

Abstract
HAX1 was identified as the gene responsible for the autosomal recessive type of severe congenital neutropenia. However, the connection between mutations in the HAX1 gene and defective granulopoiesis in this disease has remained unclear, mainly due to the lack of a useful experimental model for this disease. In this study, we generated induced pluripotent stem cell lines from a patient presenting for severe congenital neutropenia with HAX1 gene deficiency, and analyzed their in vitro neutrophil differentiation potential by using a novel serum- and feeder-free directed differentiation culture system. Cytostaining and flow cytometric analyses of myeloid cells differentiated from patient-derived induced pluripotent stem cells showed arrest at the myeloid progenitor stage and apoptotic predisposition, both of which replicated abnormal granulopoiesis. Moreover, lentiviral transduction of the HAX1 cDNA into patient-derived induced pluripotent stem cells reversed disease-related abnormal granulopoiesis. This in vitro neutrophil differentiation system, which uses patient-derived induced pluripotent stem cells for disease investigation, may serve as a novel experimental model and a platform for high-throughput screening of drugs for various congenital neutrophil disorders in the future.
Introduction
Severe congenital neutropenia (SCN) is a rare myelopoietic disorder resulting in recurrent life-threatening infections due to a lack of mature neutrophils,1 and individuals with SCN present for myeloid hypoplasia with an arrest of myelopoiesis at the promyelocyte/myelocyte stage.1,2 SCN is actually a multigene syndrome that can be caused by inherited mutations in several genes. For instance, approximately 60% of SCN patients are known to carry autosomal dominant mutations in the ELANE gene, which encodes neutrophil elastase (NE).3 An autosomal recessive type of SCN was first described by Kostmann in 1956,4 and defined as Kostmann disease. Although the gene responsible for this classical type of SCN remained unknown for more than 50 years, Klein et al. identified mutations in HAX1 to be responsible for this type of SCN in 2007.5 HAX1 localizes predominantly to mitochondria, where it controls inner mitochondrial membrane potential (Δψm) and apoptosis.6,7 Although an increase in apoptosis in mature neutrophils was presumed to cause neutropenia in HAX1 gene deficiency,5 the connection between HAX1 gene mutations and defective granulopoiesis in SCN has remained unclear.
To control infections, SCN patients are generally treated with granulocyte colony-stimulating factor (G-CSF); however, long-term G-CSF therapy associates with an increased risk of myelodysplastic syndrome and acute myeloid leukemia (MDS/AML).8,9 Although hematopoietic stem cell transplantations are available as the only curative therapy for this disease, they can result in various complications and mortality.4
Many murine models of human congenital and acquired diseases are invaluable for disease investigation as well as for novel drug discoveries. However, their use in a research setting can be limited if they fail to mimic strictly the phenotype of the human disease in question. For instance, the Hax1 knock-out mouse is characterized by lymphocyte loss and neuronal apoptosis, but not neutropenia.10 Thus, it is not a suitable experimental model for SCN. Induced pluripotent stem (iPS) cells are reprogrammed somatic cells with embryonic stem (ES) cell-like characteristics produced by the introduction of specific transcription factors,11,16 and they may substitute murine models of human disease. It is believed that iPS cell technology, which generates disease-specific pluripotent stem cells in combination with directed cell differentiation, will contribute enormously to patient-oriented research, including disease pathophysiology, drug screening, cell transplantation, and gene therapy.
In vitro neutrophil differentiation systems, which can reproduce the differentiation of myeloid progenitor cells to mature neutrophils, are needed to understand the pathogenesis of SCN better. Recently, we established a neutrophil differentiation system from human iPS cells17 as well as a serum- and feeder-free monolayer hematopoietic culture system from human ES and iPS cells.18 In this study, we generate iPS cell lines from an SCN patient with HAX1 gene deficiency and differentiate them into neutrophils in vitro. Furthermore, we corrected for the HAX1 gene deficiency in HAX1-iPS cells by lentiviral transduction with HAX1 cDNA and analyzed the neutrophil differentiation potential of these cells. Thus, this in vitro neutrophil differentiation system from patient-derived iPS cells may be a useful model for future studies in SCN patients with HAX1 gene deficiency.
Methods
Human iPS cell generation
Skin biopsy specimens were obtained from an 11-year old male SCN patient with HAX1 gene deficiency.19 This study was approved by the Ethics Committee of Kyoto University, and informed consent was obtained from the patient’s guardians in accordance with the Declaration of Helsinki. Fibroblasts were expanded in DMEM (Nacalai Tesque, Inc., Kyoto, Japan) containing 10% FBS (vol/vol, Invitrogen, Carlsbad, CA, USA) and 0.5% penicillin and streptomycin (wt/vol, Invitrogen). Generation of iPS cells was performed as described previously.12 In brief, we introduced OCT3/4, SOX2, KLF4, and cMYC using ecotropic retroviral transduction into patient’s fibroblasts expressing mouse Slc7a1. Six days after transduction, cells were harvested and re-plated onto mitotically inactive SNL feeder cells. On the following day, DMEM was replaced with primate ES cell medium (ReproCELL, Kanagawa, Japan) supplemented with basic fibroblast growth factor (5 ng/mL, R&D Systems, Minneapolis, MN, USA). Three weeks later, individual colonies were isolated and expanded.
Maintenance of cells
Control ES (KhES-1) and control iPS (253G4 and 201B6) cells were kindly provided by Drs. Norio Nakatsuji and Shinya Yamanaka (Kyoto University, Kyoto, Japan), respectively. These human ES and iPS cell lines were maintained on mitomycin-C (Kyowa Hakko Kirin, Tokyo, Japan)-treated SNL feeder cells as described previously17 and subcultured onto new SNL feeder cells every seven days.
Flow cytometric analysis
Cells were stained with antibodies as reported previously.17 Samples were analyzed using an LSR flow cytometer and Cell Quest software (Becton-Dickinson).
Neutrophil differentiation of iPS cells
In a previous study, we established a serum and feeder-free monolayer hematopoietic culture system from human ES and iPS cells.18 In this study, we modified this culture system to direct neutrophil differentiation. iPS cell colonies were cultured on growth factor-reduced Matrigel (Becton-Dickinson)-coated cell culture dishes in Stemline II hematopoietic stem cell expansion medium (Sigma-Aldrich, St. Louis, MO, USA) containing the insulin-transferrin-selenium (ITS) supplement (Invitrogen) and cytokines. iPS cells were treated with cytokines as follows: bone morphogenetic protein (BMP) 4 (20 ng/mL, R&D Systems) was added for four days and then replaced with vascular endothelial growth factor (VEGF) 165 (40 ng/mL, R&D Systems) on Day 4. On Day 6, VEGF 165 was replaced with a combination of stem cell factor (SCF, 50 ng/mL, R&D Systems), interleukin (IL)-3 (50 ng/mL, R&D Systems), thrombopoietin (TPO, 5 ng/mL, kindly provided by Kyowa Hakko Kirin), and G-CSF (50 ng/mL, also kindly provided by Kyowa Hakko Kirin). Thereafter, medium was replaced every five days.
Dead cell removal and CD45+ leukocyte separation
Floating cells were collected, followed by the removal of dead cells and cellular debris with the Dead Cell Removal kit (Miltenyi Biotec, Bergisch Gladbach, Germany). CD45+ cells were then separated using human CD45 microbeads (Miltenyi Biotec). Cell separation procedures were performed using the autoMACS Pro Separator (Miltenyi Biotec).
Statistical analysis
Statistical analysis was carried out using Student’s t-test. P<0.05 was considered statistically significant.
Results
Generation of iPS cell lines from an SCN patient with HAX1 gene deficiency
To generate patient-derived iPS cell lines, dermal fibroblasts were obtained from a male SCN patient with a homozygous 256C-to-T transition resulting in an R86X mutation in the HAX1 gene.19 These fibroblasts were reprogrammed to iPS cells after transduction with retroviral vectors encoding OCT3/4, SOX2, KLF4 and cMYC,12 and a total of 11 iPS cell clones were obtained. From these, we randomly selected three clones for propagation and subsequent analyses. One of these clones (HAX1 4F5) was generated with four factors (OCT3/4, SOX2, KLF4, and cMYC); the remaining clones (HAX1 3F3 and 3F5) were generated with three factors (OCT3/4, SOX2, and KLF4).12
All of these patient-derived iPS cell clones showed a characteristic human ES cell-like morphology (Figure 1A), and they propagated for serial passages in human ES cell maintenance culture medium. Quantitative PCR analysis showed the expression of NANOG, a pluripotent marker gene, to be comparable to that of control ES (KhES-1) and iPS (253G4 and 201B6) cells (Figure 1B). Surface marker analysis indicated that they were also positive for SSEA4, a human ES and iPS cell marker (Figure 1C). DNA sequencing analysis verified an identical mutation in the HAX1 gene in all established iPS cell clones (Figure 1D). The pluripotency of all iPS cell clones was confirmed by the presence of cell derivatives representing all three germ layers by teratoma formation after subcutaneous injection of undifferentiated iPS cells into immunocompromised NOD/SCID/γcnull mice (Figure 1E).
Figure 1.

Generation of iPS cell lines from an SCN patient with HAX1 gene deficiency. (A) Human ES cell-like morphology of HAX1-iPS cells. Scale bar: 200 μm. (B) NANOG expression in HAX1-iPS cells, control iPS cells (253G4 and 201B6), and patient-derived fibroblasts (HAX1 Fibro) compared to control ES cells (KhES1). GAPDH was used as an internal control (n = 3; bars represent SDs). (C) SSEA-4 expression analysis using flow cytometry. Gated on TRA1-85+DAPI− cells as viable human iPS (ES) cells (n = 3; bars represent SDs). (D) DNA sequencing analysis of the HAX1 gene in iPS cells. HAX1-iPS cells showed 256C>T (R86X) mutation that was found in the patient. (E) Teratoma formation from HAX1-iPS cells in the NOD/SCID/γcnull (NOG) mouse. Arrows indicate the following; Endoderm: respiratory epithelium; Mesoderm: cartilage; Ectoderm: pigmented epithelium. Scale bars: 200 μm. (A, D–E) Representative data (HAX1 3F5) are shown.
To validate the authenticity of iPS cells further, we investigated the expression of the four genes that were used for iPS cell generation. The expression level of all endogenous genes was comparable to control ES and iPS cells. On the other hand, transgene expression was largely undetectable in patient-derived iPS cell clones (Online Supplementary Figure S1A). Chromosomal analysis revealed that all patient-derived iPS cell clones maintained a normal karyotype (Online Supplementary Figure S1B). Genetic identity was shown by short tandem repeat analysis (Online Supplementary Figure S1C).
Taken collectively, these results indicate that iPS cell clones were comprised of good quality iPS cells derived from the somatic cells of an SCN patient with HAX1 gene deficiency (HAX1-iPS cells).
Maturation arrest at the progenitor level in neutrophil differentiation from HAX1-iPS cells
The paucity of mature neutrophils in the peripheral blood and a maturation arrest at the promyelocyte/myelocyte stage in the bone marrow are characteristic laboratory findings presented in the SCN patients with HAX1 gene deficiency. To investigate whether our patient-derived iPS cell model accurately replicated this disease phenotype, we assessed neutrophil differentiation from HAX1-iPS cells by using a serum- and feeder-free monolayer culture system18 with minor modifications (Online Supplementary Figure S2).
In this system, we cultured iPS cell colonies on Matrigel-coated dishes in serum-free medium supplemented with several cytokines and obtained hematopoietic cells as floating cells on approximately Day 26 of differentiation. May-Giemsa staining of floating live CD45+ cells derived from normal iPS cells showed that approximately 40% were mature neutrophils (Figure 2A and B). The remaining cells consisted of immature myeloid cells as well as a small number of macrophages. Cells of other lineages such as erythroid or lymphoid cells were not observed. On the other hand, HAX1-iPS cell-derived blood cells contained only approximately 10% mature neutrophils and approximately 50% immature myeloid cells, including myeloblasts and promyelocytes (Figure 2A and B). Flow cytometric analysis revealed that the percentage of CD34+ cells within HAX1-iPS cell-derived blood cells was significantly higher than in normal iPS cell-derived blood cells (Figure 2C), which also showed that the percentage of phenotypically immature myeloid cells was higher in HAX1-iPS cell-derived blood cells than in normal iPS cell-derived blood cells.
Figure 2.

Maturation arrest at the progenitor level in neutrophil differentiation from HAX1-iPS cells. (A) May-Giemsa staining of CD45+ cells derived from normal (253G4) and HAX1-iPS (HAX1 3F5) cells. Scale bars: 10 μm. (B) Morphological classification of CD45+ cells derived from iPS cells. Cells were classified into three groups: myeloblast and promyelocyte (MB/ProM), myelocyte and metamyelocyte (Myelo/Meta), and band and segmented neutrophils (Band/Seg) (n = 3; bars represent SDs). (C) Flow cytometric analysis of CD45+ cells derived from iPS cells. Cells gated on human CD45+ DAPI− were analyzed (n = 3; bars represent SDs; *P<0.05 compared to control iPS cells). (D) Immunocytochemical analysis of CD45+ cells derived from iPS cells (n = 3; bars represent SDs; *P<0.05 compared to control iPS cells). (E) NE staining of CD45+ cells derived from iPS cells (n = 3; bars represent SDs; *P<0.05 compared to control iPS cells). (F) Colony-forming assay of cells derived from iPS cells. On Day 16, living adherent cells were collected and cultured in methylcellulose medium (see Online Supplementary Appendix). The number of colonies generated from 1×104 cells is indicated (n = 3; bars represent SD; *P<0.05 compared to control iPS cells). (A–E) Live CD45+ cells derived from normal and HAX1-iPS cells on Day 26 of neutrophil differentiation were analyzed. Dead cells and CD45− cells were depleted using an autoMACS Pro separator (see Methods).
Immunocytochemical analysis for lactoferrin and gelatinase, which are constitutive proteins of neutrophil specific granules observed in mature neutrophils, showed that the proportion of these granule-positive cells was significantly lower in HAX1-iPS cell-derived blood cells than in normal iPS cell-derived blood cells (Figure 2D). NE is a protease stored in primary granules of neutrophilic granulocytes that are formed at the promyelocytic phase of granulocyte differentiation. ELANE mRNA expression in myeloid progenitors and the protein level of NE in plasma are markedly reduced in SCN patients with mutations in ELANE or HAX1.20 Consistent with this, the proportion of NE-positive cells was significantly lower in blood cells derived from HAX1-iPS cells than in those derived from normal iPS cells (Figure 2E). Thus, the level of functionally mature neutrophils decreased during in vitro granulopoietic differentiation of HAX1-iPS cells.
Next, we analyzed the colony-forming potential of HAX1-iPS cell-derived myeloprogenitor cells. Significantly fewer colonies, which were classified as granulocyte-macrophage (GM) or granulocyte (G) colony-forming units (CFU), were derived from HAX1-iPS cells than from control iPS cells. Furthermore, the colonies derived from HAX1-iPS cells were predominantly CFU-GM (Figure 2F). Thus, maturation arrest occurred at the clonogenic progenitor stage during in vitro neutrophil differentiation of HAX1-iPS cells.
SCN is characterized by severe neutropenia with very low absolute neutrophil counts in peripheral blood, and many SCN patients respond to G-CSF treatment.1,2 In colony-forming assays using bone marrow cells of SCN patients, primitive myeloid progenitor cells have reduced responsiveness to hematopoietic cytokines including G-CSF.21,22 Therefore, we next examined the response of HAX1-iPS cell-derived blood cells to G-CSF using a colony-forming assay. Although the number of colonies derived from HAX1-iPS cells slightly increased following the addition of G-CSF, it remained significantly lower than the number of colonies derived from control iPS cells in the absence of G-CSF (Figure 2F). These results indicate that the responsiveness of HAX1-iPS-derived blood cells to G-CSF was insufficient to restore the neutrophil count to a normal level and are consistent with the fact that the absolute neutrophil counts of SCN patients remain low following G-CSF therapy.19,21
Neutrophils derived from HAX1-iPS cells are predisposed to undergo apoptosis due to their reduced Δψm
Previous studies have shown HAX1 to localize to mitochondria6 and to mediate anti-apoptotic activity.7 Interestingly, this apoptotic predisposition of neutrophils due to their reduced Δψm was observed in HAX1-deficient patients,5 prompting us to examine apoptosis in HAX1-iPS cell-derived blood cells. Consistent with these reports, HAX1-iPS cell-derived blood cells showed a significantly higher percentage of Annexin V-positive cells than in control cells (Figure 3A). In addition, a mitochondrial membrane potential assay revealed that the percentage of cells with a low Δψm was significantly higher in HAX1-iPS cell-derived blood cells than in blood cells derived from control iPS cells (Figure 3B). By contrast, the percentage of cells with a low Δψm was similar in undifferentiated HAX1-iPS cells and undifferentiated control iPS cells (Online Supplementary Figure S3).
Figure 3.
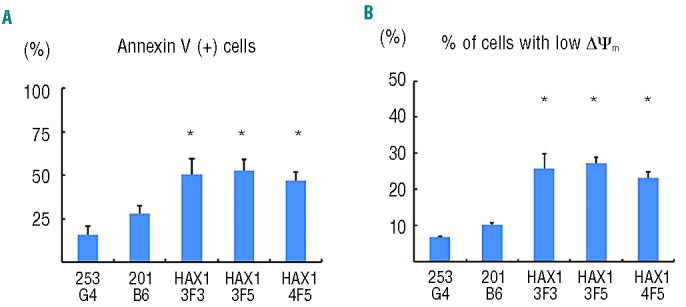
Neutrophils derived from HAX1-iPS cells are predisposed to undergo apoptosis due to their reduced Δψm. Annexin V assay (A) and mitochondrial membrane potential assay (B) of iPS cell-derived cells on Day 26 of neutrophil differentiation using flow cytometry. Cells gated on human CD45+ were analyzed (n = 3; bars represent SDs; *P<0.05 to control iPS cells).
Thus, increased apoptosis due to reduced Δψm causes defective granulopoiesis during neutrophil differentiation from HAX1-iPS cells, similar to the process observed in SCN patients with HAX1 gene deficiency.
Lentiviral transduction of HAX1 cDNA improves maturation arrest and apoptotic predisposition of HAX1-iPS cells
Because most HAX1 gene mutations in SCN patients are nonsense mutations resulting in a premature stop codon and protein truncation,23 loss of the HAX1 protein is believed to cause severe neutropenia. To uncover the pathophysiological hallmarks of this disease, we performed lentiviral transduction of HAX1 cDNA into HAX1-iPS cells.
We constructed lentiviral vectors that expressed HAX1 cDNA and EGFP as a marker gene (pCSII-EF-IEGFP; EGFP only, pCSII-EF-HAX1-IEGFP; HAX1 cDNA and EGFP) (Figure 4A). Efficient transduction of HAX1-iPS cells with these lentiviral vectors (HAX1 3F5+GFP; HAX1 3F5 transduced with pCSII-EF-IEGFP, HAX1 3F5+HAX1; HAX1 3F5 transduced with pCSII-EF-HAX1-IEGFP) was confirmed by a significant increase in HAX1 protein by Western blotting analysis (Figure 4B).
Figure 4.
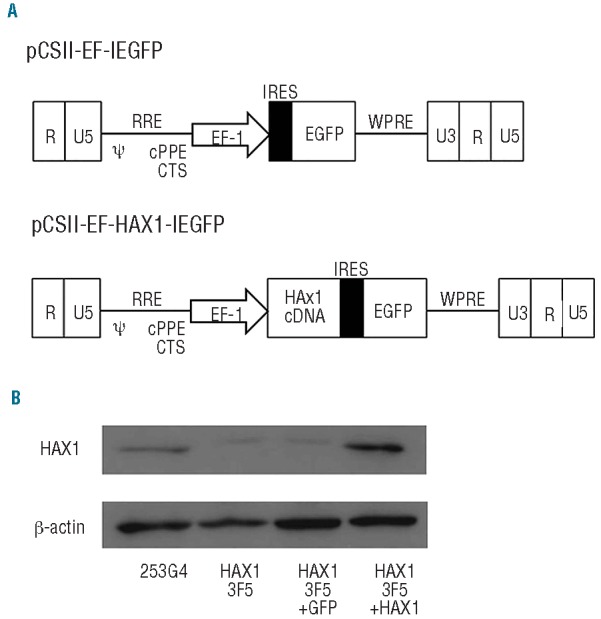
Lentiviral transduction of HAX1-iPS cells. (A) Lentiviral vector constructs with only EGFP pCSII-EF-IEGFP), and HAX1 cDNA and EGFP (pCSII-EF-HAX1-IEGFP). (B) Western blot analysis for HAX1 protein in lentivirally-transduced HAX1-iPS cells. β-actin was used as a loading control.
We then differentiated these lentiviral-transduced iPS cells into neutrophils, and examined whether defective granulopoiesis and apoptotic predisposition could be reversed. Morphologically, cells derived from HAX1 3F5+HAX1 showed a higher proportion of mature neutrophils than cells derived from HAX1 3F5+GFP and HAX1 3F5 (Figure 5A and B). Flow cytometric analysis revealed that the proportion of CD34+ cells was significantly lower in the cells derived from HAX1 3F5+HAX1 than HAX1 3F5+GFP and HAX1 3F5 (Figure 5C). Immunocytochemical analysis for lactoferrin and gelatinase showed that the proportion of these granule-positive cells in generated blood cells was significantly higher in HAX 3F5+HAX1 than in HAX13F5+GFP and HAX1 3F5 (Figure 5D). These results indicated that HAX1 cDNA increased the number of mature neutrophils in the neutrophil differentiation culture from HAX1-iPS cells in vitro. In addition, the percentage of NE-positive cells was significantly higher in cells derived from HAX1 3F5+HAX1 than in cells derived from HAX1 3F5+GFP and HAX1 3F5 (Figure 5E). Furthermore, the number of colonies derived from HAX1 3F5+HAX1 was comparable to the number derived from control cells (Figure 5F).
Figure 5.

Lentiviral transduction of HAX1 cDNA improves maturation arrest of HAX1-iPS cells. (A) May-Giemsa staining of CD45+ cells derived from HAX1 3F5+GFP and HAX1 3F5+HAX1 cells. Scale bars: 10 μm. (B) Morphological classification of CD45+ cells derived from lentivirally-transduced iPS cells. (n = 3; bars represent SDs). (C) Flow cytometric analysis of CD45+ cells derived from lentivirally-transduced iPS cells. Cells gated on GFP+ human CD45+ DAPI− were analyzed (n = 3; bars represent SDs; *P<0.05). (D) Immunocytochemical analysis of CD45+ cells derived from lentivirally-transduced iPS cells (n = 3; bars represent SDs; *P<0.05). (E) NE staining of CD45+ cells derived from lentivirally-transduced iPS cells (n = 3; bars represent SDs; *P<0.05). (F) Colony-forming assay of lentivirally-transduced cells derived from iPS cells. The number of colonies derived from 1×104 cells is indicated (n = 3; bars represent SD; *P<0.05 compared to HAX1 3F5 and HAX1 3F5+GFP). (A–E) Live CD45+ cells derived from lentivirally-transduced iPS cells on Day 26 of neutrophil differentiation were analyzed. Dead cells and CD45-cells were depleted using an autoMACS Pro separator (see Methods).
HAX1 3F5+HAX1-derived blood cells showed a significantly lower percentage of Annexin V-positive cells (Figure 6A) and a significantly lower percentage of cells with a low Δψm (Figure 6B) than HAX13F5+GFP and HAX1 3F5-derived blood cells. These results indicated that only HAX1 cDNA transduction improved defective granulopoiesis and apoptotic predisposition due to low Δψm in the neutrophil differentiation culture from HAX1-iPS cells in vitro.
Figure 6.
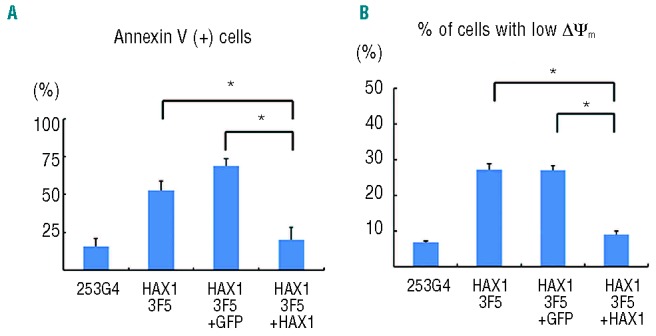
Lentiviral transduction of HAX1 cDNA prevents HAX1-iPS cells being predisposed to undergo apoptosis. Annexin V assay (A) and mitochondrial membrane potential assay (B) of lentivirally-transduced iPS cell-derived cells on Day 26 of neutrophil differentiation. Cells gated on GFP+ human CD45+ were analyzed (n = 3; bars represent SDs; *P<0.05).
Discussion
Animal models and in vitro cultures consisting of cells derived from patients are often used to investigate disease pathophysiology and to develop novel therapies. Unfortunately, Hax1 knock-out mice fail to reproduce abnormal granulopoiesis as observed in SCN patients.10 Moreover, bone marrow cells are not an ideal experimental tool because it is difficult to obtain sufficient blood cells due to the invasiveness of the aspiration procedure. Moreover, the pathophysiological mechanisms occurring during early granulopoiesis are difficult to address in primary patient samples.
Our established culture system efficiently induced directed hematopoietic differentiation, which consisted of myeloid cells at different stages of development, from various control and patient-derived HAX1-iPS cell lines. Furthermore, this in vitro neutrophil differentiation system produced sufficient myeloid cells, which enabled us to perform various types of assays. In addition, flow cytometry, a colony-forming assay, and cytostaining of HAX1-iPS cell-derived blood cells quantitatively demonstrated maturation arrest at the progenitor level and apoptotic predisposition due to low Δψm resulting in defective granulopoiesis, which were typically observed in SCN patients with HAX1 gene deficiency. Thus, our culture system may serve as a novel experimental model and a platform for high-throughput screening of drugs for neutropenia in SCN with HAX1 gene deficiency.
A colony-forming assay showed that the response to G-CSF administration correlated well with the responsiveness of SCN patients to G-CSF therapy. Defective granulopoiesis was recently reported in SCN-iPS cells with a mutation in ELANE.24 Our data showing defective granulopoiesis and reduced response to G-CSF administration are generally consistent with this report. The slight differences in CFU-G/GM colony-forming potential between this previous study and the current study might be due to differences in the causative gene (HAX1 or ELANE) or the culture system used for neutrophil differentiation, and/or to variation in the differentiation capabilities of the clones.
In our serum and feeder-free monolayer culture system, human ES and iPS cells differentiate into hematopoietic and endothelial cells via common KDR+CD34+ hemoangiogenic progenitors, which exist during early embryogenesis.18 Therefore, emergence of abnormal granulopoiesis in this system suggests that disease onset might occur at early hematopoietic stage (yolk sac or fetal liver), which would have never been addressed with patient samples.
We also showed that HAX1 cDNA transduction could reverse disease-related phenotypes such as abnormal granulopoiesis and apoptotic predisposition. Although little is known about the pathophysiology of SCN with HAX1 gene deficiency, these results clearly indicated that a loss in HAX1 protein might be the primary cause of neutropenia. These results also indicated the possibility of using patient-derived iPS cells for gene therapy; however, there are technical difficulties that would preclude these cells from being used in a clinical setting. Lentiviral vectors that randomly integrate transgenes can affect the expression of related genes, including cancer-related genes.25–28 To overcome these problems, we are required to select clones in which transgenes are integrated ‘safe harbor’ sites and highly expressed without perturbation of neighboring gene expression,29 or to take the zinc finger nuclease-mediated gene targeting approach30–32 specifically to a predesigned safe harbor site such as the AAVS1 locus,33 which has previously been shown to permit stable expression of transgenes with minimal effects on nearby genes.
The pluripotency of patient-derived iPS cells enables investigation of the pathophysiology of various organ abnormalities and/or dysfunctions. Many types of inherited bone marrow failure syndrome were characterized by multisystem developmental defects that affected the heart, kidney, skeletomuscular system, and central nervous system. Among these, neurological symptoms were frequently seen in SCN patients with HAX1 gene deficiency,19,23,34 suggesting that a loss in HAX1 may also affect neural development. Indeed, our patient also presented for epilepsy and severe delays in motor, cognitive, and intellectual development.19 In patient-derived cells, Δψm was not reduced in undifferentiated iPS cells but was reduced in differentiated neutrophils. No marked abnormalities in teratoma formation by HAX1-iPS cells were observed. These results are partially consistent with the fact that SCN patients with a HAX1 gene deficiency have only neutropenia and neurological symptoms, despite HAX1 being a ubiquitously expressed gene.6 Because some of these neurological symptoms cannot be reproduced in the currently available mouse model,10 additional studies will be necessary to address the effects of HAX1 on neural development by directed culture models of patient-derived iPS cells.
In conclusion, patient-derived iPS cell-derived myeloid cells were similar in disease presentation to SCN patients with HAX1 gene deficiency, which could be reversed by gene correction in a novel in vitro neutrophil differentiation system. This culture system will serve as a new tool to facilitate disease modeling and drug screening for congenital neutrophil disorders.
Acknowledgments
The authors would like to thank Dr. Norio Nakatsuji for providing the human ES cell line KhES-1, Dr. Shinya Yamanaka for providing human iPS cell lines 201B6 and 253G4, and Dr. Hiroyuki Miyoshi for providing pCSII-EF-MCS. We are grateful to Kyowa Hakko Kirin for providing TPO and G-CSF. We also thank the Center for Anatomical Studies, Kyoto University Graduate School of Medicine, for immunocytochemical analysis. Funding was provided by grants from the Ministry of Health, Labour and Welfare to KW, TN, and TH, a grant from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) to KW, TN, and TH, grants from the Leading Project of MEXT to TN, a grant from Funding Program for World-Leading Innovative Research and Development on Science and Technology (FIRST Program) of Japan Society for the Promotion of Science (JSPS) to TN, grants from the SENSHIN Medical Research Foundation to IK, and grants from the Fujiwara Memorial Foundation to TM. This work was also supported by the Global COE Program “Center for Frontier Medicine” from MEXT, Japan.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Welte K, Zeidler C, Dale DC. Severe congenital neutropenia. Semin Hematol. 2006;43(3):189–95 [DOI] [PubMed] [Google Scholar]
- 2.Skokowa J, Germeshausen M, Zeidler C, Welte K. Severe congenital neutropenia: inheritance and pathophysiology. Curr Opin Hematol. 2007;14(1):22–8 [DOI] [PubMed] [Google Scholar]
- 3.Dale DC, Person RE, Bolyard AA, Aprikyan AG, Bos C, Bonilla MA, et al. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000;96(7):2317–22 [PubMed] [Google Scholar]
- 4.Kostmann R. Infantile genetic agranulocytosis; agranulocytosis infantilis hereditaria. Acta Paediatr Suppl. 1956;45(Suppl 105):1–78 [PubMed] [Google Scholar]
- 5.Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA, et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet. 2007;39(1):86–92 [DOI] [PubMed] [Google Scholar]
- 6.Suzuki Y, Demoliere C, Kitamura D, Takeshita H, Deuschle U, Watanabe T. HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J Immunol. 1997;158(6):2736–44 [PubMed] [Google Scholar]
- 7.Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, Ye H, et al. K15 protein of Kaposi’s sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J Virol. 2002;76(2):802–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freedman MH, Bonilla MA, Fier C, Bolyard AA, Scarlata D, Boxer LA, et al. Myelodysplasia syndrome and acute myeloid leukemia in patients with congenital neutropenia receiving G-CSF therapy. Blood. 2000;96(2):429–36 [PubMed] [Google Scholar]
- 9.Rosenberg PS, Zeidler C, Bolyard AA, Alter BP, Bonilla MA, Boxer LA, et al. Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol. 2010;150(2):196–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chao JR, Parganas E, Boyd K, Hong CY, Opferman JT, Ihle JN. Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature. 2008;452(7183):98–102 [DOI] [PubMed] [Google Scholar]
- 11.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76 [DOI] [PubMed] [Google Scholar]
- 12.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72 [DOI] [PubMed] [Google Scholar]
- 13.Meissner A, Wernig M, Jaenisch R. Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nat Biotechnol. 2007;25(10):1177–81 [DOI] [PubMed] [Google Scholar]
- 14.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313–7 [DOI] [PubMed] [Google Scholar]
- 15.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451(7175):141–6 [DOI] [PubMed] [Google Scholar]
- 16.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–20 [DOI] [PubMed] [Google Scholar]
- 17.Morishima T, Watanabe K, Niwa A, Fujino H, Matsubara H, Adachi S, et al. Neutrophil differentiation from human-induced pluripotent stem cells. J Cell Physiol. 2011;226(5):1283–91 [DOI] [PubMed] [Google Scholar]
- 18.Niwa A, Heike T, Umeda K, Oshima K, Kato I, Sakai H, et al. A novel serum-free monolayer culture for orderly hematopoietic differentiation of human pluripotent cells via mesodermal progenitors. PLoS One. 2011;6(7):e22261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsubara K, Imai K, Okada S, Miki M, Ishikawa N, Tsumura M, et al. Severe developmental delay and epilepsy in a Japanese patient with severe congenital neutropenia due to HAX1 deficiency. Haematologica. 2007;92(12):e123–5 [DOI] [PubMed] [Google Scholar]
- 20.Skokowa J, Fobiwe JP, Dan L, Thakur BK, Welte K. Neutrophil elastase is severely down-regulated in severe congenital neutropenia independent of ELA2 or HAX1 mutations but dependent on LEF-1. Blood. 2009;114(14):3044–51 [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi M, Yumiba C, Kawaguchi Y, Tanaka Y, Ueda K, Komazawa Y, et al. Abnormal responses of myeloid progenitor cells to recombinant human colony-stimulating factors in congenital neutropenia. Blood. 1990;75(11):2143–9 [PubMed] [Google Scholar]
- 22.Konishi N, Kobayashi M, Miyagawa S, Sato T, Katoh O, Ueda K. Defective proliferation of primitive myeloid progenitor cells in patients with severe congenital neutropenia. Blood. 1999;94(12):4077–83 [PubMed] [Google Scholar]
- 23.Germeshausen M, Grudzien M, Zeidler C, Abdollahpour H, Yetgin S, Rezaei N, et al. Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood. 2008;111(10):4954–7 [DOI] [PubMed] [Google Scholar]
- 24.Hiramoto T, Ebihara Y, Mizoguchi Y, Nakamura K, Yamaguchi K, Ueno K, et al. Wnt3a stimulates maturation of impaired neutrophils developed from severe congenital neutropenia patient-derived pluripotent stem cells. Proc Natl Acad Sci USA. 2013;110(8):3023–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–9 [DOI] [PubMed] [Google Scholar]
- 26.Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12(4):401–9 [DOI] [PubMed] [Google Scholar]
- 27.Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008; 118(9):3143–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467(7313):318–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LM, et al. Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat Biotechnol. 2011;29(1):73–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou J, Sweeney CL, Chou BK, Choi U, Pan J, Wang H, et al. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood. 2011;117(21):5561–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeKelver RC, Choi VM, Moehle EA, Paschon DE, Hockemeyer D, Meijsing SH, et al. Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 2010;20(8):1133–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27(9):851–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henckaerts E, Dutheil N, Zeltner N, Kattman S, Kohlbrenner E, Ward P, et al. Site-specific integration of adeno-associated virus involves partial duplication of the target locus. Proc Natl Acad Sci USA. 2009;106(18):7571–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishikawa N, Okada S, Miki M, Shirao K, Kihara H, Tsumura M, et al. Neuro-developmental abnormalities associated with severe congenital neutropenia due to the R86X mutation in the HAX1 gene. J Med Genet. 2008;45(12):802–7 [DOI] [PubMed] [Google Scholar]