

Abstract
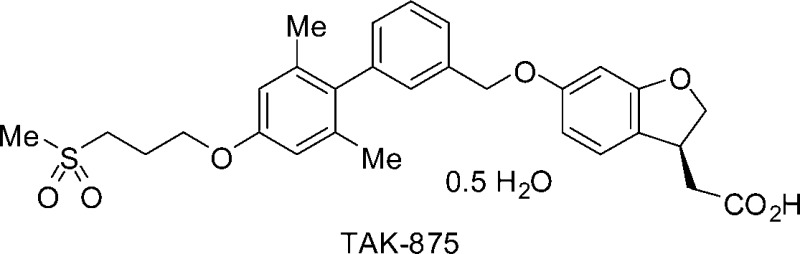
GPR40, one of the G protein-coupled receptors predominantly expressed in pancreatic β-cells, mediates enhancement of glucose-stimulated insulin secretion by free fatty acids. A potent and selective GPR40 agonist is theorized to be a safe and effective antidiabetic drug with little or no risk of hypoglycemia. Cyclization of the phenylpropanoic acid moiety of lead compound 1 produced fused phenylalkanoic acids with favorable in vitro agonist activities and pharmacokinetic profiles. Further optimization led to the discovery of dihydrobenzofuran derivative 9a ([(3S)-6-({2′,6′-dimethyl-4′-[3-(methylsulfonyl)propoxy]biphenyl-3-yl}methoxy)-2,3-dihydro-1-benzofuran-3-yl]acetic acid hemi-hydrate, TAK-875) as a potent, selective, and orally bioavailable GPR40 agonist, with a pharmacokinetic profile enabling long-acting drug efficacy. Compound 9a showed potent plasma glucose-lowering action and insulinotropic action during an oral glucose tolerance test in female Wistar fatty rats with impaired glucose tolerance. Compound 9a is currently in clinical trials for the treatment of type 2 diabetes mellitus.
Keywords: GPR40, agonist, GSIS, TAK-875, OGTT, insulin secretagogue
Type 2 diabetes mellitus is characterized by insulin resistance or reduced insulin sensitivity, combined with reduced insulin secretion from pancreatic β-cells. Persistent or uncontrolled hyperglycemia increases the risk of macrovascular and microvascular complications. Accordingly, therapeutic control of blood glucose levels is critically important in the clinical management and treatment of diabetes. Insulin secretagogues such as sulfonylureas and meglitinides are widely used for the treatment of type 2 diabetes.1 They bind to an ATP-dependent K+ channel in β-cells, causing depolarization of the plasma membrane and opening the voltage-gated Ca2+ channels leading to insulin release. Because this action is independent of extracellular blood glucose concentration, use of these agents could cause hypoglycemia,2 possibly accelerating the loss of function and apoptosis of pancreatic β-cells.3 Furthermore, the relatively unfavorable cardiovascular risk profiles of sulfonylureas as compared with metformin have been shown in a retrospective analysis encompassing over 90000 diabetics in a British general practice database.4 Therefore, administration of insulin secretagogues must be carefully controlled. Recently, agents prompting glucose-stimulated insulin secretion (GSIS) have attracted considerable attention, for example, dipeptidyl peptidase-4 (DPP-4) inhibitors or biologically stabilized analogues of glucagon-like peptide-1 (GLP-1).5
Free fatty acids (FFAs) provide an important energy source and also act as signaling molecules. FFAs have pleiotropic effects on pancreatic β-cells. Although chronic exposure to high levels of FFAs impairs β-cell function and secretory capacity (lipotoxicity), acute administration of FFAs promotes GSIS.6,7 In 2003, it was reported that GPR40 contributes to the enhancement of GSIS by FFAs.8
GPR40 was identified over 10 years ago as an orphan G protein-coupled receptor (GPCR).9 It is highly expressed in human and rodent pancreatic β-cells and β-cell lines (MIN6 or INS-1E) and is also found in several areas of the human brain.8,10,11 GPR40 couples mainly with a G protein α-subunit of the Gq family (Gαq). Several reports suggest that FFAs binding to GPR40 increase the intracellular Ca2+ concentration by a Gq-mediated pathway, leading to enhanced insulin secretion in a glucose concentration-dependent manner.8,12,13 Meanwhile, treatment of MIN6 or INS-1E cells with small interfering RNA (siRNA) specific for GPR40 prevents insulin secretion by stimulation with FFAs.8,12 This study further supported the hypothesis that GPR40 might serve as an attractive target for a novel insulin secretagogue. Thus, a potent and selective GPR40 agonist could prove to be an effective antidiabetic drug with little or no risk of hypoglycemia as observed in sulfonylurea agents. This communication describes the discovery of [(3S)-6-({2′,6′-dimethyl-4′-[3-(methylsulfonyl)propoxy]biphenyl-3-yl}methoxy)-2,3-dihydro-1-benzofuran-3-yl]acetic acid hemi-hydrate (9a, TAK-875), a potent, selective, and orally bioavailable GPR40 agonist that has been selected as a clinical candidate.
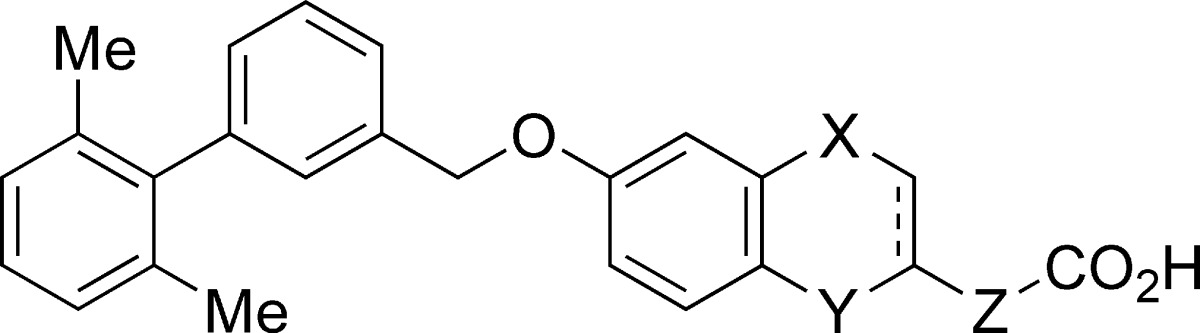
Small molecule GPR40 agonists based on scaffolds such as arylalkanoic acids and thiazolidinediones have been reported by several groups.14 We independently identified a range of synthetic agonists by ligand-based drug design. Potent agonists have also been discovered among the natural ligands, especially polyunsaturated fatty acids such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA).8,10,11 We therefore speculated that a π−π interaction might have a great impact on the receptor−ligand interaction. We selected several arylalkanoic acids from our in-house library and screened them for activity using a fluorometric imaging plate reader (FLIPR) assay. This screening identified benzyloxyphenylpropanoic acids as a promising lead series. Optimization of the lipophilic portion of the molecule led to the identification of phenylpropanoic acid 1 that showed in vitro agonist activity and in vivo efficacy in a diabetic animal model.15 While compound 1 possessed a modest pharmacokinetic (PK) profile, it appeared to be susceptible to β-oxidation at the phenylpropanoic acid moiety. Therefore, we planned to improve metabolic stability by constructing a fused structure between the ortho-position of the phenyl ring and the α- or β-position of the propanoic acid moiety (Figure 1). In addition, we anticipated that a more constrained structure distinct from the more flexible fatty acids would avoid off-target activities derived from multifunctional fatty acids.
Figure 1.

Design of fused phenylalkanoic acids.
The synthesis of fused phenylalkanoic acids 3−8 is detailed in the Supporting Information.
Agonist activities of the prepared compounds were measured by FLIPR assay in the presence of 0.1% bovine serum albumin (BSA). Binding affinities for human and rat receptors were also measured in the presence of 0.2% BSA.17 As depicted in Table 1, fused five- and six-membered ring compounds 3 and 4 exhibited comparable agonist activities (3, EC50 = 0.028 μM; 4, EC50 = 0.023 μM) to the phenylpropanoic acid 1 (EC50 = 0.0057 μM). Expansion of the fused ring to a seven-membered cycle resulted in decreased potency (5, EC50 = 0.54 μM). Replacement of the carbon atom of 3 with an oxygen atom retained potency (7, EC50 = 0.022 μM). However, benzofuran derivative 6 with an unsaturated ring showed weaker activity than 7, suggesting that a planar template might be unfavorable. Interestingly, the seven-membered analogue 8 fused with the phenyl ring at the α-position instead of the β-position of the phenylpropanoic acid moiety recovered activity (EC50 = 0.049 μM) against the human receptor. These results suggest that placement of the phenyl ring and the carboxylic group is important in determining activity properly regardless of the size of the fused ring. In our binding assay, affinity for the human receptor showed a trend similar to that observed in the FLIPR assay. Meanwhile, affinity for the rat receptor was decreased in relation to the expansion in ring size (five-membered ring 3 and 7 > six-membered ring 4 > seven-membered ring 8). Thus, the five-membered fused ring compounds (3 and 7) had the best profiles in terms of potency and species difference.
Table 1. In Vitro Activities of Fused Phenylalkanoic Acids.
| FLIPR | binding |
||||||
|---|---|---|---|---|---|---|---|
| compd | X | Y | Z | bond type | human EC50 (μM)a | human Ki (μM)b | rat Ki (μM)b |
| 3 | CH2 | bond | CH2 | single | 0.028 | 0.21 | 0.52 |
| 4 | −(CH2)2− | bond | CH2 | single | 0.023 | 0.059 | 3.7 |
| 5 | −(CH2)3− | bond | CH2 | single | 0.54 | 2.4 | >10 |
| 6 | O | bond | CH2 | double | 1.7 | >10 | >10 |
| 7 | O | bond | CH2 | single | 0.022 | 0.17 | 1.1 |
| 8 | −OCH2− | CH2 | bond | single | 0.049 | 0.27 | >10 |
| 1 | 0.0057 | 0.032 | 0.054 | ||||
All values are averages of n = 3 in the presence of 0.1% BSA.
All values are averages of n = 2 or 3 in the presence of 0.2% BSA.
The PK parameters were evaluated in rats (Table 2). Fused ring compounds (3, 4, and 7) with favorable activity profiles showed higher oral bioavailability (F) (3, 47.8%; 4, 56.9%; 7, 69.4%) than that of the phenylpropanoic acid 1 (21.5%). Likewise, these compounds (3, 4, and 7) exhibited >2.6-fold higher maximum plasma concentration (Cmax) and >3.7-fold higher plasma exposure (AUCpo, area under curve) than that of 1. These results demonstrate the utility of our strategy to improve PK profiles by cyclization of the phenylpropanoic acid moiety. One of the reasons for the desirable PK profiles of these fused ring compounds is that they might be tolerant to β-oxidation, as we speculated above.
Table 2. Pharmacokinetic Profiles for Fused Phenylalkanoic Acidsa.
| compd | Cmax (ng/mL) | AUCpo (ng h/mL) | F (%) |
|---|---|---|---|
| 3 | 285.1 | 1701.3 | 47.8 |
| 4 | 220.7 | 925.7 | 56.9 |
| 7 | 449.7 | 2357.3 | 69.4 |
| 1 | 86.0 | 249.0 | 21.5 |
Rat cassette dosing at 0.1 mg/kg, iv, and 1 mg/kg, po. Average of three rats. F means bioavailability.
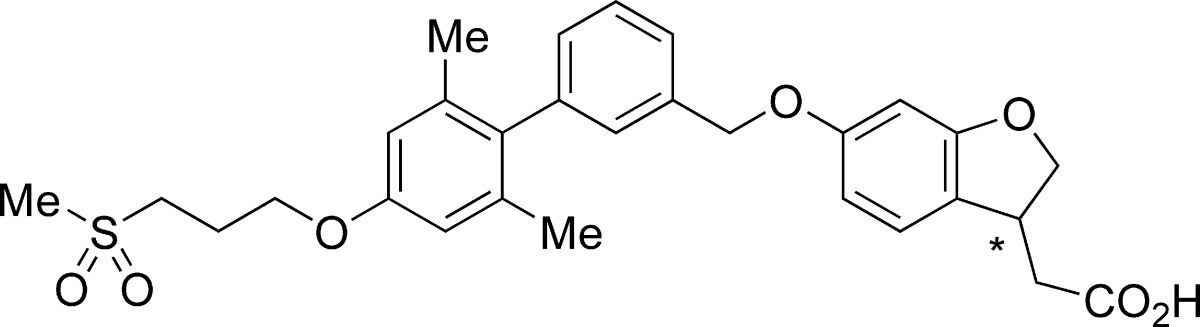
On the basis of these in vitro activities and PK profiles, we fixed the dihydrobenzofuran as a template for further investigation. Detailed structure−activity relationship (SAR) analyses revealed that the substituents at the 4′-position of the biphenyl scaffold were well-tolerated.18 These efforts led to the discovery of compound 9a.
As shown in Table 3, the (S)-enantiomer of 9a exhibited potent agonist activity (EC50 = 0.014 μM) and high binding affinity (Ki = 0.038 μM) to the human receptor, whereas weaker affinity was found toward the rat receptor as with other fused analogues. A preference for (S)-stereochemistry over (R) was observed; (S)-enantiomer 9a possesses higher affinity than (R)-enantiomer 9b. This result indicates that the configuration of the acetic acid moiety attached to the dihydrobenzofuran ring makes a significant impact on GPR40 activity, namely, the spatial configuration between the aromatic ring and the carboxylic acid moiety certainly affects the interaction with the GPR40 receptor as expected.
Table 3. In Vitro Activity Profiles of 9a and Its Enantiomer 9b.
| FLIPR | binding |
|||
|---|---|---|---|---|
| compd | stereo | human EC50 (μM)a | human Ki (μM)b | rat Ki (μM)b |
| 9a | S | 0.014 | 0.038 | 0.14 |
| 9b | R | 0.37 | 1.6 | |
The activity was measured with anhydrous 9a. The value is an average of n = 3 in the presence of 0.1% BSA.
All values are averages of n = 2 or 3 in the presence of 0.2% BSA.
GPR40 belongs to a family of FFAs binding GPCRs, which includes GPR40, GPR41, GPR43, and GPR120.19,20 While GPR41 and GPR43 are activated by short-chain FFAs, GPR40 and GPR120 are activated by medium- to long-chain FFAs and some eicosanoids. As shown in Table 4, compound 9a exhibited excellent agonist potency selectivity for GPR40 receptor over other members of the FFA receptor family (for which EC50 > 10 μM). Our medicinal chemistry efforts, moving away from the fatty acid-like structure and reducing lipophilicity, resulted in acquiring specificity for the GPR40 receptor.
Table 4. Selectivity Profile for 9aa.
| compd | human GPR40 EC50 (μM)b | human GPR41 EC50 (μM)c | human GPR43 EC50 (μM)c | human GPR120 EC50 (μM)d |
|---|---|---|---|---|
| 9a | 0.014 | >10 | >10 | >10 |
The activities were measured with anhydrous 9a.
The value is an average of n = 3 in the presence of 0.1% BSA.
All values are averages of n = 2 in the presence of 0.5% BSA.
The value is average of n = 4 in the presence of 0.1% BSA.
Several modeling studies of GPR40 have been published.21,22 To verify the interaction between the GPR40 and the synthetic agonist 9a, a modeling study was performed. At the time of this study, bovine rhodopsin structures were the only but attractive templates for homology modeling of family A GPCRs. GPR40 also belongs to the cluster of family A GPCRs, so we chose the X-ray crystallographic structure of bovine rhodopsin as a template. Our method for homology modeling and ligand docking is detailed in the Supporting Information. Figure 2 illustrates a docking model of 9a inside a GPR40 homology model. In this model, three groups of interactions are observed. The first group involves hydrophilic interactions. The carboxylate of 9a forms two hydrogen bonds with Arg183 (TM5) and one bond with Arg258 (TM7). In addition, Arg258 (TM7) forms a π−π interaction with the phenyl ring of the dihydrobenzofuran ring, and the carbamoyl moiety of Asn244 (TM6) locks the arginine residue of Arg258 (TM7) into a fixed conformation. The second group is formed by aromatic and hydrophobic interactions located around the 2′,6′-dimethylbiphenyl moiety, including Tyr12 (TM1), Leu67 (TM2), Trp72 (E-I loop), Phe82 (TM3), and Leu262 (TM7). The third group consists of polar residues at the space between TM1 and TM7, including Ser8 (TM1) and Lys259 (TM7). This modeling study proposed that 9a exerts its GPR40 activity utilizing a number of different interactions effectively.
Figure 2.
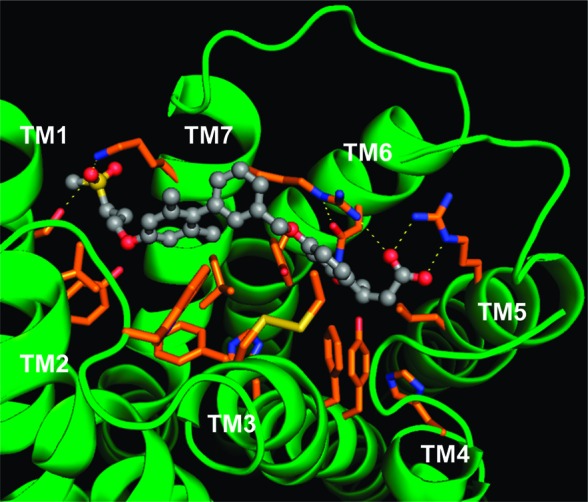
Docking model of GPR40 in complex with 9a (gray).
The PK profile of 9a was studied in rats and dogs (Table 5). Compound 9a showed low plasma clearance (CL) and low volume of distribution (Vd(ss)), resulting in sustained plasma half-lives (iv t1/2λ: rat, 4.7 h; dog, 5.9 h) in each species. In addition, oral administration of 9a exhibited rapid absorption, high Cmax, and high plasma exposure with high bioavailabilities (rat, 76.0%; dog, 92.4%). These results encouraged us to investigate 9a further as a candidate for clinical development.
Table 5. PK Parameters for 9a (Hemi-hydrate) in Fasted Rats and Dogsa.
| iv |
po |
|||||||
|---|---|---|---|---|---|---|---|---|
| species | CL (mL/h/kg) | Vd(ss) (mL/kg) | t1/2λ (h) | t1/2λ (h) | Cmax (μg/mL) | Tmax (h) | AUCpo, 0−24 h (μg h/mL) | F (%) |
| rat | 34.16 | 208.49 | 4.7 | 4.1 | 5.77 | 1.0 | 65.00 | 76.0 |
| dog | 29.79 | 224.67 | 5.9 | 7.5 | 3.29 | 2.0 | 29.45 | 92.4 |
Administered at a dose of 1 mg/kg, iv; 3 mg/kg, po, in rats. Administered at a dose of 0.5 mg/kg, iv; 1 mg/kg, po, in dogs. The values for Cmax and AUC were expressed as equivalent of anhydrous 9a. Data are expressed as mean values (rats, n = 3; dogs, n = 4).
Compound 9a was assessed for its ability to improve glucose intolerance in female Wistar fatty rats, a model developing obesity and obesity-related features such as impaired glucose tolerance, hyperinsulinemia, and hyperlipidemia.23 Single oral dosing of 9a (0.3−3 mg/kg) reduced the blood glucose excursion (Figure 3A) and augmented insulin secretion (Figure 3C) during an oral glucose tolerance test (OGTT), when the compound was administered 1 h before an oral glucose challenge. The area under the curve of plasma glucose (AUC0−120 min) and plasma insulin (AUCpre−30 min) showed that the minimum effective dose was 1 mg/kg (Figure 3B, D, respectively). Because in vitro binding affinity of 9a for human GPR40 was stronger than that for rat GPR40 and its agonist activity measured on FLIPR was also potent (Table 3), 9a could be a potent insulinotropic drug in patients with type 2 diabetes.
Figure 3.

Effects of 9a during an OGTT in female Wistar fatty rats. Panels A and C show time-dependent changes of plasma glucose (PG) and plasma insulin after oral administration of 9a followed by 1 g/kg oral glucose challenge, respectively. Data in panels B and D represent incremental AUC0−120 min of PG levels and incremental AUCpre−30 min of plasma insulin levels shown in panels A and C, respectively. Values are means ± SDs (n = 6). #p ≤ 0.025 as compared with control by one-tailed Williams' test.
In conclusion, we discovered compound 9a, a structurally novel GPR40 agonist showing potent and selective GPR40 agonist activity in vitro, plasma glucose lowering and insulinotropic efficacy in a rat model of diabetes, and an excellent PK profile. Our isosteric conversion of the phenylpropanoic acid moiety developed an efficient strategy to retain potency and improve the PK profile in rats and dogs. Compound 9a is currently undergoing human clinical trials. Detailed studies of SAR and pharmacological evaluation will be reported elsewhere.
Acknowledgments
We thank Dr. Shuji Kitamura, Dr. Nozomu Sakai, and Junichi Sakamoto for helpful discussions; Shinichi Satou for performing the agonist selectivity assays; and Masayuki Yamashita, Katsuhiko Miwa, Miki Ueda, Katsuaki Oda, Shozo Yamamoto, and Mitsutaka Tanaka for preparations of the key intermediate and analyses of enantiomeric excess.
Supporting Information Available
Experimental procedures and analytical data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Rendell M. The role of sulphonylureas in the management of type 2 diabetes mellitus. Drugs 2004, 64, 1339–1358. [DOI] [PubMed] [Google Scholar]
- Burge M. R.; Sood V.; Sobhy T. A.; Rassam A. G.; Schade D. S. Sulphonylurea-induced hypoglycaemia in type 2 diabetes mellitus: a review. Diabetes, Obes. Metab. 1999, 1, 199–206. [DOI] [PubMed] [Google Scholar]
- Maedler K.; Carr R. D.; Bosco D.; Zuellig R. A.; Berney T.; Donath M. Y. Sulfonylurea induced β-cell apoptosis in cultured human islets. J. Clin. Endocrinol. Metab. 2005, 90, 501–506. [DOI] [PubMed] [Google Scholar]
- Tzoulaki I.; Molokhia M.; Curcin V.; Little M. P.; Millett C. J.; Ng A.; Hughes R. I.; Khunti K.; Wilkins M. R.; Majeed A.; Elliott P. Risk of cardiovascular disease and all cause mortality among patients with type 2 diabetes prescribed oral antidiabetes drugs: retrospective cohort study using UK general practice research database. Br. Med. J. 2009, 339, b4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker D. J.; Nauck M. A. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [DOI] [PubMed] [Google Scholar]
- Shimabukuro M.; Zhou Y.-T.; Levi M.; Unger R. H. Fatty acid-induced β-cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 2498–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry J. D.; Dobbins R. L. Fatty acids, lipotoxicity and insulin secretion. Diabetologia 1999, 42, 128–138. [DOI] [PubMed] [Google Scholar]
- Itoh Y.; Kawamata Y.; Harada M.; Kobayashi M.; Fujii R.; Fukusumi S.; Ogi K.; Hosoya M.; Tanaka Y.; Uejima H.; Tanaka H.; Maruyama M.; Satoh R.; Okubo S.; Kizawa H.; Komatsu H.; Matsumura F.; Noguchi Y.; Shinohara T.; Hinuma S.; Fujisawa Y.; Fujino M. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 2003, 422, 173–176. [DOI] [PubMed] [Google Scholar]
- Sawzdargo M.; George S. R.; Nguyen T.; Xu S. J.; Kolakowski L. F.; Odowd B. F. A cluster of four novel human G protein-coupled receptor genes occurring in close proximity to CD22 gene on chromosome 19q13.1. Biochem. Biophys. Res. Commun. 1997, 239, 543–547. [DOI] [PubMed] [Google Scholar]
- Briscoe C. P.; Tadayyon M.; Andrews J. L.; Benson W. G.; Chambers J. K.; Eilert M. M.; Eillis C.; Elshourbagy N. A.; Goetz A. S.; Minnick D. T.; Murdock P. R.; Sauls H. R.; Shabon U.; Spinage L. D.; Strum J. C.; Szekeres P. G.; Tan K. B.; Way J. M.; Ignar D. M.; Wilson S.; Muir A. I. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003, 278, 11303–11311. [DOI] [PubMed] [Google Scholar]
- Kotarsky K.; Nilsson N. E.; Flodgren E.; Owman C.; Olde B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem. Biophys. Res. Commun. 2003, 301, 406–410. [DOI] [PubMed] [Google Scholar]
- Shapiro H.; Shachar S.; Sekler I.; Hershfinkel M.; Walker M. D. Role of GPR40 in fatty acid action on the β cell line INS-1E. Biochem. Biophys. Res. Commun. 2005, 335, 97–104. [DOI] [PubMed] [Google Scholar]
- Fujiwara K.; Maekawa F.; Yada T. Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet β-cells: mediation by PLC and L-type Ca2+ channel and link to insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E670–E677. [DOI] [PubMed] [Google Scholar]
- For recent reviews, see Bharate S. B.; Nemmani K. VS.; Vishwakarma R. A. Progress in the discovery and development of small-molecule modulators of G-protein-coupled receptor 40 (GPR40/FFA1/FFAR1): An emerging target for type 2 diabetes. Expert Opin. Ther. Pat. 2009, 19, 237–264. [DOI] [PubMed] [Google Scholar]
- Manuscript in preparation on lead discovery process.
- The binding assay system was established using a synthetic ligand. The [3H]-labeled ligand 3-[4-({2′,6′-dimethyl-6-[(4-3H)phenylmethoxy]biphenyl-3-yl}methoxy)phenyl]propanoic acid was prepared from the corresponding iodide with tritium gas.
- Detailed SAR studies will be reported elsewhere.
- Brown J. A.; Goldsworthy S. M.; Barnes A. A.; Eilert M. M.; Tcheang L.; Daniels D.; Muir A. I.; Wigglesworth M. J.; Kinghorn I.; Fraser N. J.; Pike N. B.; Strum J. C.; Steplewski K. M.; Murdock P. R.; Holder J. C.; Marshall F. H.; Szekeres P. G.; Wilson S.; Ignar D. M.; Foord M., S.; Wise A.; Dowell S. J. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [DOI] [PubMed] [Google Scholar]
- Hirasawa A.; Tsumaya K.; Awaji T.; Katsuma S.; Adachi T.; Yamada M.; Sugimoto Y.; Miyazaki S.; Tsujimoto G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [DOI] [PubMed] [Google Scholar]
- Tikhonova I. G.; Sum C. S.; Neumann S.; Thomas C. J.; Raaka B. M.; Costanzi S.; Gershengorn M. C. Bidirectional, iterative approach to the structural delineation of the functional “Chemoprint” in GPR40 for agonist recognition. J. Med. Chem. 2007, 50, 2981–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sum C. S.; Tikhonova I. G.; Neumann S.; Engel S.; Raaka B. M.; Costanzi S.; Gershengorn M. C. Identification of residues important for agonist recognition and activation in GPR40. J. Biol. Chem. 2007, 282, 29248–29255. [DOI] [PubMed] [Google Scholar]
- Ikeda H.; Shino A.; Matsuo T.; Iwatsuka H.; Suzuoki Z. A new genetically obese-hyperglycemic rat (Wistar fatty). Diabetes 1981, 30, 1045–1050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.