

Abstract
Three distinct immature T-cell acute lymphoblastic leukemia entities have been described including cases that express an early T-cell precursor immunophenotype or expression profile, immature MEF2C-dysregulated T-cell acute lymphoblastic leukemia cluster cases based on gene expression analysis (immature cluster) and cases that retain non-rearranged TRG@ loci. Early T-cell precursor acute lymphoblastic leukemia cases exclusively overlap with immature cluster samples based on the expression of early T-cell precursor acute lymphoblastic leukemia signature genes, indicating that both are featuring a single disease entity. Patients lacking TRG@ rearrangements represent only 40% of immature cluster cases, but no further evidence was found to suggest that cases with absence of bi-allelic TRG@ deletions reflect a distinct and even more immature disease entity. Immature cluster/early T-cell precursor acute lymphoblastic leukemia cases are strongly enriched for genes expressed in hematopoietic stem cells as well as genes expressed in normal early thymocyte progenitor or double negative-2A T-cell subsets. Identification of early T-cell precursor acute lymphoblastic leukemia cases solely by defined immunophenotypic criteria strongly underestimates the number of cases that have a corresponding gene signature. However, early T-cell precursor acute lymphoblastic leukemia samples correlate best with a CD1 negative, CD4 and CD8 double negative immunophenotype with expression of CD34 and/or myeloid markers CD13 or CD33. Unlike various other studies, immature cluster/early T-cell precursor acute lymphoblastic leukemia patients treated on the COALL-97 protocol did not have an overall inferior outcome, and demonstrated equal sensitivity levels to most conventional therapeutic drugs compared to other pediatric T-cell acute lymphoblastic leukemia patients.
Introduction
During normal T-cell development, early T-cell precursors (ETPs) migrate to the thymus to differentiate into mature T cells.1,2 T-cell acute lymphoblastic leukemias (T-ALL) represent malignant counterparts of thymocytes that have arrested at specific developmental stages that are coupled to specific patterns of T-cell receptor rearrangements.3 Developmental arrest seems dependent on the presence of so-called “type A mutations”, which activate either T-ALL oncogenes such as TAL1, LMO2, TLX3, TLX1, NKX2-1/NKX2-2 or fusion proteins that activate HOXA genes.4–6 For TLX oncoproteins, it has recently been found that these can directly interfere with TRA@ rearrangements by binding to ETS1 on the Eα enhancer resulting in a block of active transcription, histone modification-dependent chromatin opening and rearrangements resulting in a developmental arrest.7
Various studies have identified T-ALL entities that arrest at an extremely immature developmental stage. Using transcriptome analysis, it was first described as the LYL1 subgroup based on the appreciation of high LYL1 expression.8 Three years later, the immature subgroup was also identified by unsupervised cluster analysis and expressed an early thymocyte profile.9 Coustan-Smith and co-workers identified the ETP-ALL subtype that is characterized by a distinct ETP gene-expression profile and immunophenotype.10,11 Using unsupervised transcriptome analysis, in 2011 we described that immature T-ALL cluster patients are characterized by rearrangements of either MEF2C or MEF2C-regulating transcription factors.6 Another immature T-ALL entity, described in 2010, is characterized by absence of bi-allelic deletions of the T-cell receptor gamma gene locus (TRG@) (and denoted as ABD cases) possibly representing early maturation arrest before the onset of T-cell receptor rearrangements.12
ETP-ALL, as first described by Coustan-Smith and colleagues, predicts poor outcome for patients treated on St. Jude (XIII, XIV and XV) or AIEOP ALL-2000 protocols.10 Although immature T-cell development arrest was identified as a poor prognostic factor for T-ALL before,13–18 that study identified a uniform entity that expresses a gene signature like early thymic progenitor cells.10,11 In children, the incidence of ETP-ALL is approximately 13% of all T-ALL cases, but varies among different cohorts: St Jude Children’s Research Hospital study (17 ETP-ALL out of 135 T-ALL patients), AIEOP ALL-2000 study (13 of 100 cases), COGP9404 and DFCI00-01 (14 of 40 cases), the Tokyo Children’s Cancer Study group L99-15 (5 of 91 cases) and the Shanghai Children’s Medical Center study (12 of 72 cases).10,11,19,20 ETP-ALL patients (as predicted by the mouse ETP gene signature)10 that were treated on the Children’s Oncology Group Study (COG) P9404 or Dana-Farber Cancer Institute (DFCI) 00-01 protocols, or ETP-ALL patients based on the ETP-ALL immunophenotype in the Japanese L99-15 study or the Shanghai study, were also related with poor outcome.12,19,20 For adult T-ALL patients treated on German ALL multicenter study group (GMALL) protocols, the incidence of immunophenotypic ETP-ALL was 7.4% and was also associated with poor outcome, like the inferior outcome for early T-ALL.21 Molecular analysis by whole-genome sequencing revealed that most ETP-ALL cases harbor loss-of-function alterations in regulators of hematopoietic and lymphoid development (RUNX1, IKZF1, ETV6, GATA3 and EP300) or in components of the polycomb repressor complex 2 (PRC2).11 The gene-expression profile includes many genes that are expressed in both normal and malignant hematopoietic stem cells, suggesting that ETP-ALL represents an immature leukemia with stem cell and myeloid features.11,12 Accordingly, recurrent mutations in myeloid-specific oncogenes (e.g. IDH1, IDH2, DNMT3A, FLT3, NRAS), were identified in immature22 or ETP-ALL T-ALL patients while having a low incidence of NOTCH1-activating mutations.11,23 At this stage, it remains unknown whether ETP-ALL cases are related to the AML entity that has C/EBPA hypermethylation with expression of T-lineage markers and/or T-ALL mutations like NOTCH1 mutations.24,25
In the study of Gutierrez and co-workers, ABD T-ALL was associated with a poor response to induction chemotherapy, 5-year event-free survival and overall survival in pediatric T-ALL patients who were treated using the COG P9404 or DFCI 00-01 protocol.12 Similar results were described for ABD T-ALL in children treated on Taiwanese TPOG-ALL-97/2002 protocols,26 as well as for pediatric T-cell lymphoblastic lymphoma patients.27
In the present study, we investigated the extent to which ETP-ALL, immature cluster T-ALL and ABD overlap by comparing gene expression and immunophenotypic profiles of the ETP-ALL and immature cluster cases and determining the ABD status of these cases. Our findings strongly suggest that, based on gene expression, ETP-ALL and immature cluster T-ALL represent a single entity in which ABD is a subgroup. Furthermore, classifying ETP-ALL cases based purely on the previously proposed ETP immunophenotype significantly underestimates the number of actual patients with an immature cluster/ETP-ALL gene expression profile.
Methods
Patient samples
For this study, we used diagnostic samples from 117 patients for whom gene expression data were available. These patients had enrolled in the Dutch Childhood Oncology Group (DCOG) ALL-7/8 (n=19)28,29 and ALL-9 (n=26) protocols,30 together with 72 patients who were enrolled in the German Co-Operative Study Group for Childhood Acute Lymphoblastic Leukemia study (COALL-97).31 Seventeen COALL patients underwent bone marrow transplantation due to non-response to therapy at Day 29. T-ALL was defined as being positive for TdT, CD2, cytoplasmic CD3 (CyCD3) and/or CD7. The median follow-up times for the DCOG and COALL patients were 63 and 52 months, respectively. Each patient’s parents or legal guardian provided informed consent to use excess diagnostic material for research purposes as approved by the Institutional Review Board/Ethics committee of the Erasmus MC Rotterdam and in accordance to the Declaration of Helsinki. Leukemic cells were harvested from blood or bone marrow samples and enriched as described previously.32 Enriched samples contained over 90% leukemic cells.
Statistical analysis
Statistical analysis was performed using the PASW Statistics 1 software program. Pearson’s χ2 test was performed to test for statistically significant differences in the distribution of nominal data; if fewer than 5 patients were tested in the individual groups, the Fisher’s exact test was used instead (as indicated in the corresponding tables). Statistical significance for continuous distributed data was tested using the Mann-Whitney U-test. Differences between patient populations with respect to relapse-free survival (RFS) and event-free survival (EFS) were tested using the log rank test. An event for EFS is defined as relapse, lack of response to induction therapy, death in remission due to toxicity, or the development of a secondary malignancy. Two-sided P≤0.05 was considered significant.
Results
ETP-ALL, immature cluster (MEF2C), and ABD T-ALL patients
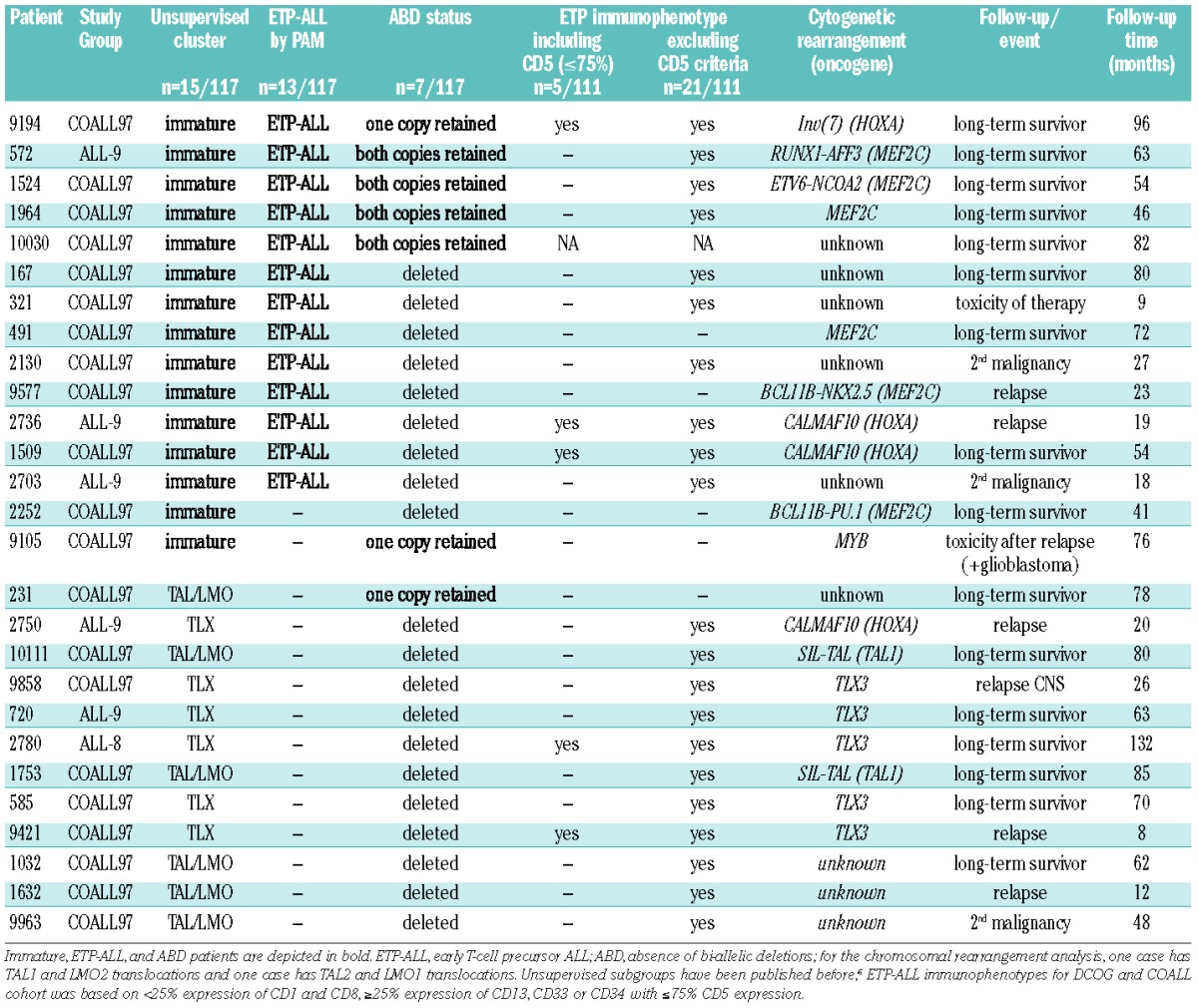
We investigated whether immature T-ALL cluster cases (15 cases), as previously identified using our unsupervised clustering approach,6 displayed ETP-ALL or ABD immature features consistent with two previous studies.10,12 For this gene expression cohort, made up of 117 pediatric T-ALL patient samples, prediction analysis of microarrays (PAM) predicted immature cluster cases as ETP-ALL based on the 100, 200 or 500 most significant probe sets from the human ETP-ALL gene signature11 (Online Supplementary Table S1). Most significant up- and down-regulated probe sets from the ETP-ALL gene signature are strongly enriched as assessed by GSEA analysis in immature cluster and non-immature cluster cases, respectively (Online Supplementary Figure S1). By using PAM analysis, 13 of 15 immature cluster cases were consistently predicted as ETP-ALL based on these ETP-ALL probe sets, while none of the remaining 102 non-immature cluster cases were predicted (P<0.001) (Online Supplementary Figure S2). This implies that ETP-ALL and the immature cluster represent single or strongly overlapping T-ALL entities (Table 1 and Online Supplementary Table S2). Two out of 15 immature cluster cases were not predicted by the ETP-ALL gene signature. Seven patients (6%) were identified as ABD cases based on the preservation of non-rearranged TRG@ loci as detected by RQ-PCR12 (Table 1). Six of the 7 ABD cases were immature cluster cases.6 and 5 out of these 6 also had an ETP signature (Online Supplementary Figure S3). Thus, approximately 40% (5 of 13 or 6 of 15) of immature cluster/ETP-ALL cases have retained non-rearranged TRG@ loci.
Table 1.
Characteristics of immature T-ALL, ETP-ALL (predicted ETP-ALL), ABD patients and patients that have an ETP-ALL immunophenotype.
Relation to immunophenotype
ETP-ALL cases were originally defined by the absence of both CD1 and CD8 (present in fewer than 5% of leukemic cells), absent or weak expression of CD5 (in ≤75% of total cells or ≥10-fold lower than in normal T cells), or the expression of one or more of the markers CD117, CD34, HLA-DR, CD13, CD33, CD11b and CD65 (in ≥25% of total cells).10 We next defined whether ETP-ALL gene signature positive cases met with the immunophenotypic criteria as originally proposed for ETP-ALL.10 In the present study, historic flow cytometry data were not obtained on the gated leukemic cell population and various markers were missing, so we used a simplified ETP-ALL immunophenotype as being CD1 negative, CD8 negative, weak CD5 (≤75%) or CD5 negative with positive expression of CD34 or CD13/33.10 We identified only 5 samples out of the 111 patients for whom both immunophenotype and gene-expression data were available that had such an immunophenotype, and only 3 of these 5 cases expressed an ETP-ALL signature (P=0.01) (Table 2). Leaving out the CD5 marker led to the identification of 10 out of 13 ETP-ALL but also identified 11 out of 98 T-ALL cases that lack an ETP-ALL gene expression signature (P=0.01) (Table 2).
Table 2.
Immunophenotypic markers predicting for immature T-ALL cases with an ETP-ALL gene expression profile.
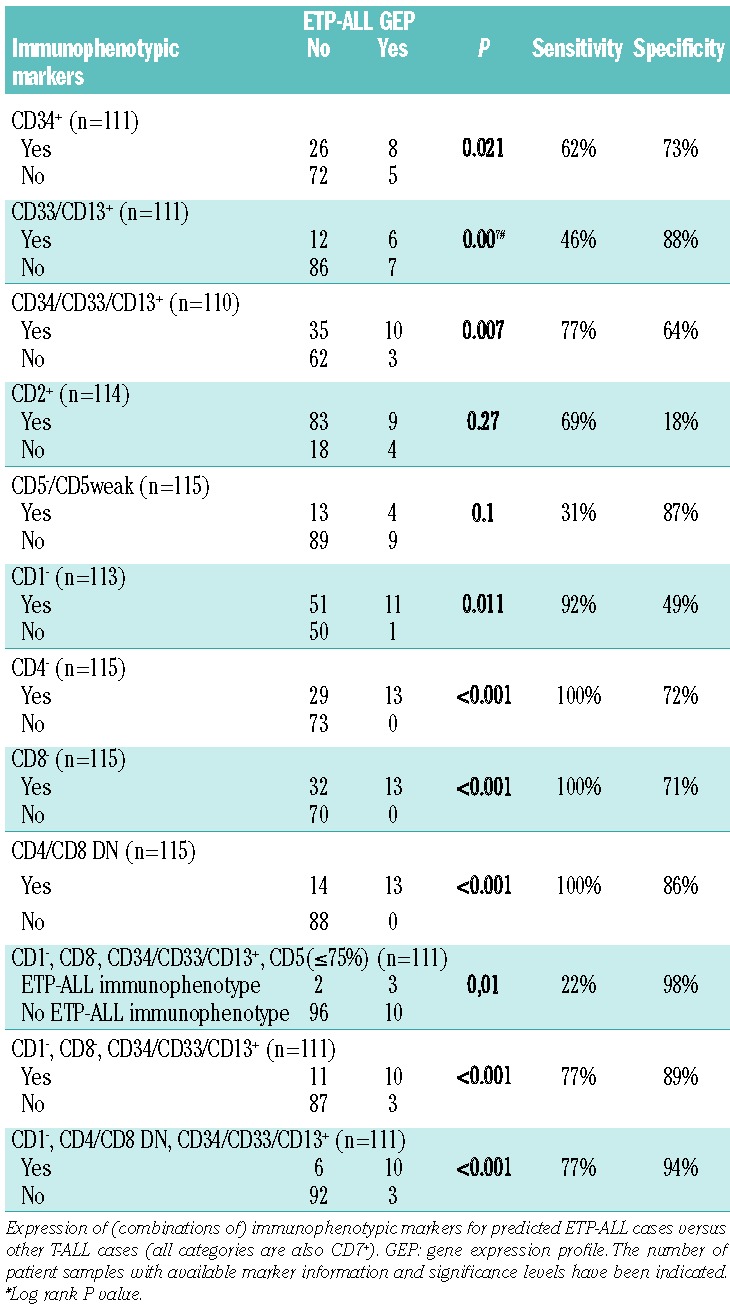
We then investigated which other immunophenotypic markers or combinations of them could be defined that most strongly associates with cases expressing the ETP-ALL gene signature (Table 2). This analysis showed that these ETP-ALL cases strongly associated with absence of CD4 expression in addition to absence of CD8. Inclusion of absence of CD4 expression in addition to absence of expression of CD1 and CD8 but presence of expression of CD34 and/or CD13/33 markers (i.e. CD1−, CD4/CD8 DN, CD34/CD33/CD13+) predicted 10 out of 13 cases with an ETP-ALL gene signature compared to only 6 out of 98 T-ALL cases that lacked an ETP-ALL gene signature, resulting in 77% sensitivity and 94% specificity levels (Table 2). This immunophenotype also strongly associated with the immature cluster cases; 10 of the 15 immature cluster cases had this immunophenotype compared to only 6 of the 96 non-immature cluster cases (P<0.001; sensitivity 67%, specificity 94%) (Online Supplementary Table S3). Also, 4 out of 7 ABD cases had such an immunophenotype compared to 12 out of 104 non-ABD patients (P=0.008) (Online Supplementary Table S4).
Clinical and molecular-genetic features
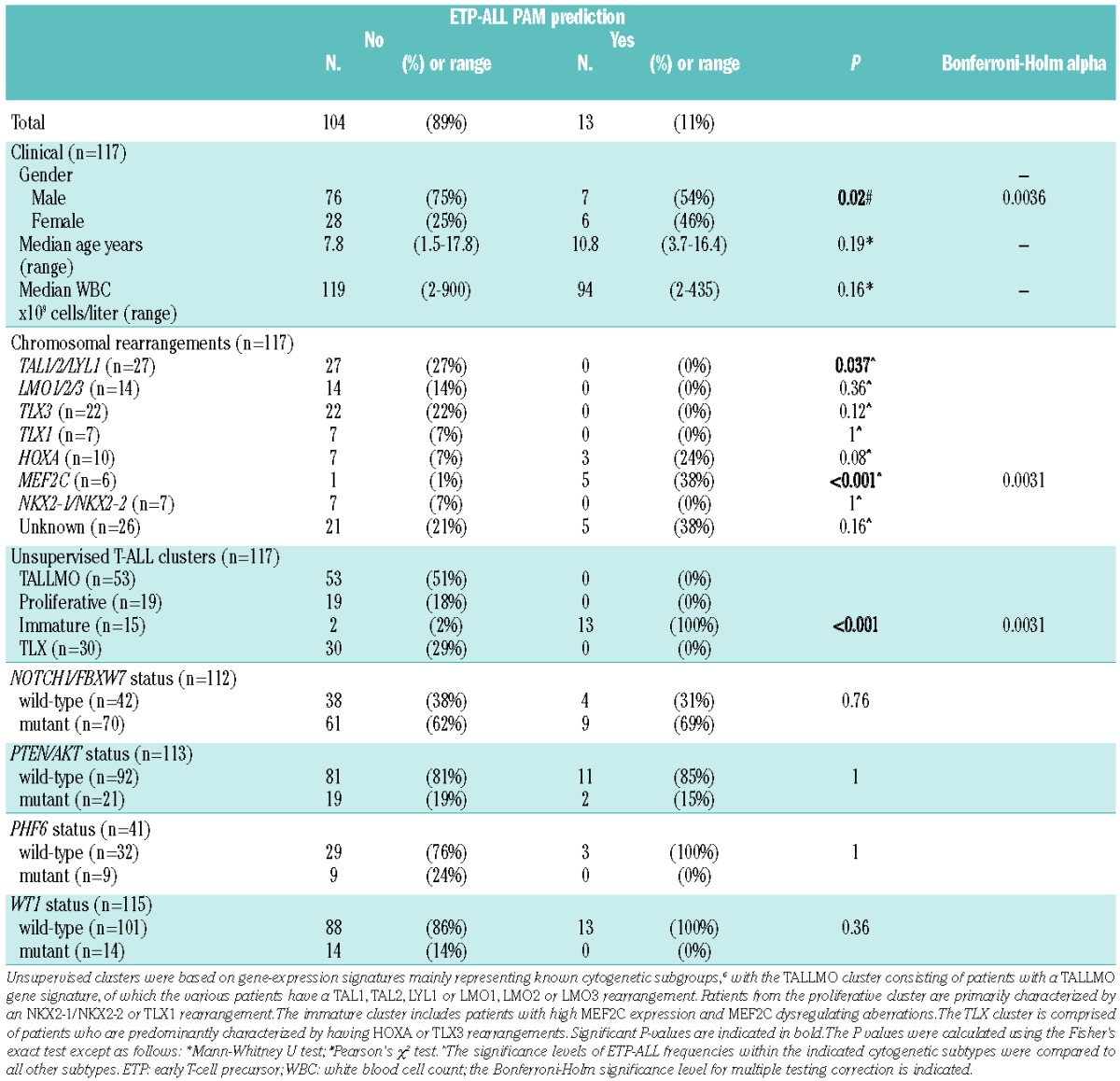
About half of predicted ETP-ALL patients were female, but did not associate with age. White blood cell (WBC) counts tended to be lower for both ETP-ALL cases and immature cluster cases, and were significantly lower for ABD cases (15×109 vs. 129×109 cells/L; P=0.002) (Table 3 and Online Supplementary Tables S3 and S4). Predicted ETP-ALL patients lacked rearrangements in TAL1, LMO2, TLX3, TLX1, MYB or NKX2-1/NKX2-2 genes (Tables 1 and 3). However, 3 of the 13 ETP-ALL cases had HOXA-dysregulating events.9,33 These same 3 cases also represent immature cluster cases and have an immature immunophenotype with expression of the myeloid marker CD33.6 MEF2C-dysregulating mechanisms (including ETV6-NCOA2 (1 case), RUNX1-AFF3 (1 case), NKX2-5 (1 case) and PU.1/SPI1 (1 case) translocations) or an MEF2C rearrangement (2 cases) were originally identified in 6 out of 15 immature cluster cases.6 Five of these 6 cases expressed the human ETP-ALL gene signature (Tables 1 and 3) (P<0.001). The PU.1/(SPI1) immature cluster case was the only MEF2C-dysregulated case that was not predicted by the human ETP-ALL gene signature. No genetic aberrations have been identified yet for 5 ETP-ALL cases. With respect to other recurrent T-ALL mutations, the incidences of NOTCH1-activating mutations (NOTCH1/FBXW7) and PTEN/AKT mutations in the immature cluster, ETP-ALL and ABD cases did not differ significantly from other T-ALL patients (Table 3 and Online Supplementary Tables S3 and S4). Similar results were obtained with respect to PHF6 and WT1 mutations. ETV6 and RUNX1 mutations, as previously associated with early T-ALL22 or immunophenotypic ETP-ALL,11 were only identified in 2 and one case out of 71 COALL-97 T-ALL patients, respectively (data not shown), but none of these mutant cases expressed an ETP-ALL gene signature.
Table 3.
Overall clinical, immunophenotypic and molecular cytogenetic properties of predicted ETP-ALL and non-ETP-ALL patients.
The ETP-ALL cases in our cohort were associated with high MEF2C, LMO2 and LYL1 expression levels (P<0.0001, P<0.0001 and P=0.0002, respectively) (Figure 1A, C and D). LMO2 and LYL1 were previously identified as direct target genes for MEF2C.6 MEF2C-dysregulating events were identified in both ETP-ALL cases with and without ABD characteristics: 3 ABD ETP-ALL cases had MEF2C-dysregulating events compared to 2 non-ABD ETP-ALL cases (Table 1). Cytogenetic defects underlying the other 2 ETP-ALL cases with ABD-characteristics include a HOXA case due to an inversion on chromosome 7 and an unknown case. Two other ABD cases did not have an ETP-ALL profile: one had an MYB translocation and had already been identified as an immature cluster case whereas the other belonged to the TALLMO cluster for which the underlying genetic defect remains unknown (Table 1 and Online Supplementary Table S4). Linear Models for MicroArray (LIMMA) data analysis revealed that no genes were differentially expressed between the ABD and non-ABD ETP-ALL cases, suggesting that these cases likely represent a single disease entity. Although the expression of LYL1 was higher in the ABD cases than in the non-ABD ETP-ALL cases, significance was lost following correction for multiple testing. ERG1 or BAALC levels, previously linked to adult immature T-ALL/ETP-ALL and poor outcome in some,34,35 but not all,36 studies were not significantly elevated in our pediatric ETP-ALL series.
Figure 1.

ETP-ALL patients express high MEF2C, LMO2 and LYL1 levels but not ERG or BAALC. The expression of (A) MEF2C (probe set 239966_at), (B) LMO1 (probe set 206718_at), (C) LMO2 (probe set 204249_s_at), (D) LYL1 (probe set 210044_s_at), (E) ERG (probe set 1563392_at), and (F) BAALC (probe set 222780_s_at) was based on VSN normalized microarray gene expression data for ETP-ALL (gray squares) and non-ETP-ALL cases (white squares). For the ETP-ALL subgroup, the ABD and non-ABD cases are indicated separately.
Both pediatric and adult ETP-ALL cases are characterized by the expression of hematopoietic stem cell signature genes.11,12 ETP-ALL may, therefore, resemble a stem cell-like leukemia with myeloid and lymphoid features. Consistent with this hypothesis, the gene signature of our immature cluster6 (Online Supplementary Table S5) was significantly enriched for genes (probe sets) that are expressed in sorted hematopoietic stem cells, early erythroid precursor cells, and B-cell fractions as established by Novershtern and co-workers37 (Online Supplementary Table S6). Remarkably, genes that are typically down-regulated during normal T-cell development were enriched in cases with the immature cluster signature. Also B-cell genes were significantly enriched, possibly reflecting the early status of ETP-ALL as an entity that has not yet committed to T-cell development. This is further strengthened by strong enrichment of genes that are expressed in normal MMP-ETP-DN2A immature stages rather than genes from later T-cell development stages beyond DN2B (Online Supplementary Figure S4A and B). It was also reported that ETP-ALL cases express myeloid signature genes.11,22 We, therefore, tested whether ETP-ALL cases would be enriched for differentially expressed genes of AML cases with hypermethylation or mutation of C/EBPA. Although gene set enrichment (GSEA) results overall were not significant, possibly due to the limited number of probe sets, most of these up- or down-regulated signature genes were strongly enriched in immature cluster/ETP-ALL cases (Online Supplementary Figure S4C and D). These data confirm that immature cluster/ETP-ALL is a stem cell-like leukemia that arises at the decision point between early myeloid and lymphoid development.
Relation to outcome
ETP-ALL has been associated with extremely poor outcome in several studies.10,12,19 Therefore, we investigated the outcome of immature cluster/ETP-ALL patients in the COALL-97 protocol: 72 of the 117 patients for which gene expression signatures were available had been enrolled in the COALL-97 protocol, and these 72 included 11 of the 15 immature cluster patients and 10 of the 13 predicted ETP-ALL cases. Surprisingly, ETP-ALL patients were not significantly different from the other T-ALL patients with respect to relapse-free survival (5-year RFS 89±11% vs. 71±7%, respectively; P=0.31) or event-free survival (5-year EFS 70±15% vs. 61±7%, respectively; P=0.66) (Figure 2). Moreover, no differences were detected in the 5-year overall survival (OS) curves (data not shown). Also with respect to the immature cluster cases, the ABD cases and the cases with an ETP-ALL immunophenotype as defined by our criteria, no differences in the RFS or EFS curves were identified (Online Supplementary Figure S5A–C). For ETP-ALL patients, high-dose cytarabine has been suggested to improve outcome of ETP-ALL patients.11 As the COALL-97 protocol is a high-dose cytarabine-containing treatment, this may be one of the reasons for the relatively good outcome of ETP-ALL and ABD patients in this study compared to other studies.10,12,19,26 However, we could not demonstrate differential sensitivity levels to various drugs including cytarabine for ETP-ALL patients in our in vitro cytotoxicity assay (Online Supplementary Figure S6).
Figure 2.
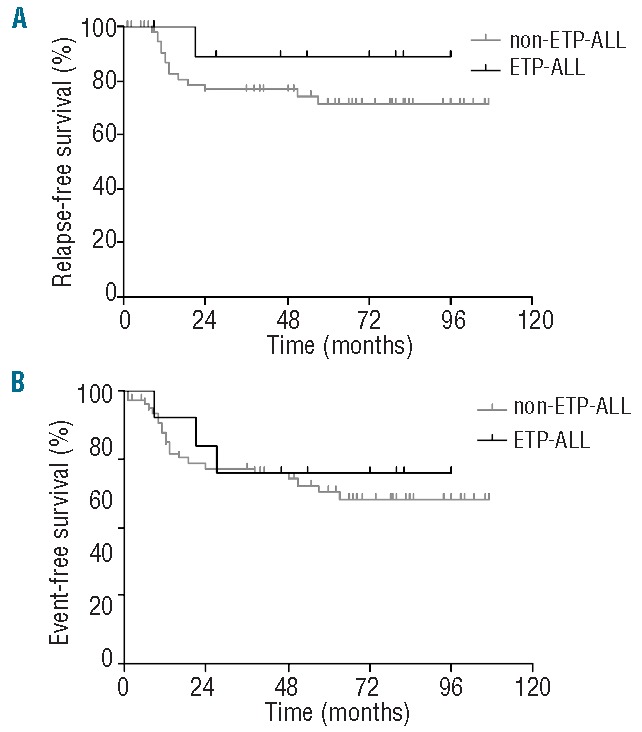
ETP-ALL patients who were treated using the COALL-97 protocol were not associated with poor outcome. (A) Relapse-free survival (RFS) and (B) event-free survival (EFS) curves were generated for the COALL pediatric T-ALL patients. Shown are the RFS and EFS curves for ETP-ALL cases (black line) versus non-ETP-ALL cases (gray line).6 Vertical tick marks represent individual cases for which no further follow-up data is available.
Discussion
In this study, we found that 13 of our 15 previously reported immature cluster cases6 are consistently predicted by PAM based on the human ETP-ALL expression signature,10,11 strongly suggesting that ETP-ALL and immature cluster cases represent a single entity. Consistent with previous observations,12 we found that ABD T-ALL patients represent a subset of the immature cluster/ETP-ALL cases. Six out of 7 ABD patients belong to the immature cluster, where 5 out of 7 cases had an ETP-ALL signature. So, approximately 40% of immature cluster cases retain non-rearranged TRG@ loci, and perhaps represent an even more immature entity among immature cluster/ETP-ALL cases. This entity may closely overlap with the FLT3 mutant adult ETP-ALL cases that fail to demonstrate monoclonal TCR rearrangements38 in line with expected results for ABD ETP-ALL patients. Although we identified some genes using our microarray analysis that were differentially expressed between the ABD and non-ABD immature cluster/ETP-ALL cases (including higher levels of LYL1 in ABD cases), none of these differences remained significant after correcting for multiple testing. The total number of ABD and non-ABD immature cluster/ETP-ALL patients in our cohort may have been insufficient to reveal subtle differences in gene expression levels. On the other hand, we did not detect differences in oncogenic rearrangement types between the ABD and non-ABD immature cluster/ETP-ALL cases. We also found that both ABD-ETP-ALL as well as non-ABD ETP-ALL entities are associated with MEF2C-dysregulating mechanisms, and both entities lack TAL1, TLX1 and NKX2-1/NKX2-2 rearrangements that were previously associated with other T-ALL subgroups.6 A low white blood cell count was significantly associated with both the ETP-ALL and ABD cases. Taken together, based on expression profiles, genetic data and clinical findings, ABD and non-ABD ETP-ALL cases seem to reflect a single ETP-ALL entity. In line with other studies,11,12 immature cluster/ETP-ALL cases are enriched for stem-cell gene signatures. ETP-ALL cases are also strongly enriched for genes that are normally up-regulated in MMP-ETP-DN2A immature T-cell subsets, but do not express genes that are normally expressed in T-cell subsets beyond the DN2B stage.39 Although overall enrichment results for AML with inactivated C/EBPA expression25 were not significant, most of the up- or down-regulated signature genes for that AML cluster were concordantly up- or down-regulated in immature cluster/ETP-ALL cases. Lack of significance may be due to the limited size of the gene signatures. Alternatively, it may be due to the fact that this cluster contains 2 AML entities of which only C/EBPA-hypermethylated AML cases have T-ALL characteristics.25 These data imply that immature cluster/ETP-ALL in C/EBPA-hypermethylated AML cases may be closely-related disease entities that need further investigation. To date, exome sequencing of ETP-ALL patient samples has identified mutations in genes that regulate hematopoietic and lymphoid development, cytokine and/or Ras signaling pathways and the polycomb repressor complex 2 (PRC2), reflecting important chromatin-modifying enzymes and myeloid-leukemia associated oncogenes. In addition, there is a lower prevalence of NOTCH1-activating mutations in ETP-ALL patient samples.11,21–23
Our immature cluster/ETP-ALL cases lack PHF6 and WT1 mutations, and no differences were observed with respect to the frequency of NOTCH1-activating mutations or PI3K/AKT-activating events between the immature cluster/ETP-ALL cases and the other T-ALL cases. The low incidence of ETV6 and RUNX1 mutations may reflect the lower incidence of mutations in children as compared to adult leukemia patients.40 Although none of these mutations have previously been explicitly associated with outcome in our relatively limited number of patient samples, additional mutation screens using an expanded series of patient samples are required to determine whether our immature cluster/ETP-ALL samples differ from the spectrum of mutations identified in the St. Jude’s and COG patient samples.11,12 In contrast to adult T-ALL studies,34–36 we did not observe low ERG or low BAALC expression in our immature cluster/ETP-ALL samples in line with results for adult T-ALL patients that enrolled on GRAALL protocols.36
With respect to our immature T-ALL cluster, we previously identified a variety of distinct genetic rearrangements that result in the activation of oncogenes (e.g. NKX2-5, PU.1, and MEF2C), RUNX1, or ETV6 fusion products, all of which converge on the activation of MEF2C.6 Although MEF2C seems to be activated in multiple immature cluster/ETP-ALL samples for which the underlying genetic defect has not yet been identified, some cases had low levels of MEF2C expression, suggesting that alternate pathogenic pathways may also be affected in ETP-ALL. The expression of LMO2 and LYL1 was significantly elevated in immature cluster/ETP-ALL samples, and both of these genes were previously identified as direct target genes for MEF2C.6 Unlike previous findings,8 immature cluster/ETP-ALL patients lack LYL1 rearrangements as assessed by FISH. The only LYL1-rearranged case in our cohort has a TALLMO gene signature, consistent with the high homology between TAL1 and LYL1 oncoproteins.41
Coustan-Smith and colleagues originally described an ETP-ALL immunophenotype that was associated with T-ALL cases that were predicted by a mouse ETP-like signature.10 This immunophenotype includes multiple markers and may, therefore, be less useful for identifying ETP-ALL cases in retrospective studies for which, in general, relatively fewer parameters are available. With respect to our cohort, immunophenotypic parameters were measured on the bulk of mononuclear cells following Ficoll gradient centrifugation. The leukemic population may, therefore, be contaminated with low numbers of normal cells. We then set the positivity threshold for various markers to 25% or over. Using the immunophenotypic data, we characterized ETP-ALL cases by a simplified ETP-ALL immunophenotype, that apart from expressing CD7, also expressed CD34 and/or myeloid markers CD13 and/or CD33, no or weak (<75%) expression of CD5 in the absence of CD1 and CD8. Only 5 of 117 patients were identified in our study that met this criterion and only 3 of these samples had an ETP-ALL gene signature. In the study of Gutierrez et al.,12 only one of 14 cases as identified by the mouse ETP-like gene signature has such an ETP-ALL immunophenotype.10 Also in the original ETP-ALL study of Coustan-Smith and co-workers,10 only 9 of the 14 initial cases that were identified using the mouse ETP gene signature had a bona fide ETP-ALL immunophenotype. In all of these instances, the current ETP-ALL immunophenotype may severly underestimate the actual number of ETP-ALL cases that express an ETP-ALL gene signature. For our cohort, the CD34+ and/or CD13/33+, CD1− CD4− and CD8− immunophenotype in addition to CD7 positivity most closely associated with cases that expressed the human ETP-ALL gene signature, with a sensitivity level of 77% and a specificity level of 94%.
Predicted ETP-ALL patients based on the human ETP-ALL gene signature who were treated on the COALL-97 protocol did not show a worse outcome in comparison to non-ETP-ALL patients in contrast to some other studies.10,19–21 We also did not observe a worse outcome for immature cluster cases, ABD patients and immunophenotypic ETP-ALL patients (regardless of the inclusion of CD5 data). As ETP-ALL cases express human hematopoietic stem cell gene signatures and early myeloid-associated gene signatures,11,22 treatment with high-dose cytarabine as included in AML treatment protocols has been suggested to improve the outcome or to increase the cure rate of ETP-ALL patients.11 High-dose cytarabine has been incorporated into the COALL-97 treatment protocol, and this may explain the relatively good outcome in contrast to various other studies.10,19–21 So far, in vitro cytotoxicity data for various conventional therapeutic drugs, including cytarabine, failed to reveal differences in sensitivity levels for immature cluster/ETP-ALL compared to other T-ALL patient samples.
In conclusion, the expression of the ETP-ALL gene signature and clustering in the immature T-ALL cluster following unsupervised cluster analysis are highly overlapping and point to a single ETP-ALL entity with respect to biology and genetics. We found no evidence to suggest that ABD and non-ABD cases reflect distinct entities among ETP-ALL cases. Different ETP-ALL patient populations may be identified based on immunophenotypic or gene expression data (ETP-ALL gene signature) and could explain differences in outcome between our and other studies. In this study, samples with an ETP-ALL gene signature correlated best with a CD34/13/33+, CD1− and CD4/CD8 double negative immunophenotype. ETP-ALL cases in the COALL-97 study were not associated with a poor outcome compared to other T-ALL cases. High-dose cytarabine as incorporated in the COALL-97 protocol may have improved the outcome for ETP-ALL patients. Limited numbers of ETP-ALL cases have been investigated in various studies so far. Better definitions for ETP-ALL based on immunophenotypic and gene expression data and clinical outcome require a systematic review as part of a large international meta-analysis.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by the The Children Cancer Free Foundation, Stichting Kinderen Kankervrij (KiKa): project ns. KIKA2007-012 (LZ), KIKA2008-029 (KC-B and WKS), and KIKA2010-82 (YL) and the Dutch Cancer Society Project n. AMC2008-4265 (JB-G). AG is supported by NIH grant CA167124, DOD grant CA120215, an award from the William Lawrence Blanche Hughes Foundation, and is a Research Fellow of the Gabrielle’s Angel Foundation for Cancer Research.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Kueh HY, Rothenberg EV. Regulatory gene network circuits underlying T cell development from multipotent progenitors. Wiley Interdiscip Rev Syst Biol Med. 2012;4(1):79–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson PK, Zuniga-Pflucker JC. On becoming a T cell, a convergence of factors kick it up a Notch along the way. Semin Immunol. 2011;23(5):350–9 [DOI] [PubMed] [Google Scholar]
- 3.Asnafi V, Beldjord K, Boulanger E, Comba B, Le Tutour P, Estienne MH, et al. Analysis of TCR, pT alpha, and RAG-1 in T-acute lymphoblastic leukemias improves understanding of early human T-lymphoid lineage commitment. Blood. 2003;101(7):2693–703 [DOI] [PubMed] [Google Scholar]
- 4.Meijerink JP. Genetic rearrangements in relation to immunophenotype and outcome in T-cell acute lymphoblastic leukaemia. Best Pract Res Clin Haematol. 2010;23(3):307–18 [DOI] [PubMed] [Google Scholar]
- 5.Van Vlierberghe P, Pieters R, Beverloo HB, Meijerink JP. Molecular-genetic insights in paediatric T-cell acute lymphoblastic leukaemia. Br J Haematol. 2008;143(2):153–68 [DOI] [PubMed] [Google Scholar]
- 6.Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011;19(4):484–97 [DOI] [PubMed] [Google Scholar]
- 7.Dadi S, Le Noir S, Payet-Bornet D, Lhermitte L, Zacarias-Cabeza J, Bergeron J, et al. TLX homeodomain oncogenes mediate T cell maturation arrest in T-ALL via interaction with ETS1 and suppression of TCRalpha gene expression. Cancer Cell. 2012;21(4):563–76 [DOI] [PubMed] [Google Scholar]
- 8.Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1(1):75–87 [DOI] [PubMed] [Google Scholar]
- 9.Soulier J, Clappier E, Cayuela JM, Regnault A, Garcia-Peydro M, Dombret H, et al. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood. 2005;106(1):274–86 [DOI] [PubMed] [Google Scholar]
- 10.Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10(2):147–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012; 481(7380):157–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutierrez A, Dahlberg SE, Neuberg DS, Zhang J, Grebliunaite R, Sanda T, et al. Absence of Biallelic TCR{gamma} Deletion Predicts Early Treatment Failure in Pediatric T-Cell Acute Lymphoblastic Leukemia. J Clin Oncol. 2010;28(24):3816–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asnafi V, Buzyn A, Thomas X, Huguet F, Vey N, Boiron JM, et al. Impact of TCR status and genotype on outcome in adult T-cell acute lymphoblastic leukemia: a LALA-94 study. Blood. 2005;105(8):3072–8 [DOI] [PubMed] [Google Scholar]
- 14.Garand R, Voisin S, Papin S, Praloran V, Lenormand B, Favre M, et al. Characteristics of pro-T ALL subgroups: comparison with late T-ALL. The Groupe d’Etude Immunologique des Leucemies. Leukemia. 1993;7(2):161–7 [PubMed] [Google Scholar]
- 15.Marks DI, Paietta EM, Moorman AV, Richards SM, Buck G, DeWald G, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood. 2009;114(25):5136–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiel E, Kranz BR, Raghavachar A, Bartram CR, Loffler H, Messerer D, et al. Prethymic phenotype and genotype of pre-T (CD7+/ER−)-cell leukemia and its clinical significance within adult acute lymphoblastic leukemia. Blood. 1989;73(5):1247–58 [PubMed] [Google Scholar]
- 17.Uckun FM, Gaynon PS, Sensel MG, Nachman J, Trigg ME, Steinherz PG, et al. Clinical features and treatment outcome of childhood T-lineage acute lymphoblastic leukemia according to the apparent maturational stage of T-lineage leukemic blasts: a Children’s Cancer Group study. J Clin Oncol. 1997;15(6):2214–21 [DOI] [PubMed] [Google Scholar]
- 18.Vitale A, Guarini A, Ariola C, Mancini M, Mecucci C, Cuneo A, et al. Adult T-cell acute lymphoblastic leukemia: biologic profile at presentation and correlation with response to induction treatment in patients enrolled in the GIMEMA LAL 0496 protocol. Blood. 2006;107(2):473–9 [DOI] [PubMed] [Google Scholar]
- 19.Inukai T, Kiyokawa N, Campana D, Coustan-Smith E, Kikuchi A, Kobayashi M, et al. Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children’s Cancer Study Group Study L99-15. Br J Haematol. 2012;156(3):358–65 [DOI] [PubMed] [Google Scholar]
- 20.Ma M, Wang X, Tang J, Xue H, Chen J, Pan C, et al. Early T-cell precursor leukemia: a subtype of high risk childhood acute lymphoblastic leukemia. Front Med. 2012;6(4):416–20 [DOI] [PubMed] [Google Scholar]
- 21.Neumann M, Heesch S, Gokbuget N, Schwartz S, Schlee C, Benlasfer O, et al. Clinical and molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL with a high frequency of FLT3 mutations. Blood Cancer J. 2012;2(1):e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, Haydu JE, Rigo I, Hadler M, et al. ETV6 mutations in early immature human T cell leukemias. J Exp Med. 2011; 208(13):2571–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neumann M, Heesch S, Schlee C, Schwartz S, Gokbuget N, Hoelzer D, et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood. 2013;121(23):4749–52 [DOI] [PubMed] [Google Scholar]
- 24.Terriou L, Ben Abdelali R, Roumier C, Lhermitte L, de Vos J, Cornillet P, et al. C/EBPA methylation is common in T-ALL but not in M0 AML. Blood. 2009;113(8):1864–6; author reply 6 [DOI] [PubMed] [Google Scholar]
- 25.Wouters BJ, Jorda MA, Keeshan K, Louwers I, Erpelinck-Verschueren CA, Tielemans D, et al. Distinct gene expression profiles of acute myeloid/T-lymphoid leukemia with silenced CEBPA and mutations in NOTCH1. Blood. 2007;110(10):3706–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang YL, Hsiao CC, Chen HY, Lin KH, Jou ST, Chen JS, et al. Absence of biallelic TCRgamma deletion predicts induction failure and poorer outcomes in childhood T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer. 2012;58(6):846–51 [DOI] [PubMed] [Google Scholar]
- 27.Callens C, Baleydier F, Lengline E, Ben Abdelali R, Petit A, Villarese P, et al. Clinical impact of NOTCH1 and/or FBXW7 mutations, FLASH deletion, and TCR status in pediatric T-cell lymphoblastic lymphoma. J Clin Oncol. 2012;30(16):1966–73 [DOI] [PubMed] [Google Scholar]
- 28.Kamps WA, Bokkerink JP, Hahlen K, Hermans J, Riehm H, Gadner H, et al. Intensive treatment of children with acute lymphoblastic leukemia according to ALL-BFM-86 without cranial radiotherapy: results of Dutch Childhood Leukemia Study Group Protocol ALL-7 (1988–1991). Blood. 1999;94(4):1226–36 [PubMed] [Google Scholar]
- 29.Kamps WA, Bokkerink JP, Hakvoort-Cammel FG, Veerman AJ, Weening RS, van Wering ER, et al. BFM-oriented treatment for children with acute lymphoblastic leukemia without cranial irradiation and treatment reduction for standard risk patients: results of DCLSG protocol ALL-8 (1991–1996). Leukemia. 2002;16(6):1099–111 [DOI] [PubMed] [Google Scholar]
- 30.Veerman AJ, Kamps WA, van den Berg H, van den Berg E, Bokkerink JP, Bruin MC, et al. Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997–2004). Lancet Oncol. 2009;10(10):957–66 [DOI] [PubMed] [Google Scholar]
- 31.van Grotel M, Meijerink JP, van Wering ER, Langerak AW, Beverloo HB, Buijs-Gladdines JG, et al. Prognostic significance of molecular-cytogenetic abnormalities in pediatric T-ALL is not explained by immunophenotypic differences. Leukemia. 2008;22(1):124–31 [DOI] [PubMed] [Google Scholar]
- 32.Stam RW, den Boer ML, Meijerink JP, Ebus ME, Peters GJ, Noordhuis P, et al. Differential mRNA expression of Ara-C-metabolizing enzymes explains Ara-C sensitivity in MLL gene-rearranged infant acute lymphoblastic leukemia. Blood. 2003;101(4):1270–6 [DOI] [PubMed] [Google Scholar]
- 33.Bergeron J, Clappier E, Cauwelier B, Dastugue N, Millien C, Delabesse E, et al. HOXA cluster deregulation in T-ALL associated with both a TCRD-HOXA and a CALM-AF10 chromosomal translocation. Leukemia. 2006;20(6):1184–7 [DOI] [PubMed] [Google Scholar]
- 34.Baldus CD, Burmeister T, Martus P, Schwartz S, Gokbuget N, Bloomfield CD, et al. High expression of the ETS transcription factor ERG predicts adverse outcome in acute T-lymphoblastic leukemia in adults. J Clin Oncol. 2006;24(29):4714–20 [DOI] [PubMed] [Google Scholar]
- 35.Baldus CD, Martus P, Burmeister T, Schwartz S, Gokbuget N, Bloomfield CD, et al. Low ERG and BAALC expression identifies a new subgroup of adult acute T-lymphoblastic leukemia with a highly favorable outcome. J Clin Oncol. 2007;25(24):3739–45 [DOI] [PubMed] [Google Scholar]
- 36.Ben Abdelali R, Asnafi V, Leguay T, Boissel N, Buzyn A, Chevallier P, et al. Pediatricinspired intensified therapy of adult T-ALL reveals the favorable outcome of NOTCH1/FBXW7 mutations, but not of low ERG/BAALC expression: a GRAALL study. Blood. 2011;118(19):5099–107 [DOI] [PubMed] [Google Scholar]
- 37.Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144(2):296–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neumann M, Coskun E, Fransecky L, Mochmann LH, Bartram I, Sartangi NF, et al. FLT3 mutations in early T-cell precursor ALL characterize a stem cell like leukemia and imply the clinical use of tyrosine kinase inhibitors. PLoS One. 2013;8(1):e53190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mingueneau M, Kreslavsky T, Gray D, Heng T, Cruse R, Ericson J, et al. The transcriptional landscape of alphabeta T cell differentiation. Nat Immunol. 2013;14(6):619–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Homminga I, Vuerhard MJ, Langerak AW, Buijs-Gladdines J, Pieters R, Meijerink JP. Characterization of a pediatric T-cell acute lymphoblastic leukemia patient with simultaneous LYL1 and LMO2 rearrangements. Haematologica. 2012;97(2):258–61 [DOI] [PMC free article] [PubMed] [Google Scholar]