

Abstract

Inhibition of mitotic kinesins represents a novel approach for the discovery of a new generation of anti-mitotic cancer chemotherapeutics. We report here the discovery of the first potent and selective inhibitor of centromere-associated protein E (CENP-E) 3-chloro-N-{(1S)-2-[(N,N-dimethylglycyl)amino]-1-[(4-{8-[(1S)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl}phenyl)methyl]ethyl}-4-[(1-methylethyl)oxy]benzamide (GSK923295; 1), starting from a high-throughput screening hit, 3-chloro-4-isopropoxybenzoic acid 2. Compound 1 has demonstrated broad antitumor activity in vivo and is currently in human clinical trials.
Keywords: CENP-E, inhibitor, GSK923295, mitotic kinesin
Kinesins are a superfamily of motor proteins that utilize the energy from ATP hydrolysis to transport cellular cargoes along microtubules.1−3 Mitotic kinesins are a functional class of kinesins essential for mitotic spindle assembly and function during cell division.4,5 Perturbation of mitotic kinesin activity results in cell cycle arrest in mitosis and subsequent cell death.6 The past decade has seen an increased interest in the development of small-molecule inhibitors of mitotic kinesins as a new generation of antimitotic agents.7−10 Since these agents specifically target dividing cells, mitotic kinesin inhibitors have the potential of capturing the therapeutic benefits of antimicrotubule (MT) agents, such as the taxanes, while minimizing toxicities on nondividing cells, thereby mitigating side effects such as peripheral neuropathies.6−10 The most advanced mitotic kinesin inhibitors in clinical development target kinesin spindle protein (KSP or HsEg5), a mitotic kinesin required for spindle pole separation during prometaphase.7−13 Centromere-associated protein E (CENP-E) is a mitotic kinesin directly involved in coupling the mechanics of mitosis with the mitotic checkpoint signaling machinery, regulating the cell-cycle transition from metaphase to anaphase.14−17 During mitosis, CENP-E is localized to the region of mitotic chromosomes responsible for interaction with spindle microtubules and it is essential for prometaphase chromosome movements that contribute to metaphase chromosome alignment. Disruption of CENP-E function using a variety of methods, including antibody microinjection and ablation of gene expression with siRNA, induces mitotic arrest and a cellular phenotype characterized by misaligned chromosomes arrayed on bipolar spindles, and leads to subsequent cell death.18−23
From a high-throughput screen of a 700K-member small molecule compound library looking for inhibitors of the microtubule-stimulated ATPase activity of CENP-E, we identified a low-molecular-weight fragment (benzoic acid 2, Figure 1) with a biochemical IC50 of 6.7 μM and no detectable cellular effect at 40 μM. Although we were unable to pursue a typical fragment based optimization approach utilizing X-ray crystallography or NMR,24 the good ligand binding efficiency25,26 (ΔG/number of non-hydrogen atoms, LE = 0.50), selectivity vs other kinesins, and structural features amenable to rapid creation of analogues made 2 an attractive starting point for further optimization. Reasoning that the lack of cellular activity might be due to the poor permeability related to the presence of the carboxylate, a small library of amide analogues was prepared by coupling 2 with a set of amino acid derivatives bearing a variety of side chains and different C-terminal capping groups.27 Representative examples are shown in Table 1. Compounds 3g and 3h, which contain a benzyl group side chain and a primary or methyl amide C-terminus, were found to have IC50 values similar to screening hit 2. The simple glycine amide analogue (3a) was inactive, as were analogues with side chains containing a simple alkyl chain (3b), H-bond donors or acceptors (3c and 3d), or benzyl group homologues (3e and 3f). A tertiary amide (3i) and methyl ester (3j) at the C-terminus also rendered compounds inactive. Carboxylic acid analogue 3k retained some potency but was 10-fold less active than 3g.
Figure 1.

Structures of 1 and 2.
Table 1. Biochemical Activity of Benzamide Analogues.
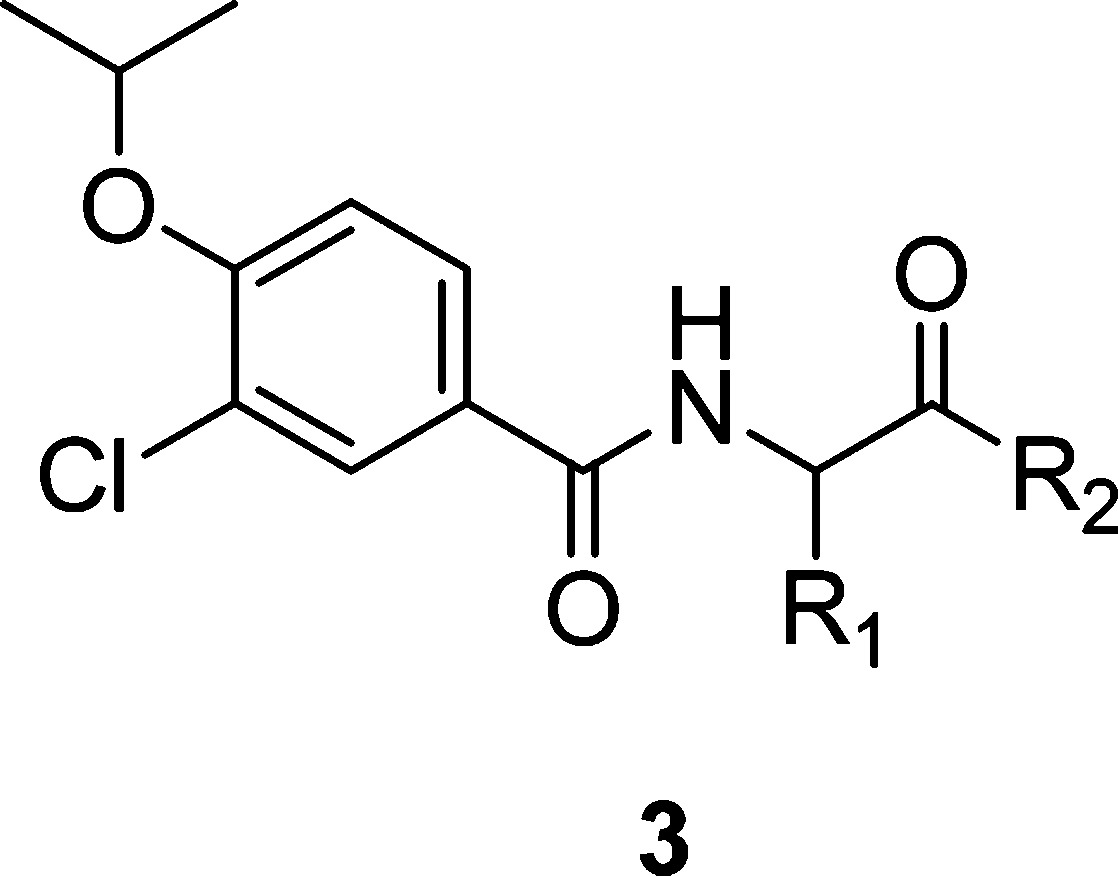
| compd | R1 | R2 | CENP-E IC50 (μM) |
|---|---|---|---|
| 3a | H | NHMe | >100 |
| 3b | iPr | NHMe | >100 |
| 3c | CH2OH | NHMe | >100 |
| 3d | CH2CONH2 | NHMe | >100 |
| 3e | Ph | NHMe | >100 |
| 3f | CH2CH2Ph | NHMe | >100 |
| 3g | CH2Ph | NHMe | 6.5 |
| 3 h | CH2Ph | NH2 | 5.4 |
| 3i | CH2Ph | N(Me)2 | >100 |
| 3j | CH2Ph | OMe | >100 |
| 3k | CH2Ph | OH | 61 |
With the identification of the phenylalaninamide as a new active scaffold, we explored the substitution on the side chain phenyl group to introduce further structural diversity. A systematic Topliss scan28 revealed that substitution was tolerated roughly equally at all positions with either electron-withdrawing or electron-donating groups (4a−4f) as shown in Table 2. Substitution with a larger phenyl group was dramatically more sensitive. A phenyl ring appended at the 4-position (4g) improved the biochemical potency by 10-fold whereas the same substitution at the 2- and 3-positions (4h and 4i) dramatically attenuated potency. Encouragingly, 4g also showed the first sign of antiproliferative effect in the SKOV-3 human ovarian carcinoma cell line with an IC50 of 6.2 μM. In light of these results, we investigated heterocyclic substitution at the 4-position of the phenyl ring as a means to optimize physicochemical properties while further improving biochemical and cellular activity. An imidazolyl group linked via the 2- or 4-position was found to be the most active among a variety of five- and six-membered heterocycles explored. In going from a modestly potent methyl substituent (4j) to a bulky tert-butyl group (4k and 4l) or fusion with a phenyl ring (i.e., benzimidazolyl, 4m), biochemical and cellular potencies were further improved.
Table 2. Biological Activity of Phenylalanine Amide Analogues.
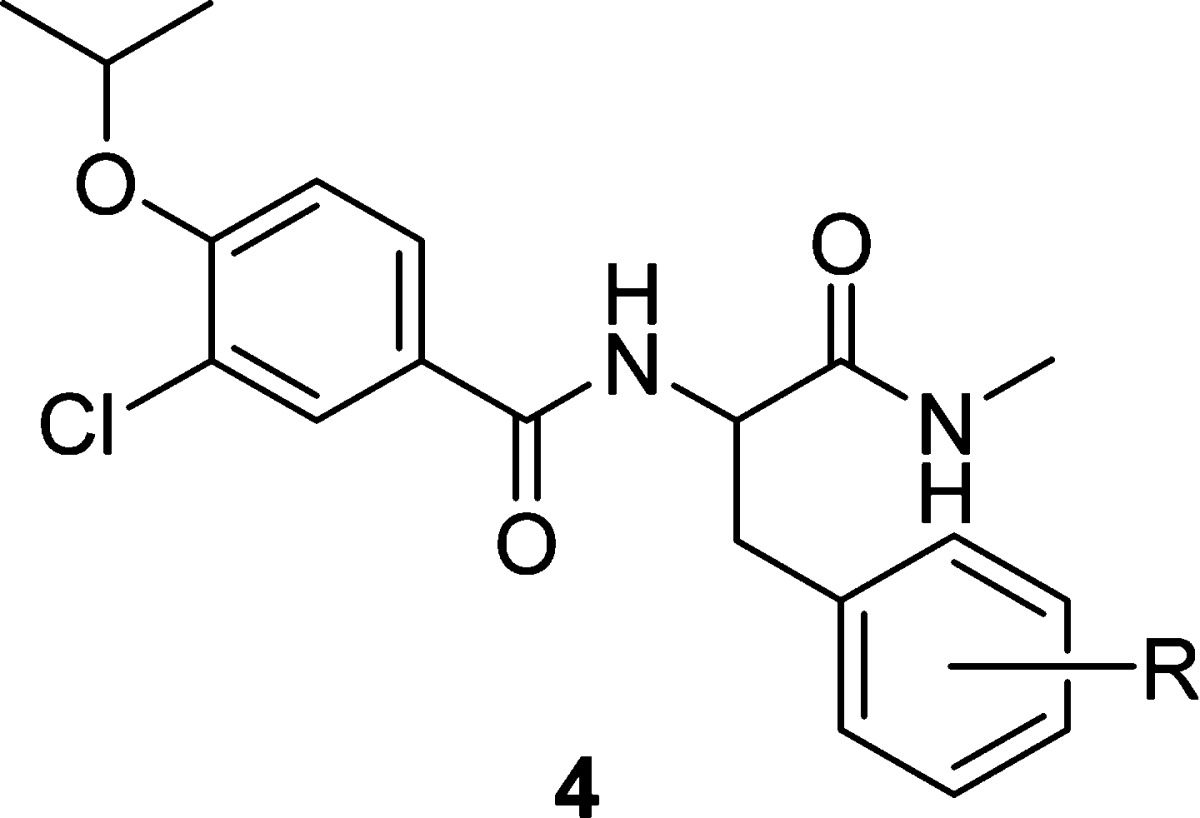
| compd | R | CENP-E IC50 (μM) | SKOV-3 IC50 (μM) |
|---|---|---|---|
| 3g | H | 6.5 | >25 |
| 4a | 4-OH | 3.3 | >25 |
| 4b | 3-OH | 6.2 | >25 |
| 4c | 2-OH | 8.8 | >25 |
| 4d | 4-F | 9.7 | >25 |
| 4e | 4-Cl | 7.2 | >25 |
| 4f | 4-Me | 5.4 | >25 |
| 4g | 4-Ph | 0.36 | 6.2 |
| 4h | 3-Ph | 36 | >25 |
| 4i | 2-Ph | >100 | >25 |
| 4j | 4-(2-Me)-imidazol-4-yl | 1.0 | >25 |
| 4k | 4-(2-tBu)-imidazol-4-yl | 0.13 | 1.2 |
| 4l | 4-(4-tBu)-imidazol-2-yl | 0.19 | 4.4 |
| 4m | 4-benzimidazol-2-yl | 0.066 | 3.1 |
Having a potent compound (4m) in hand, we examined whether there was a stereochemical preference for binding to CENP-E. Both (S)- and (R)-enantiomers of 4m were synthesized using enantiomerically pure starting materials, and final chiral purity was >98% ee by chiral HPLC for each antipode. As shown in Table 3, the (S)-enantiomer (5a) was >400 times more potent than the (R)-enantiomer (5b), demonstrating a pronounced stereochemical bias at the binding pocket for the (S)-antipode. This stereoselectivity translated well into cellular activity as cells treated with 5a displayed the characteristic mitotic arrest phenotype (bipolar spindles with misaligned chromosomes), whereas 5b showed no effect, providing strong support for an on-target mechanism for inhibition of cell proliferation based on the inhibition of CENP-E motor protein.
Table 3. Stereoselectivity of Phenylalanine Amide Analogues.
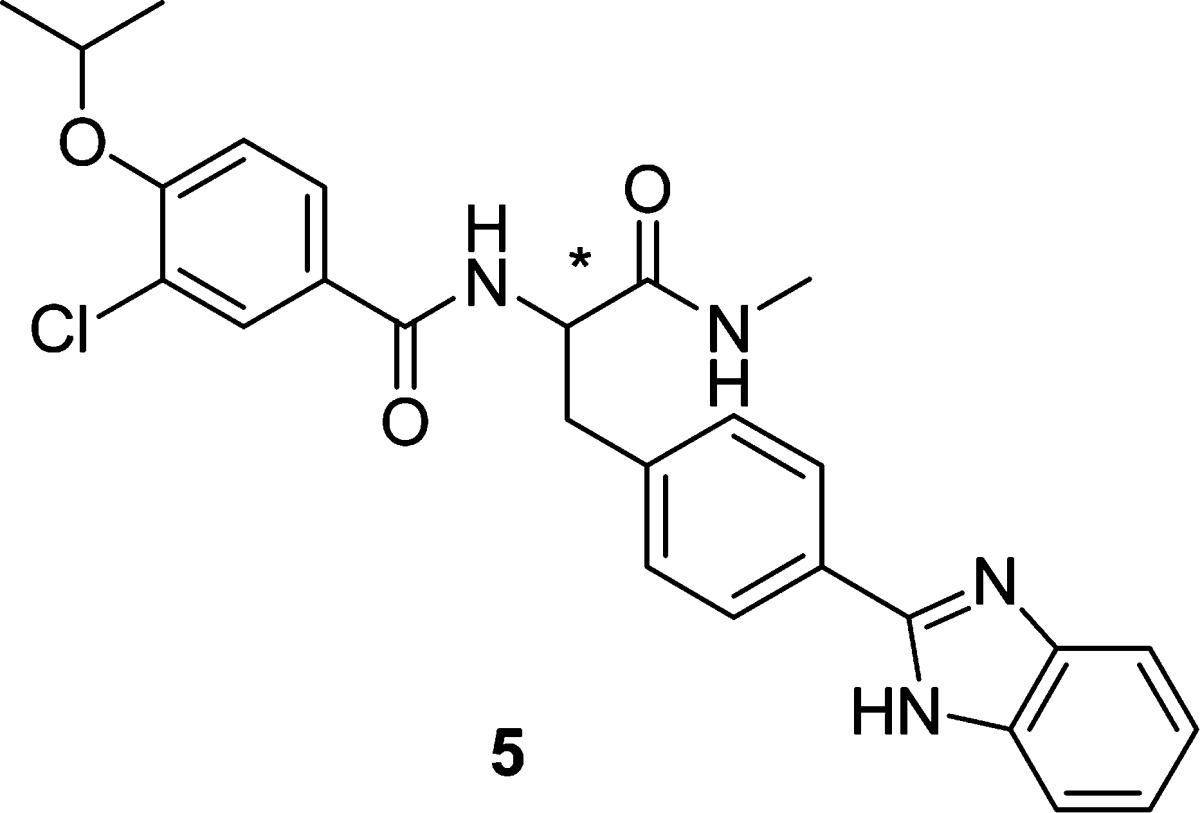
| compd | stereo | CENP-E IC50 (μM) | SKOV-3 IC50 (μM) |
|---|---|---|---|
| 5a | S | 0.032 | 2.0 |
| 5b | R | 13.8 | >20 |
Although the biochemical IC50 was greatly improved upon incorporation of a biaryl group in the side chain, the cellular activity of these compounds was still 10−100-fold below their biochemical potency. Our efforts shifted to modification of the C-terminus in an attempt to narrow this differential. Reduced C-terminal groups such as hydroxymethyl (6a and 6b), aminomethyl (6c) and aminoethyl (6e) gave modest improvements to both the biochemical and cellular potency (Table 4). Remarkably, however, a one-carbon extension to a hydroxyethyl group (6d) significantly improved cellular potency (SKOV-3 IC50 = 94 nM), with only a modest improvement in biochemical activity. Detailed mechanistic studies of 6d and 4g revealed that there was a significant difference in their modes of inhibition. Steady-state kinetic studies showed that 6d was uncompetitive with ATP, while 4g was ATP-competitive. Since the ATP concentration in cells is much higher than that used in the biochemical assay (500 μM), this could help explain the disparity between the biochemical and cellular activities for ATP-competitive inhibitors such as 4g.
Table 4. Effect of C-Terminal Modification on Biological Activity.
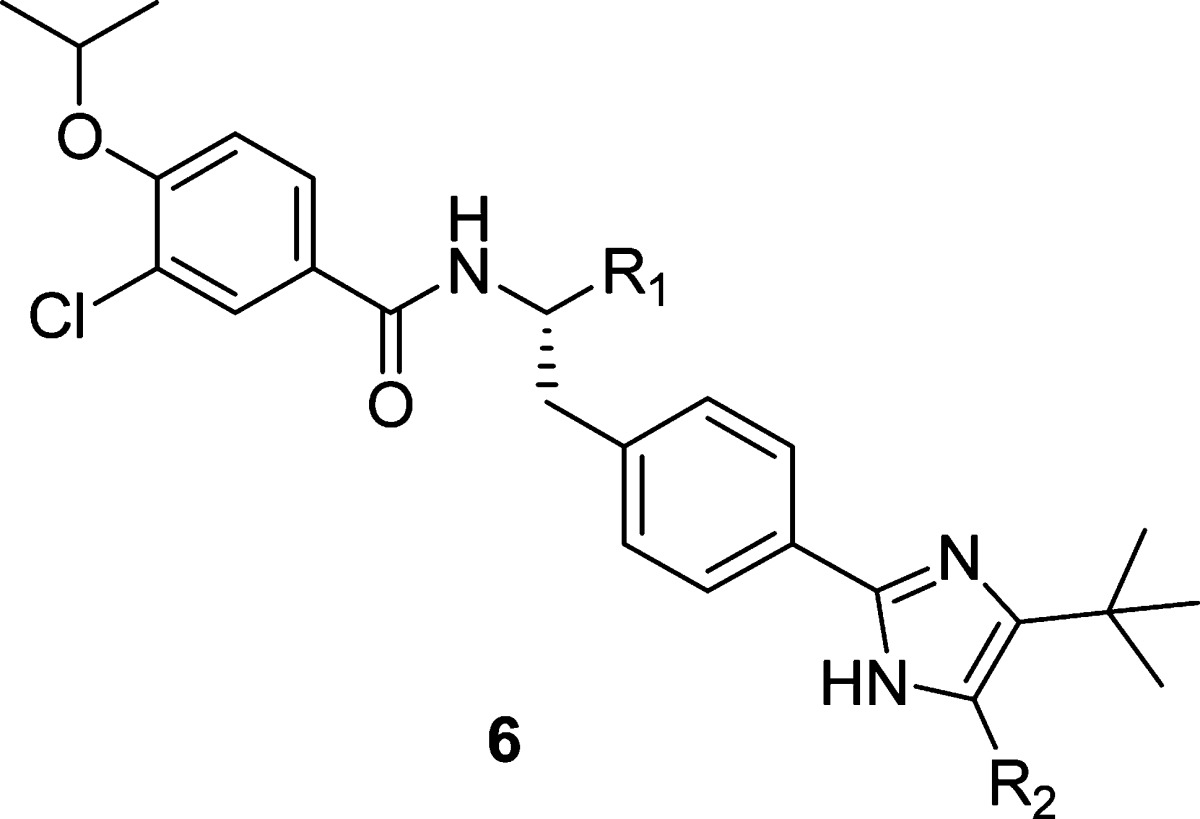
| compd | R1 | R2 | CENP-E IC50 (μM) | SKOV-3 IC50 (μM) |
|---|---|---|---|---|
| 4l | CONHCH3 | H | 0.19 | 4.4 |
| 6a | CH2OH | H | 0.056 | 2.7 |
| 6b | CH2OH | Me | 0.043 | 1.1 |
| 6c | CH2NH2 | Me | 0.050 | 1.4 |
| 6d | CH2CH2OH | Me | 0.022 | 0.094 |
| 6e | CH2CH2NH2 | Me | 0.10 | 7.2 |
We postulate that both 4g and 6d interact with the enzyme in an allosteric binding cleft adjacent to the ATP binding site and that the relatively minor structural modification perturbs the ATP pocket such that a change in inhibitor modality is observed.29 Most importantly, the improved cellular activity proved to be general with the incorporation of the hydroxyethyl into the inhibitor template and fueled further exploration.
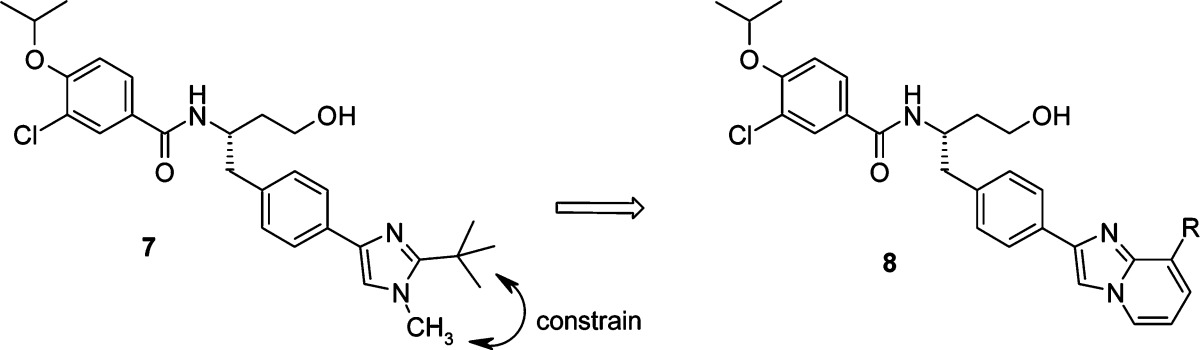
Maintaining the hydroxyethyl moiety, we further explored substitution of the side chain phenyl group. Transposition of one of the imidazole nitrogens to give isomeric imidazole 7 showed no advantage. However, constraining the methyl and tert-butyl groups to form an imidazopyridine ring (8a) resulted in a significant increase in biochemical and cellular activity (Table 5). Unfortunately, 8a was much less soluble,30 and since our goal was to identify a drug that could be administered intravenously, maximizing aqueous solubility was crucial. Encouragingly, the addition of a hydroxyl group (8b) to the imidazopyridine side chain restored the solubility with no attenuation of biochemical and cellular potency.31
Table 5. Biological Activity of Selected Imidazopyridine Analogues.
| compd | R | CENP-E IC50 (nM) | SKOV-3 IC50 (nM) | sol30 (μM) |
|---|---|---|---|---|
| 7 | 56 | 41 | 23 | |
| 8a | Me | 3 | 15 | 1 |
| 8b | (S)-CH(CH3)OH | 7 | 14 | 21 |
Although significant gains in enzyme and cellular potency had been realized, the rat pharmacokinetic (PK) profiles of these analogues were characterized by high clearances and short half-lives. In an attempt to rationalize the underlying poor PK, the in vitro metabolism of selected analogues in rat and human hepatocytes and microsomes was investigated. The results indicated that hydroxylation of the biaryl side chain and oxidation of the primary hydroxyl group were the most prevalent metabolic pathways, and derivatives that incorporated modifications likely to reduce or block these putative metabolic sites were targeted.
Modification of the side chain, including the incorporation of fluorine atoms at various positions on the side chain aryl groups, had little effect on PK.32 We next examined alternatives to the C-terminus hydroxyethyl group, keeping in mind that maintaining the ATP-uncompetitive mechanism of inhibition would be necessary for retention of good cellular activity. Although various cyclic and acyclic groups maintained high levels of enzyme and cellular potency, substituted glycinamides also provided a moderate boost in exposure coupled with significantly improved solubility. These efforts ultimately led to the identification of GSK923295 (1), a potent inhibitor of human CENP-E.27,33,34
Under steady-state kinetic conditions, 1 behaves as an uncompetitive inhibitor of both ATP and MT (Table 6). To ascertain its kinesin selectivity, 1 was evaluated against a panel of mitotic human kinesins and showed only minimal inhibitory activity (<25%) at 50 μM.
Table 6. Biological Activity Profile of 1.
| hCENP-E Ki | 3 nM | |
| mode of inhibition | ATP | uncompetitive |
| MT | uncompetitive | |
| kinesin selectivitya | <25% inhibition @ 50 μM | |
| cell IC50 | SKOV-3 | 22 nM |
| Colo205 | 22 nM | |
Panel: KSP, Kif1A, MKLP1, RabK6, HSET, MCAK, Kif4.
Compound 1 exhibits inhibition of cell proliferation in a broad panel of human solid tumor and hematological cell lines and induces mitotic arrest leading to apoptosis and cell death.33,34 Consistent with CENP-E inhibition, cells treated with the drug display a phenotype characterized by bipolar mitotic spindles with misaligned chromosomes.
Compound 1 also showed dose-dependent activity in a Colo205 human tumor xenograft efficacy model.35−37 Mice were administered 1 as a single dose three consecutive days per week for two weeks (6 total doses) resulting in significant effects with tumor regression observed for the top doses (125 and 250 mg kg−1) (Figure 2). In other studies, 1 showed broad spectrum activity against a range of human tumor xenografts in mice.36
Figure 2.
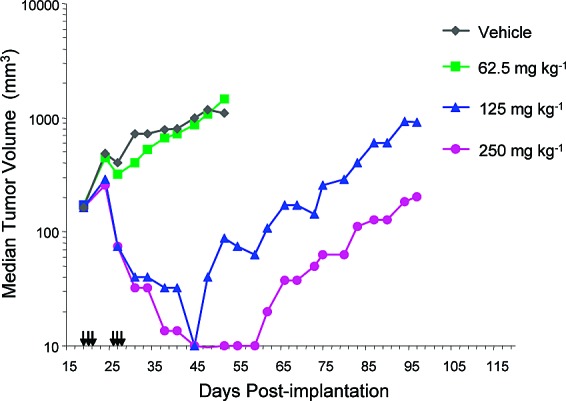
Efficacy of 1 in Colo205 human tumor xenograft study in mice. Arrows denote dosing days.
Compound 1 showed an overall profile suitable for clinical development, including a solubility of 5 mg mL−1 in 0.1 M acetate buffer at pH 5.6. Compound 1 exhibited half-lives in the rat and dog of 1.3 and 2.1 h, respectively. This has translated to dose-proportional PK profile in humans with a mean terminal elimination half-life of ∼12 h.38
In summary, starting from a fragment based screening hit, benzoic acid 2, SAR studies culminated in the discovery of 1, a highly potent and selective inhibitor of CENP-E. Inhibition of CENP-E with 1 induces mitotic arrest in human tumor cells and tumor regressions in vivo. CENP-E inhibition is expected to have a beneficial effect on cancer therapy, and 1 is being evaluated in human clinical trials for the treatment of cancer.39
Acknowledgments
We thank all members of the much larger CENP-E Program Team at Cytokinetics and GlaxoSmithKline for their contributions.
Abbreviations
CENP-E, centromere-associated protein E; KSP, kinesin spindle protein; LE, ligand efficiency; MT, microtubules.
Supporting Information Available
Experimental procedures and analytical data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Vale R. D. The Molecular Motor Toolbox for Intracellular Transport. Cell 2003, 112, 467–480. [DOI] [PubMed] [Google Scholar]
- Miki H.; Setou M.; Kaneshiro K.; Hirokawa N. All Kinesin Superfamily Protein, KIF, Genes in Mouse and Human. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 7004–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein L. S.; Philip A. V. The Road Less Travelled: Emerging Principles of Kinesin Motor Utilization. Annu. Rev. Cell Dev. Biol. 1999, 15, 141–183. [DOI] [PubMed] [Google Scholar]
- Bergnes G.; Brejc K.; Belmont L. Mitotic Kinesins: Prospects for Antimitotic Drug Discovery. Curr. Top. Med. Chem. 2005, 5, 127–145. [DOI] [PubMed] [Google Scholar]
- Sharp D. J.; Rogers G. C.; Scholey J. M. Microtubule Motors in Mitosis. Nature 2000, 407, 41–47. [DOI] [PubMed] [Google Scholar]
- Wood K. W.; Cornwell W. D.; Jackson J. R. Past and Future of the Mitotic Spindle as an Oncology Target. Curr. Opin. Pharmacol. 2001, 1, 370–377. [DOI] [PubMed] [Google Scholar]
- Matsuno K.; Swada J.; Asai A. Therapeutic Potential of Mitotic Kinesin Inhibitors in Cancer. Expert Opin. Ther. Pat. 2008, 18, 253–274. [Google Scholar]
- Jackson J. R.; Patrick D. R.; Dar M. M.; Huang P. S. Targeted Anti-Mitotic Therapies: Can We Improve on Tubulin Agents?. Nat. Rev. Cancer 2007, 7, 107–117. [DOI] [PubMed] [Google Scholar]
- Giannis A.; Sarli V. Inhibitors of Mitotic Kinesins: Next-Generation Antimitotics. ChemMedChem 2006, 1, 293–298. [DOI] [PubMed] [Google Scholar]
- Qian X.; Wolf A. A; Bergnes G. Progress on Mitotic Kinesin Inhibitors as Anti-Cancer Therapeutics. Annu. Rep. Med. Chem. 2006, 41, 263–274 and references cited therein. [Google Scholar]
- Cox C. D.; Coleman P. J.; Breslin M. J.; Whitman D. B.; Garbaccio R. M.; Fraley M. E.; Buser C. A.; Walsh E. S.; Hamilton K.; Schaber M. D.; Lobell R. B.; Tao W.; Davide J. P.; Diehl R. E.; Abrams M. T.; South V. J.; Huber H. E.; Torrent M.; Prueksaritanont T.; Li C.; Slaughter D. E.; Mahan E.; Fernandez-Metzler C.; Yan Y.; Kuo L. C.; Kohl N. E.; Hartman G. D. Kinesin Spindle Protein (KSP) Inhibitors. 9. Discovery of (2S)-4-(2,5-Difluorophenyl)-N-[(3R,4S)-3-fluoro-1-methylpiperidin-4-yl]-2-(hydroxymethyl)-N-methyl-2-phenyl-2,5-dihydro-1H-pyrrole-1-carboxamide (MK-0731) for the Treatment of Taxane-Refractory Cancer. J. Med. Chem. 2008, 51, 4239–4252. [DOI] [PubMed] [Google Scholar]
- Jiang C.; You Q.; Li Z.; Guo Q. Kinesin Spindle Protein Inhibitors as Anticancer Agents. Expert Opin. Ther. Pat. 2006, 16, 1517–1532 and references cited therein. [Google Scholar]
- Coleman P. J.; Fraley M. E. Inhibitors of the Mitotic Kinesin Spindle Protein. Expert Opin. Ther. Pat. 2004, 14, 1659–1667. [Google Scholar]
- Wood K. W.; Chua P.; Sutton D.; Jackson J. R. Centromere-Associated Protein E: a Motor that Puts the Brakes on the Mitotic Checkpoint. Clin. Cancer Res. 2008, 14, 7588–7592. [DOI] [PubMed] [Google Scholar]
- Cleveland D. W.; Mao Y.; Sullivan K. F. Centromeres and Kinetochores: from Epigenetics to Mitotic Checkpoint Signalling. Cell 2003, 112, 407–421. [DOI] [PubMed] [Google Scholar]
- Weaver B. A. A.; Bonday Z. Q.; Putkey F. R.; Kops G. J. P. L.; Silk A. D.; Cleveland D. W. Centromere-Associated Protein E is Essential for the Mammalian Mitotic Checkpoint to Prevent Aneuploidy due to Single Chromosome Loss. J. Cell. Biol. 2003, 162, 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood K. W.; Sakowicz R.; Goldstein L. S.; Cleveland D. W. CENP-E is a plus End-Directed Kinetochore Motor Required for Metaphase Chromosome Alignment. Cell 1997, 91, 357–366. [DOI] [PubMed] [Google Scholar]
- Schaar B. T.; Chan G. K.; Maddox P.; Salmon E. D.; Yen T. J. CENP-E Function at Kinetochores is Essential for Chromosome Alignment. J. Cell Biol. 1997, 139, 1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.; Abrieu A.; Zheng Y.; Sullivan K. F.; Cleveland D. W. CENP-E Forms a Link between Attachment of Spindle Microtubules to Kinetochores and the Mitotic Checkpoint. Nat. Cell Biol. 2000, 2, 484–491. [DOI] [PubMed] [Google Scholar]
- McEwen B. F.; Chan G. K.; Zubrowski B.; Savoian M. S.; Sauer M. T.; Yen T. J. CENP-E is Essential for Reliable Bioriented Spindle Attachment, but Chromosome Alignment can be Achieved via Redundant Mechanisms in mammalian cells. Mol. Biol. Cell 2001, 12, 2776–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanudji M.; Shoemaker J.; L’Italien L.; Russell L.; Chin G.; Schebye X. M. Gene Silencing of CENP-E by Small Interfering RNA in HeLa cells Leads to Missegregation of Chromosomes after a Mitotic Delay. Mol. Biol. Cell 2004, 15, 3771–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C.; Zhao J.; Bibikova M.; Leverson J. D.; Bossy-Wetzel E.; Fan J. B.; Abraham R. T.; Jiang W. Functional Analysis of Human Microtubule-Based Motor Proteins, the Kinesins and Dyneins, in Mitosis/Cytokinesis Using RNA Interference. Mol. Biol. Cell 2005, 16, 3187–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y.; Desai A.; Cleveland D. W. Microtubule Capture by CENP-E Silences BubR1-Dependent Mitotic Checkpoint Signaling. J. Cell Biol. 2005, 170, 873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M.; Chessari G.; Tisi D.; Woodhead A. J. Recent Developments in Fragment-Based Drug Discovery. J. Med. Chem. 2008, 51, 3661–3680. [DOI] [PubMed] [Google Scholar]
- Reynolds C. H.; Tounge B. A.; Bembenek S. D. Ligand Binding Efficiency: Trends, Physical Basis, and Implications. J. Med. Chem. 2008, 51, 2432–2438. [DOI] [PubMed] [Google Scholar]
- Abad-Zapatero C.; Metz J. T. Ligand Efficiency Indices as Guideposts for Drug Discovery. Drug Discovery Today 2005, 10, 464–9. [DOI] [PubMed] [Google Scholar]
- Qian X.; McDonald A. I.; Zhou H.-J.; Ashcraft L. W.; Yao B.; Jiang H.; Huang J. K. C.; Wang J.; Morgans D. J. Jr.; Morgan B. P.; Bergnes G.; Dhanak D.; Knight S. D.; Adams N. D.; Parrish C. A.. Preparation of amino acid-related compounds for treating cellular proliferative diseases; WO2005107762, 2005.
- Topliss J. G. A Manual Method for Applying the Hansch Approach to Drug Design. J. Med. Chem. 1977, 20, 463–469. [DOI] [PubMed] [Google Scholar]
- Evidence for the proposed binding site was consistent with site directed mutagenesis and photolabeling experiments. Details of these studies will be the focus of a future publication. For another example of allosteric ATP-competitive kinesin inhibitors, see: Parrish C. A.; Adams N. D.; Auger K. R.; Burgess J. L.; Carson J. D.; Chaudhari A. M.; Copeland R. A.; Diamond M. A.; Donatelli C. A.; Duffy K. J.; Faucette L. F.; Finer J. T.; Huffman W. F.; Hugger E. D.; Jackson J. R.; Knight S. D.; Luo L.; Moore M. L.; Newlander K. A.; Ridgers L. H.; Sakowicz R.; Shaw A. N.; Sung C. M.; Sutton D.; Wood K. W.; Zhang S.; Zimmerman M. N.; Dhanak D. Novel ATP-Competitive Kinesin Spindle Protein Inhibitors. J. Med. Chem. 2007, 50, 4939–4952. [DOI] [PubMed] [Google Scholar]
- Solubility was measured routinely using a high throughput HPLC based method. Compounds were dissolved in DMSO and diluted with pH 7.4 phosphate buffer, resulting in a final mixture that contained 2% DMSO and a theoretical maximum compound concentration of 200 μM.
- The corresponding alcohol diastereomer, R = (R)-CH(CH3)OH, was ∼10-fold less active (data not shown).
- Only minor modifications to the 3-chloro-4-isopropoxybenzamide portion of the template were tolerated. Further details of these additional SAR studies will be the subject of a future publication.
- Sutton D.; Gilmartin A. G.; Kusnierz A. M.; Sung C. M.; Luo L.; Carson J. D.; Laquerre S. G.; Cornwell W.; King A. G.; Knight S. D.; Dhanak D.; Bergnes G.; Qian X.; Chua P.; Wood K. W.; Huang P. S.; Weber B. L.; Copeland R. A.; Jackson J. R.. A Potent and Selective Inhibitor of the Mitotic Kinesin CENP-E (GSK923295A) Demonstrates a Novel Mechanism of Inhibiting Tumor Cell Proliferation and Shows Activity Against a Broad Panel of Human Tumor Cell Lines in vitro; AACR-NCI-EORTC International Conference on Molecular Targets and Cancer, San Francisco, CA, October 2007. [Google Scholar]
- Hu Z.; Kuo W.; Das D.; Ziyad S.; Gu S.; Bhattacharya S.; Wyrobek A.; Wang N. J.; Felier H. S.; Wooster R.; Weber B.; Gray J. W.. Small Molecular Inhibitor of the Centromere-Associated Protein E (CENP-E), GSK923295A Inhibits Cell Growth in Breast Cancer Cells; 2009 American Association of Cancer Research (AACR), Denver, CO, April 2009. [Google Scholar]
- Johnson B. M.; Sutton D.; Fleming R. A.; Lager J. J.. Exploratory Methods for Assessment of Therapeutic Exposures and Schedules of GSK923295A, a Novel Mitotic Checkpoint Inhibitor; 2009 American Association of Cancer Research (AACR), Denver, CO, April 2009. [Google Scholar]
- Sutton D.; Diamond M.; Faucette L.; Giardiniere M.; Zhang S.; Vidal J.; Dhanak D.; Knight S.; Bergnes G.; Qian X.; Wood K.; Morgans D.; Huang P.; Jackson J.. GSK-923295, a Potent and Selective CENP-E Inhibitor, Has Broad Spectrum Activity Against Human Tumor Xenografts in Nude Mice; Annual Meeting of the American Association for Cancer Research (AACR), Los Angeles, CA, April 2007. [Google Scholar]
- Complete tumor regressions correspond to >50% decrease in volume relative to pre-treament for at least three consecutive measurements.
- Fleming R. A.; Holen K. D.; Cyr T. L.; Johnson B. M.; Gauvin J. L. S.; Lager J. J.; Williams B.; Alberti D. B.; Weber B. L.; Grilley-Olson J. E.; Chung V. A.. Phase I Dose Escalation and Pharmacokinetic Study of the Novel Mitotic Checkpoint Inhibitor GSK923295A in Patients with Solid Tumors; EORTC-NCI-AACR International Conference on Molecular Targets and Cancer, Geneva Palexpo, Geneva, Switzerland, October 2008. [Google Scholar]
- http://clinicaltrials.gov.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.