovary (CHO) cell line coexpressing recombinant human mGluR2 and a promiscuous G-protein, Gα16, using a fluorescent imaging plate reader (FLIPR) assay. Functional potency was expressed as the EC50 for potentiating a submaximal concentration of glutamate (EC20). In this assay, the newly synthesized oxazolobenzimidazole core was found to exhibit a 7-fold more potent EC50 than the corresponding leading oxazolidinone 2 even without the previously identified potency-enhancing CN group present (Figure 3).19
Figure 3.

Comparison of properties between 2 and 3.
Despite this initial success in potency optimization and the discovery of a novel mGluR2 PAM scaffold, oxazolobenzimidazole 3 possessed poor physical properties as highlighted by high logD and negligible aqueous solubility. Pharmacokinetic experiments in dog and rat further indicated that oxazolobenzimidazole 3 was a high clearance compound with a short half-life and poor microsomal stability. Given these challenges, further optimization was required to render this second generation series “druglike”, orally bioavailable, and amenable to in vivo efficacy studies.
In addition to our desire to lower logD and improve aqueous solubility, we set out to identify and impede sites of oxidative metabolism as an added strategy to improve pharmacokinetics. Using human liver microsomes and liquid chromatography−mass spectrometry analysis, primary metabolites were identified for 3 and indicated extensive oxidation on both ends of the molecule. Specifically, oxidation of both the tert-butyl group and the benzimidazole were prevalent. As a direct result of these observations, polar, electron-withdrawing substituents were envisioned at both ends of the molecule to simultaneously improve logD and solubility and to block metabolism.
An alternative synthetic route was devised that enabled the coupling of modified tert-butylphenols to a late stage oxazolobenzimidazole intermediate. Among the numerous phenols incorporated in this series, 11 and 16 proved to be uniquely valuable, and their synthesis is described below (Scheme 2). 2-Bromo-5-(trisiopropylsilyloxy)pyridine (8, R = TIPS) was reacted with an excess of copper(I) cyanide and the tert-butyl Grignard reagent to directly install the tert-butyl group in modest yield.22 Subsequent deprotection of the silyl ether 10 using tetrabutylammonium fluoride provided 2-tert-butyl-5-hydroxypyridine 11 in good yield. Alternatively, the analogous bromide 9 (R = Me) underwent metal halogen exchange and trap with dimethylacetamide to yield ketone 12. Trifluoromethane anion addition to 12 and mesylation provided intermediate 14 in low yield but on a large scale. Mesylate 14 was then reacted with trimethylaluminum to form the quaternary center in pyridylether 15.23 Sodium ethane thiolate in dimethyl formamide (DMF) at elevated temperature was utilized to provide the pyridyl phenol 16 in good yield.
Scheme 2. Synthesis of Phenols 11 and 14.

Reagents and conditions: (a) t-BuMgCl, CuCN, THF, −78 to 23 °C. (b) TBAF, THF, 27%. (c) n-BuLi, dimethylacetamide, −40 to 23 °C, 20%. (d) CF3SiMe3, LiOAc, DMF, 86%. (e) NaH, DMF then MsCl, 94%. (f) Me3Al, CH2Cl2, 0 to 23 °C, 37%. (g) NaSEt, DMF, 150 °C, 64%.
These phenols were appended to the oxazolobenzimidazole core (Scheme 3). Relying on extensive SAR established in the oxazolidinone-based PAMs,19 the inclusion of a nitrile group into the oxazolobenzimidazole was predicted to improve metabolic stability and potency. Thus, silyl-protected glycidol (17) was heated with 2-chloro-5-cyanobenzimidazole (18) in the presence of cesium carbonate under microwave irradiation to give the desired oxazolobenzimidazole as a mixture of nitrile regioisomers. This mixture of regioisomers required separation prior to silyl deprotection affording the oxazolobenzimazole alcohol 19 in high yield. Mitsunobu etherification with phenols 11 and 16 gave the desired oxazolobenzimidazoles 20 and 21, and this route proved amenable to multigram synthesis.
Scheme 3. Synthesis of Oxazolobenzimidazoles 20 and 21.

Reagents and conditions: (a) Cs2CO3, DMSO, 130 °C. (b) Separation of CN regioisomers, 36% desired isomer. (c) Et3N-3HF, MeCN, 37 °C, 73%. (d) PS-PPh3, DIAD, CH2Cl2, 65−85%.
The FLIPR EC50 values of glutamate potentiation were measured for 20 and 21 and were found to be 29 and 33 nM, respectively (Figure 4). It is important to note that these structural series are selective potentiators of mGluR2 and do not show activity (PAM, agonist, and antagonist) against other mGlu receptors (mGluRs 1, 3, 4, 5, and 6). On the basis of the synthesis of other analogues, the general SAR established that incorporation of the pyridyl nitrogen caused a 10-fold loss in potency as measured by FLIPR, and this was balanced by a 3-fold potency enhancement brought by the nitrile group. Polarity was generally not tolerated around the phenol, and the bulky tert-butyl group likely provided a hydrophobic shielding effect to allow for the polar pyridine nitrogen without further loss in potency. These two groups proved symbiotic as will later be shown. The trifluoromethyl group was a neutral substitution for methyl in FLIPR potency, but its impact was further recognized in subsequent assays.
Figure 4.

In vitro properties for oxazolobenzimidazoles 20 and 21.
Modest agonism was observed for 20 (EC50 = 700 nM) and 21 (EC50 = 730 nM); however, the percent activation relative to a maximum response to glutamate was <30% for both optimized oxazolobenzimidazoles. The cooperativity of these leads to shift glutamate potency was measured in FLIPR. This “left shift” of gluatmate's potency in FLIPR was reported as a fold shift, and a 1 μM concentration of oxazolobenzimidazoles 20 and 21 produced an increase in the EC50 of glutamate by 9- and 14-fold, respectively.
The standard assays to measure physical properties were also employed, and in comparison to the leading oxazolobenzimidazole 3, these optimized PAMs showed significantly improved measured log D values and aqueous solubility. One potential limitation in this series was the consistent observation of high plasma protein binding, which may limit the available free drug in the brain upon dosing. While P-gp susceptibility was not observed for most members of this structural class, it was confirmed that neither of these structures were P-gp substrates for rat or human.
On the basis of these in vitro assays, oxazolobenzimidazoles 20 and 21 are among the more potent mGluR2 PAMs published in the literature. Additionally, with the strategic installation of the nitrile and pyridine nitrogen, this series gained druglike physical properties while retaining potential for CNS penetration. These alterations led to an improvement in pharmacokinetic properties.
Oxazolobenzimidazoles 20 and 21 were dosed as single compounds to both rat and dog in standard IV/PO dosing protocols (Table 1). The installation of the polar, electron-withdrawing groups into leading compound 3 produced improvements in clearance, the area under the curve (AUC), and oral bioavailability.
Table 1. Pharmacokinetic Properties of Oxazolobenzimidazoles 3, 18, and 19.
| compd | species | dose (mpk) | route | vehicle | n | half-life (h) | AUC (μM h) | clearance (mL/min/kg) | oral F (%) | volume distribution (L/kg) |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | dog | 0.5 | iv | DMSO | 2 | 2.1 | 0.78 | 45.9 | 3.40 | |
| 3 | rat | 2 | iv | DMSO | 2 | 1.5 | 1.02 | 103 | 6.90 | |
| 20 | dog | 0.5 | iv | DMSO | 2 | 1.7 | 14.8 | 1.67 | 0.22 | |
| 20 | dog | 1 | po | 20% Vit E/TPGS | 2 | 9.48 | 32.1 | |||
| 20 | rat | 2 | iv | DMSO | 2 | 0.5 | 3.64 | 29.0 | 1.50 | |
| 20 | rat | 30 | po | 20% Vit E/TPGS | 2 | 27.2 | 49.7 | |||
| 21 | dog | 0.5 | iv | DMSO | 2 | 2.0 | 2.38 | 8.85 | 2.05 | |
| 21 | dog | 1.0 | po | 20% Vit E/TPGS | 2 | 3.64 | 76.5 | |||
| 21 | rat | 2 | iv | DMSO | 2 | 3.3 | 9.11 | 9.05 | 2.65 | |
| 21 | rat | 10 | po | 20% Vit E/TPGS | 2 | 27.3 | 60.0 |
When compared to the parent compound 3, oxazolobenzimidazoles 20 and 21 had improved clearance and AUC in iv-dosed experiments. These reduced clearances, along with the improved physical properties of these novel mGluR2 PAMs, led to appreciable oral bioavailability in both species. Interestingly, when metabolite identification was again carried out for 20, the primary site of oxidation was the tert-butyl group. mGluR2 PAM 21 was designed to minimize this metabolic pathway via the installation of the trifluoromethyl group, and this change led to further improvements in pharmacokinetics. The achievement of oral bioavailability enabled the use of oral dosing for the assessment of efficacy in a rat behavioral model of antipsychotic activity.
Clinically approved typical and atypical antipsychotics and mGluR2/3 agonists have been found to block the hyperlocomotive effects in rodents produced by psychostimulants, such as PCP, and this assay is believed to be predictive of antipsychotic potential. This model was therefore used here to evaluate the effect of the mGluR2 PAM 20 (Figure 5). Sprague−Dawley rats were habituated for 30 min and then dosed with an oxazolobenzimidazole (20) or vehicle and returned to the chambers. After another 30 min, rats were dosed with either PCP (5.0 mg/kg, sc) or vehicle (sc), and the activity was recorded for 90 min. Following behavioral testing, plasma and CSF were collected from a subset of animals to determine drug levels in these matrices.
Figure 5.
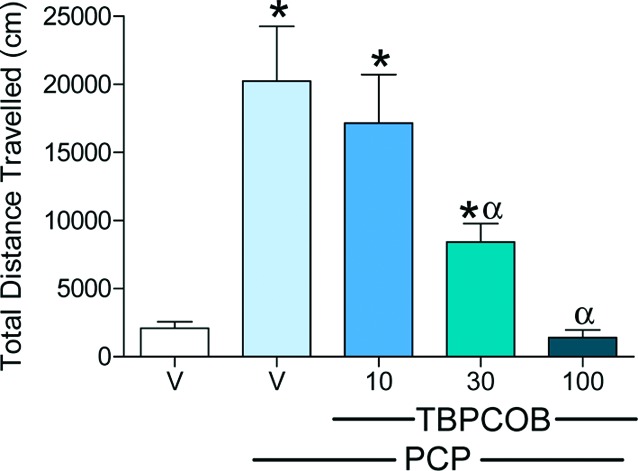
Locomotor response to PCP (5.0 mpk; sc) over the 90 min session as a function of treatment with TBPCOB 20 (10, 30, or 100 mpk; po) or vehicle (20% Vit E TPGS) in male Sprague−Dawley rats (250−300 g) that had been pair-housed in standard laboratory conditions. Rats were placed in activity monitors and, after 30 min of habituation, given either vehicle or 20 and placed back in activity monitors. After an additional 30 min, animals were given an injection of saline or PCP. Distance traveled over the following 90 min was summed.
The mGluR2 selective PAM tert-butyl phenyloxymethyl cyano oxazolobenzimidazole (TBPCOB, 20) fully inhibited the effect of PCP at 100 mg/kg po, a dose that resulted in plasma and CSF levels of 86 and 1.5 μM, respectively. The intermediate dose of 30 mg/kg also significantly attenuated the PCP response (CSF = 550 nM), whereas the 10 mg/kg dose failed to produce a significant effect (CSF = 70 nM). Importantly, this stands as one of a few published examples of mGluR2 PAM efficacy15−17,24−26 in a rodent model sensitive to clinically therapeutic antipsychotics and is notable given the full efficacy observed with oral dosing. These data support the notion that a selective mGluR2 PAM has antipsychotic potential similar to that seen with an mGluR2/3 agonist.27 The observed in vivo efficacy of TBPCOB (20) is believed to be driven by mGluR2 potentiation given that intrinsic agonism was determined to be low (23% maximial activation and weak EC50 = 700 nM). Rat FLIPR potency was also obtained for 20 (EC50 = 42 nM, 77% max) and found to be similar to the human potency in further support of this notion. In addition, TBPCOB (20) showed no activity against other mGluR's and did not have binding affinity for targets that could produce inhibition of PCP-induced hyperlocomotion in this rat behavioral model (D2 dopamine or 5-HT2A receptors).
In summary, an HTS-derived lead oxazolidinone was transformed into a novel series of oxazolobenzimidazole mGluR2 PAMs using a ring-constraint strategy. While potency was significantly enhanced, the physical properties and pharmacokinetics of this new series were initially poor. A combined strategy of improving physical properties and blocking sites of metabolism led to dramatic improvements in the overall profile of these optimized compounds. Robust efficacy in the rat hyperlocomotion model was observed with oral dosing of TBPCOB (20), and importantly, full inhibition of the PCP-induced hyperactivity was achievable. Oxazolobenzimidazoles 20 and 21 therefore represent optimized leads with druglike properties that have potential therapeutic value for the treatment of schizophrenia.
Acknowledgments
We thank Joan Murphy and Dr. Charles Ross III for HR-MS analysis.
Supporting Information Available
Additional experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Regier D. A.; Narrow W. E.; Rae D. S.; Manderscheid R. W.; Locke B. Z.; Goodwin F. K. The de Facto US Mental and Addictive Disorders Service System: Epidemiologic Catchment Area Prospective 1-Year Prevalence Rates of Disorders and Services. Arch. Gen. Psychiatry 1993, 50, 85. [DOI] [PubMed] [Google Scholar]
- Tandon R.; Keshavan M. S.; Nasrallah H. A. Schizophrenia, “Just the Facts”: what we know in 2008 part 1: overview. Schizophr. Res. 2008, 100, 4. [DOI] [PubMed] [Google Scholar]
- Tandon R.; Keshavan M. S.; Nasrallah H. A. Schizophrenia, “just the facts” what we know in 2008. 2. Epidemiology and etiology. Schizophr. Res. 2008, 102, 1. [DOI] [PubMed] [Google Scholar]
- Murphy B. P.; Chung Y. C.; Park T. W.; McGorry P. D. Pharmacological treatment of primary negative symptoms in schizophrenia: a systematic review. Schizophr. Res. 2006, 88, 5. [DOI] [PubMed] [Google Scholar]
- Lieberman J. A.; Stroup T. S.; McEvoy J. P.; Swartz M. S.; Rosenheck R. A.; Perkins D. O.; Keefe R. S.; Davis S. M.; Davis C. E.; Lebowitz B. D.; Severe J.; Hsiao J. K. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 2005, 353, 1209. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega L. E.; Schaffhauser H.; Campbell U. C. Metabotropic Glutamate Receptors: Potential Drug Targets for the Treatment of Schizophrenia. Curr. Drug Targets: CNS Neurol. Dis. 2002, 1, 261. [DOI] [PubMed] [Google Scholar]
- Carmell J.; Perry K. W.; Salhoff C. R.; Monn J. A.; Schoepp D. D. The Potent, Selective mGlu2/3 Receptor Agonist LY379268 Increases Extracellular Levels of Dopamine, 3,4-Dihydroxyphenylacetic Acid, Homovanillic Acid, and 5-Hydroxyindole-3-Acetic Acid in the Medial Prefrontal Cortex of the Freely Moving Rat. J. Neurochem. 2000, 75, 1147. [DOI] [PubMed] [Google Scholar]
- Macek T. A.; Winder D. G.; Gereau R. W.; Ladd C. O.; Coon P. J. Differential involvement of group II and group III mGluRs as autoreceptors at lateral and medial perforant path synapse. J. Neurophysiol. 1996, 76, 3798. [DOI] [PubMed] [Google Scholar]
- Patil S. T.; Zhang L.; Martenyi F.; Lowe S. L.; Jackson K. A.; Andreev B. V.; Avedisova A. S.; Bardenstein L. M.; Gurovich I. Y.; Morozova M. A.; Mosolov S. N.; Neznanov N. G.; Reznik A. M.; Smulevich A. B.; Tochilov V. A.; Johnson B. G.; Monn J. A.; Scheopp D. D. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat. Med. 2007, 13, 1102. [DOI] [PubMed] [Google Scholar]
- Wooley M. L.; Pemberton D. J.; Bate S.; Corti C.; Jones D. M. The mGlu2 but not the mGlu3 receptor mediates the actions of the mGluR2/3 agonist, LY379268, in mouse models predictive of antipsychotic activity. Psycopharmacology 2008, 196, 431. [DOI] [PubMed] [Google Scholar]
- Fell M. J.; Svensson K. A.; Johnson B. G.; Schoepp D. D. Evidence for the Role of Metabotropic Glutamate (mGlu)2 Not mGlu3 Receptors in the Preclinical Antipsychotic Pharmacology of the mGlu2/3 Receptor Agonist (−)-(1R,4S,5S,6S)-4-Amino-2-sulfonylbicyclo[3.1.0]hexane-4,6-dicarboxylic Acid (LY404039). J. Pharmacol. Exp. Ther. 2008, 326, 209. [DOI] [PubMed] [Google Scholar]
- Conn J. P.; Chirstopolous A.; Lindsley C. W. A Novel Selective Positive Allosteric Modulator of Metabotropic Glutamate Receptor Subtype 5 Has in Vivo Activity and Antipsychotic-Like Effects in Rat Behavioral Models. Nat. Rev. Drug Discovery 2009, 8, 41. [DOI] [PubMed] [Google Scholar]
- Cube R. V.; Vernier J. M.; Hutchinson J. H.; Gardner M. F.; James J. K.; Rowe B. A.; Schaffhauser H.; Daggett L.; Pinkerton A. B.. 3-(2-Ethoxy-4-{4-[3-hydroxy-2-methyl-4-(3-methylbutanoyl)phenoxy]butoxy}phenyl)propanoic acid: a brain penetrant allosteric potentiator at the metabotropic glutamate receptor 2 (mGluR2). Bioorg. Med. Chem. Lett. 2005, 14, 2389 and references therein. [DOI] [PubMed] [Google Scholar]
- Duplantier A. J.; Efremov I.; Candler J.; Doran A. C.; Ganong A. H.; Haas J. A.; Hanks A. N.; Kraus K. G.; Lazzaro J. T.; Lu J.; Maklad N.; McCarthy S. A.; O'Sullivan T. J.; Rogers B. N.; Siuciak J. A.; Spraklin D. K.; Zhang L. B. 3-Benzyl-1,3-oxazolidin-2-ones as mGluR2 positive allosteric modulators: Hit-to lead and lead optimization. Bioorg. Med. Chem. Lett. 2009, 19, 2524. [DOI] [PubMed] [Google Scholar]
- Tresadern G.; Cid J. M.; Macdonald G. J.; Vega J. A.; de Lucas A. I.; Garcia A.; Matesanz E.; Linares M. L.; Oehlrich D.; Lavreysen H.; Biesmans I.; Trabanco A. A.. Scaffold hopping from pyridones to imidazo [1, 2-a] pyridines. New positive allosteric modulators of metabotropic glutamate 2 receptor. Bioorg. Med. Chem. Lett. 2009, 175. [DOI] [PubMed] [Google Scholar]
- Fraley M. E.Positive allosteric modulators of the metabotropic glutamate receptor 2 for the treatment of schizophrenia. Expert Opin. Ther. Pat. 2009, 19, 1259 and references therein. [DOI] [PubMed] [Google Scholar]
- Pin J. P; Parmentier M. L.; Prezeau L. Positive Allosteric Modulators for γ-Aminobutyric AcidB Receptors Open New Routes for the Development of Drugs Targeting Family 3 G-Protein-Coupled Receptors. Mol. Pharmacol. 2001, 60, 881. [DOI] [PubMed] [Google Scholar]
- Brnardic E. J.; Fraley M. E.; Garbaccio R. M.; Layton M. E.; Sanders J. M.; Culberson C.; Jacobson M. A.; Magliaro B. C.; Hutson P. H.; O'Brien J. A.; Huszar S. L.; Uslaner J. M.; Fillgrove K. L.; Tang C.; Kuo Y.; Sur S. M.; Hartman G. D.. 3-Aryl-5-phenoxymethyl-1, 3-oxazolidin-2-ones as Positive Allosteric Modulators of mGluR2 for the Treatment of Schizophrenia: Hit-to-Lead Efforts. Bioorg. Med. Chem. Lett. 2010, 3129. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615. [DOI] [PubMed] [Google Scholar]
- Benigni F.; Trevisan L. Synthesis of a new nitrogenous heterocycle, 2H,3H-benzimidazo[1,2-b]oxazole. Farmaco, Edizione Scientifica 1974, 12, 936. [PubMed] [Google Scholar]
- Bell T. W.; Hu L. Y.; Patel S. V. Alkylation of heteroaryl halides by 2:1 Grignard reagent/copper(I) mixtures. Synthesis of alkylated octahydrodibenzo[b,j][1,10]phenanthrolines. J. Org. Chem. 1987, 52, 3847. [Google Scholar]
- Tanaka H.; Shishido Y. Synthesis of aromatic compounds containing a 1,1-dialkyl-2-trifluoromethyl group, a bioisostere of the tertalkyl moiety. Bioorg. Med. Chem. Lett. 2007, 17, 6079. [DOI] [PubMed] [Google Scholar]
- Pinkerton A. B.; Vernier J.-M.; Schaffhauser H.; Rowe B. A.; Campbell U. C.; Rodriguez D. E.; Lorrain D. S.; Baccei C. S.; Daggett L. P.; Bristow L. J. Phenyl-tetrazolyl acetophenones: discovery of positive allosteric potentiatiors for the metabotropic glutamate 2 receptor. J. Med. Chem. 2004, 47, 4595. [DOI] [PubMed] [Google Scholar]
- Galici R.; Echermendia N. G.; Rodriguez A. L.; Conn J. P. A Selective Allosteric Potentiator of Metabotropic Glutamate (mGlu) 2 Receptors Has Effects Similar to an Orthosteric mGlu2/3 Receptor Agonist in Mouse Models Predictive of Antipsychotic Activity. J. Pharmacol. Exp. Ther. 2005, 315, 1181. [DOI] [PubMed] [Google Scholar]
- Galici R.; Jones C. K.; Hemstapat K.; Nong Y.; Echemendia N. G.; Williams L. C.; de Paulis T.; Conn J. P. Biphenyl-indanone A, a Positive Allosteric Modulator of the Metabotropic Glutamate Receptor Subtype 2, Has Antipsychotic- and Anxiolytic-Like Effects in Mice. J. Pharmacol. Exp. Ther. 2006, 318, 173. [DOI] [PubMed] [Google Scholar]
- Rorick-Kehn L. M.; Johnson B. G.; Knitowski K. M.; Salhoff C. R.; Witkin J. M.; Perry K. W.; Griffey K. I.; Tizzano J. P.; Monn J. A.; McKinzie D. L.; Schoepp D. D. In vivo pharmacological characterization of the structurally novel, potent, selective mGlu2/3 receptor agonist LY404039 in animal models of psychiatric disorders. Psychopharmacology 2007, 193, 121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.