

Abstract
In hematopoietic stem cell transplantation for hemophagocytic lymphohistiocytosis, high transplant-related mortality after busulfan-based myeloablative regimens has been observed. Conditioning regimens with reduced toxicity based on melphalan or treosulfan are promising alternatives. We retrospectively analyzed hematopoietic stem cell transplantations in 19 hemophagocytic lymphohistiocytosis patients after conditioning with fludarabine, treosulfan, alemtuzumab, with or without thiotepa. Overall and disease-free survivals were 100% (follow up 7–31 months). Two patients required second transplant (1 after haploidentical transplantation). In 6 patients, overall donor chimerism dropped below 75% and prompted donor lymphocyte infusions. Administration of donor lymphocytes or second transplantation were significantly more frequent after transplantation from a human leukocyte antigen mismatched (9/10) versus matched (10/10) donor (P=0.018). The toxicity profile was favorable, with one veno-occlusive disease, one grade 3 graft-versus-host disease after donor lymphocyte infusion, and 2 severe viral infections (1 influenza, 1 Epstein Barr virus). In conclusion, the treosulfan-based regimen in hemophagocytic lymphohistiocytosis is effective with low toxicity and gives excellent overall and disease-free survival rates. In the future, the incidence of mixed chimerism, particularly after human leukocyte antigen mismatched donor transplants, needs to be addressed.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory condition presenting with fever, hepatosplenomegaly, and cytopenia. Untreated, the full picture of HLH is fatal. In most cases of hereditary HLH, hematopoietic stem cell transplantation (HSCT) must follow a remission induction therapy to prevent reactivations. Busulfan-based myeloablative conditioning regimens for HSCT in HLH are associated with high transplant-related mortality and particularly high incidence of veno-occlusive disease (VOD).1,2 Melphalan-based reduced intensity conditioning (RIC) regimens have substantially improved overall survival.3–6 Remaining issues are a high rate of mixed chimerism requiring donor lymphocyte infusions (DLI) with a substantial rate of subsequent graft-versus-host disease (GvHD) and fatal infections due to prolonged immunosuppression.5 We hypothesized that treosulfan (with addition of thiotepa in selected patients) might be more lymphotoxic and myelosuppressive than melphalan and still produce less adverse effects than myeloablative busulfan.
Methods
In this retrospective multicenter study, we assessed survival, engraftment, donor chimerism, disease reactivation, and adverse events after HSCT. Inclusion criteria were: 1) a known hereditary defect predisposing to HLH, i.e. familial HLH (FHL2-5), X-linked lymphoproliferative syndrome (XLP) type 1, deficiency of X-linked inhibitor of apoptosis (XIAP), Griscelli syndrome 2, and Chediak-Higashi syndrome, or if no genetic defect was identifiable, a degranulation defect as determined by the CD107 assay in resting NK cells;7 2) the administration of an HSCT conditioning regimen containing fludarabine (150–180 mg/m2 or 5–6 mg/kg), treosulfan (42 g/m2 or 36 g/m2 if <12 kg), and facultative thiotepa (10 mg/kg or 7 mg/kg if <12 kg). Alemtuzumab was administered at a dose of 0.3–1.0 mg/kg. Addition of thiotepa was dependent on center preference, in particular in patients at risk, most importantly absence of full remission as defined elsewhere2 and less frequently an HLA-mismatched (9/10) donor transplant. The decision of centers to apply this regimen was based on favorable experience with treosulfan-based conditioning in other diseases.8 Patients from centers applying different regimens, e.g. use of ATG or melphalan, were not included. Any patient age, donor type, or stem cell source was eligible. Central nervous system involvement was defined as published earlier.9 Nineteen patients from 9 German centers (6 patients from 1 center, 3 patients from 1, 2 patients each from 3, and 1 patient each from 4 centers), who underwent HSCT between June 2010 and December 2012, were analyzed.
Statistics and ethical considerations
Differences in distribution were compared in a 2-step approach. 1) Three potential risk factors (donor type, remission, and administration of thiotepa) for post-HSCT cellular therapy (DLI, second HSCT, stem cell boost) were each tested in a univariate model using two-tailed Fisher’s exact test with calculation of the odds ratio as estimator of effect. The variable with the poorest correlation as judged by the odds ratio and the P value was excluded from further analyses. Second, a multivariate analysis with the remaining variables was performed with a logistic regression model, calculating odds ratio, 95% confidence interval, and P value. P <0.05 was considered significant. Calculations were performed using PASW statistics 18 (IBM SPSS). One haploidentical HSCT was excluded from this analysis. Patients or legal guardians had given their written informed consent in accordance with the Declaration of Helsinki. The study was approved by the ethical review board of the Hamburg Chamber of Physicians.
Results
Patients’ characteristics, conditioning regimen and GvHD prophylaxis
Patients’ and HSCT characteristics are listed in Table 1. The cohort included 6 different known genetic defects. Median age at HSCT was 3.9 years (range 0.3–22 years), with 6 infants (< 12 months) and 2 young adults (≥ 18 years). Particular features were FHL5-related enteropathy in 2 patients, XIAP deficiency related chronic inflammatory bowel disease (IBD) in 2 patients, and Hodgkin’s disease two years prior to HSCT in one patient. Most patients had received the HLH2004 protocol as first-line therapy. Two patients were transplanted before the appearance of HLH; one patient with Griscelli syndrome type 2 and one patient with XIAP deficiency for whom HSCT was performed for intractable inflammatory bowel disease. Select aspects of 3 XIAP deficiency patients10 and 5 FHL5 patients11 are included in other reports.
Table 1.
Patients’ characteristics.
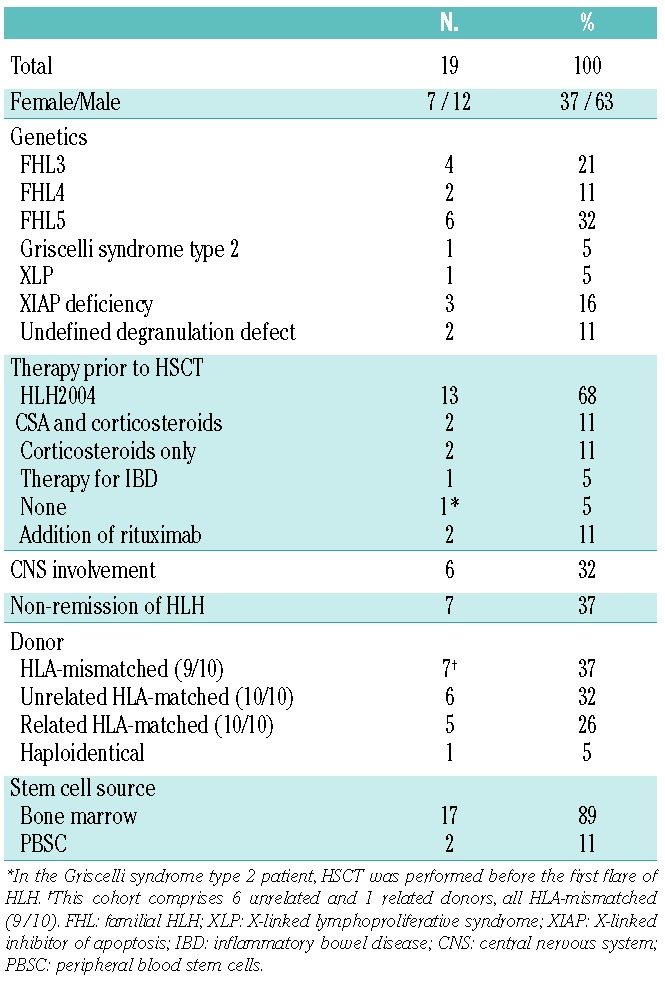
A fludarabine dose of 150 mg/m2 or 5 mg/kg was administered to 16 patients from Day −7 until Day −3 (±1 day). Three patients received 160–180 mg/m2 or 6 mg/kg from Day −8 until Day −3 (±1 day). All patients received treosulfan at a dose of 42 g/m2 (or 36 g/m2 if <12 kg) from Day −6 until Day −4 (±1 day). Thiotepa at a dose of 10 mg/kg (or 7 mg/kg if <12 kg), given on Day −3 (±1 day) divided in two doses, was added in 14 patients.
Alemtuzumab was administered in all patients: 0.8–1.0 mg/kg for HLA-mismatched donor (9/10) transplants and/or patients in non-remission, n=10; 0.3–0.4 mg/kg in HLA-matched (10/10) sibling donor transplants for patients in remission, n=4; 0.5–1.0 mg/kg for HLA-matched (10/10) unrelated donor transplants for patients in remission, n=5. In the patient who received a CD34+ selected haploidentical graft, alemtuzumab was given primarily to treat persistent HLH activity and prevent rejection. Cumulative alemtuzumab doses of 0.4 mg/kg or over were divided in daily doses of 0.2 mg/kg/d (n=16), 4 of which starting with 0.05 or 0.1 mg/kg/d; 0.3 mg/kg were administered in daily doses of 0.1 mg/kg/d (n=3). In 17 patients, the antibody was given prior to HSCT (first day ranging from −10 to −4, last day ranging from −7 to −1) and in another 2, alemtuzumab was infused until Day +1.
The following drug combinations for post-HSCT GvHD prophylaxis were applied: ciclosporin A (CSA) and methotrexate (n=9, in 1 of whom CSA was substituted by MMF on Day 49 due to poor tolerability); CSA and MMF (n=7); CSA only (n=2); tacrolimus and MMF due to poor tolerability of CSA during primary HLH therapy (n=1), and MMF only in 1 haploidentical HSCT with a CD34+ selected graft. CSA and tacrolimus administration started on Day −1, targeting at trough levels of 80–180 μg/L and 8–12 μg/L, respectively, which were terminated at a median of 105, range 35–703 days. MTX was infused on Days +1, +3 and +6 at a dose of 10 mg/m2. MMF was given from Day −1 at a dose of 45 mg/kg/d or 1200 mg/m2/d.
Overall outcome
Both overall survival and disease-free survival were 100% with a follow up of 7–31 months (median 16 months). The last Lansky/Karnofsky score was 90–100% in 18 patients and 60% in one patient due to pre-existing neurological deficits. However, the FHL5-related enteropathy persisted in both patients, as described previously.11 In both patients with IBD due to XIAP-deficiency, inflammatory activity ceased after HSCT.
Engraftment, chimerism, immune reconstitution, and subsequent cell therapy
Primary leukocyte engraftment (>1000/μL) was successful in all patients at a median of 20 days (range 11–62 days). Two patients required a second HSCT, one at Day 33 for early graft rejection after haploidentical HSCT (CD34 positive-selected peripheral stem cell graft) and one at Day 125 for secondary graft failure. In the remaining 17 patients, overall donor chimerism was complete (>95%) in 5 patients, dropped to 75–95% in 6 (3 of which returned to full chimerism and 3 remained stable after reduction of immunosuppression), and decreased to a nadir of 20–74% in 6 patients. In the latter, DLI led to stabilization of donor chimerism in 3 (1 of which with a single stem cell boost) and reversion to full donor chimerism in 2 patients; one patient is scheduled for DLI. At last follow up (median 408, range 196–847 days), overall donor chimerism was complete in 10, 75–95% in 3, and 20–74% in 4 patients. CD3+ T-cell counts over 500/μL were achieved after a median of 162 days (n=12).
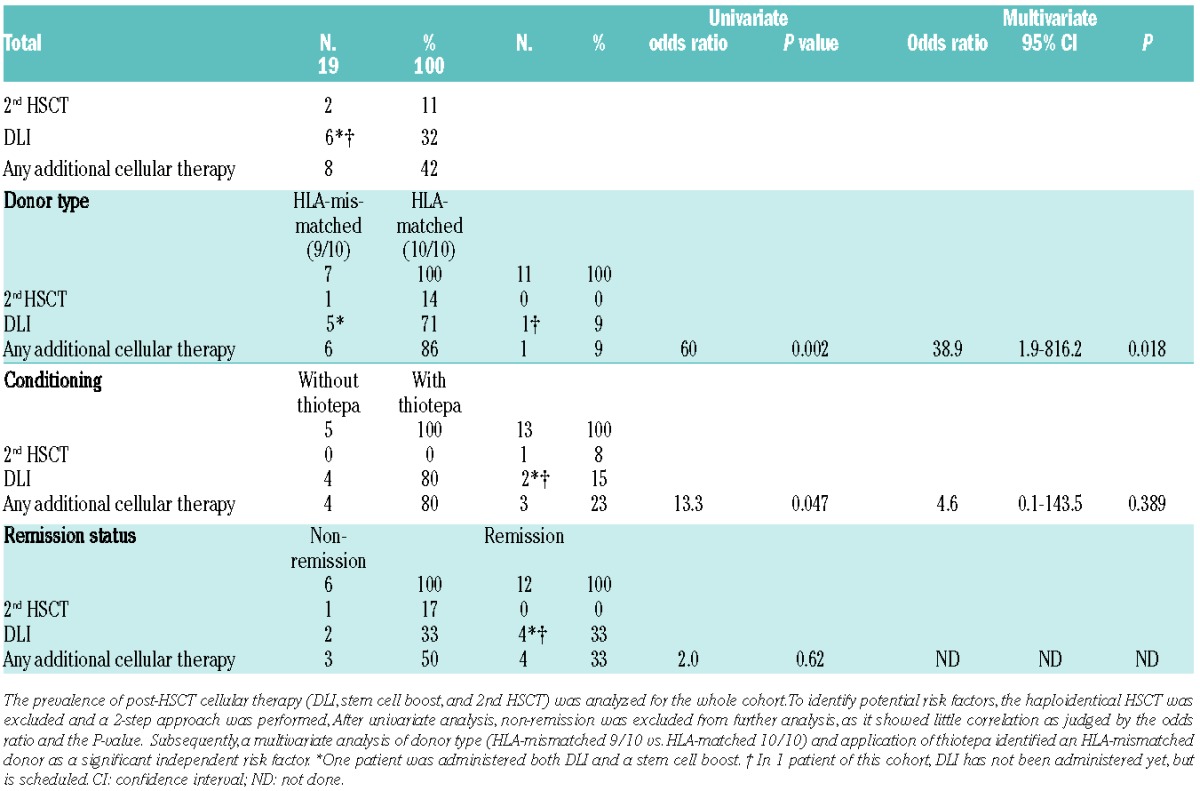
DLI, stem cell boost, or second HSCT were required in 42% of patients in our cohort (Table 2). Importantly, 5 out of 6 patients requiring DLI and one patient with secondary graft failure originated from the group of 7 patients with an HLA-mismatched (9/10) donor, and the graft rejection occurred after haploidentical HSCT. The haploidentical HSCT was excluded from the evaluation of potential risk factors for post-HSCT cellular therapy (Table 2). In the univariate analysis (donor type (HLA-matched 10/10 vs. HLA-mismatched 9/10), administration of thiotepa, and remission status), non-remission was identified as not significant (P=0.62). While the univariate analysis rendered P values of 0.002 for an HLA-mismatched (9/10) donor and 0.047 for a conditioning regimen without thiotepa, the subsequent multivariate model showed only for the risk factor donor type a significant effect (P=0.018) with an odds ratio of 38.9. The effect of thiotepa (odds ratio 4.6) was not significant in the multivariate analysis (P=0.389).
Table 2.
Comparison of risk factors for second HSCT, DLI, and stem cell boost.
Toxicity, infection and GvHD
Sixteen patients received either defibrotide12 (n=9) and/or heparin (n=9) prophylaxis for veno-occlusive disease (VOD). Only one hepatic VOD developed in the haploidentical HSCT, despite defibrotide. Further serious adverse events included life threatening hemorrhage (n=2), mucositis grade 3 or 4 (n=2), and skin toxicity grade 3 (n=3, 2 of whom had received thiotepa). Five bacteremias were detected, one of which with clinical septicemia, and 3 possible fungal infections. Post-HSCT Epstein-Barr viremia occurred in 2 patients, requiring rituximab in one. Systemic cytomegalovirus (n=3), herpes simplex virus (n=3), adenovirus (n=1), human herpes virus 6 (n=1), and influenza A (n=2) were detected, leading to major complications only in one case of influenza (pulmonary hemorrhage, extracorporal membrane oxygenation). BK virus was found in the urine in 3 and herpes zoster developed in 3 patients. Intubation was necessary in 3 patients (influenza, bleeding, and septicemia). Only one acute grade 3 GvHD after DLI (skin, gastointestinal, liver) and 4 grade 1 or 2 GvHD occurred, none resulted in long-term sequelae or chronic GvHD.
Discussion
The overall and disease-free survivals of 100% (n=19) in this cohort suggest the treosulfan-based conditioning as an alternative to the mostly melphalan-based RIC, which gave similar results with overall survival of 84% (n=25)4 and 92% (n=26).5 In a study on umbilical cord blood transplantation in HLH patients after RIC including melphalan and low-dose total body irradiation in some cases, overall survival was 91% (n=11).6 A broad spectrum of underlying genetic defects is covered in the analysis, demonstrating the efficacy in various subtypes of hereditary HLH, including XIAP-deficiency for which overall survival was reported to be poor in a previous study, particularly in patients in non-remission: 14% after myeloablative conditioning (n=7) and 50% after RIC with fludarabin, melphalan, and alemtuzumab (n=10).10 Treosulfan has previously been proven to be a safe and effective conditioning agent for malignant and non-malignant disease.8,13,14 However, previous experience with treosulfan, fludarabine, and alemtuzumab in HLH was anecdotal. In a series of immunodeficiencies, 3 of 4 HLH patients died,15 whereas 2 patients in another cohort are long-term survivors.16
The rate of additional cellular therapy after first HSCT (including second HSCT, administration of DLI, and stem cell boost) was high in the cohort with HLA-mismatched (9/10) donors (86%) while it was only 9% if the donor was HLA-matched (10/10) (P=0.018). This finding particularly points to the need to optimize the conditioning regimen for HLA-mismatched HSCT. Since at least in the univariate test, sustained engraftment in patients who had received thiotepa was superior, the addition of this drug to the regimen may be an option. However, this finding did not reach the level of significance in the multivariate model and thus requires further confirmation. In the literature, melphalan-based RIC resulted in mixed chimerism in 6 of 21 (29%)4 and 14 of 26 (54%)5 surviving patients. In a cohort with cord blood SCT after melphalan-based RIC, 2 of 10 (20%) surviving patients required second transplant and another 2 (20%) had mixed chimerism and late graft failure.6 To date, the degree of donor chimerism required in humans to prevent recurrence of HLH is not entirely clear.
Serotherapy in HSCT is a matter of debate. We have decided to use alemtuzumab because it was reported to induce remission after failure of primary HLH therapy17 and had successfully been used in previous RIC regimens for HLH.4,5 A beneficial effect of alemtuzumab in HSCT for HLH is assumed due to the wide distribution of CD52, including T cells and antigen-presenting cells, potentially suppressing remaining disease activity, which is relevant in particular for patients in non-remission.5 Despite slow T-cell reconstitution, the rate of relevant complications due to virus reactivations and infections was low. The previously reported high incidence of viral infections after alemtuzumab18 has been attributed to an estimated halflife of 15–21 days, implying that active antibodies may persist for several months.19 Given the low incidence of acute and chronic GvHD in the present study, a dose reduction of alemtuzumab in HLA-mismatched donor transplants may be another option to reduce the rate of mixed chimerism. A different approach may be the application of alemtuzumab three weeks prior to HSCT.5
Unlike most other immunodeficiencies, HLH is associated with severe complications in HSCT after myeloablative conditioning due to the hyperinflammatory nature of the condition. As in previous reports on RIC in HLH,3,5 the rate of VOD was low in our analysis. However, it must be kept in mind that defibrotide and/or heparin were given as VOD prophylaxis. Defibrotide has been shown to protect against VOD after myeloablative conditioning if at least one risk factor for VOD was present; the risk factors included HLH as underlying condition and the use of busulfan or melphalan.12 The occurrence of grade 3 skin toxicities in 3 patients is likely to be due to the use of treosulfan and thiotepa, and underscores the importance of attention to skin care, including frequent washing during application of thiotepa.
Pre-HSCT characteristics of patients in the present cohort were comparable to most previous reports. Median age at HSCT (3.9 years) in the present cohort was in the same range as in the recent report5 on melphalan-based RIC (5 years). Patients in earlier reports were younger at HSCT (6–14 months).2,3,20 This may reflect the increasing awareness of hereditary forms of HLH beyond infancy.21 The rate of patients in non-remission (37% in this report) was 8–47% in other reports1–3,5,6,20 and the incidence of CNS involvement at any time prior to transplant (32% in this report) is similar to most of these other reports1–3,5,6 at 23–41%; in only one study was this rate substantially higher at 80%.20
In conclusion, fludarabine, treosulfan, alemtuzumab, and thiotepa represent a conditioning regimen with a high rate of overall and disease-free survival and low toxicity in the complication-prone group of patients with hereditary HLH. In recipients of a graft from an HLA-mismatched donor, the rate of primary complete donor chimerism requires improvement, e.g. by inclusion of thiotepa and/or adjustment of serotherapy.
Acknowledgments
We thank Stephan Ehl for immunological workup and Udozur Stadt for genetic studies that both helped to identify hereditary disease, Sandra Standke for support in data management, and Jan Felix Kersten for support in statistical analyses.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Baker KS, Filipovich AH, Gross TG, Grossman WJ, Hale GA, Hayashi RJ, et al. Unrelated donor hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Bone Marrow Transplant. 2008;42(3):175–80 [DOI] [PubMed] [Google Scholar]
- 2.Horne A, Janka G, Maarten Egeler R, Gadner H, Imashuku S, Ladisch S, et al. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. 2005;129(5):622–30 [DOI] [PubMed] [Google Scholar]
- 3.Cooper N, Rao K, Gilmour K, Hadad L, Adams S, Cale C, et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis. Blood. 2006;107(3):1233–6 [DOI] [PubMed] [Google Scholar]
- 4.Cooper N, Rao K, Goulden N, Webb D, Amrolia P, Veys P. The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and Langerhans cell histiocytosis. Bone Marrow Transplant. 2008;42 (Suppl 2):S47–50 [DOI] [PubMed] [Google Scholar]
- 5.Marsh RA, Vaughn G, Kim MO, Li D, Jodele S, Joshi S, et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. 2010;116(26):5824–31 [DOI] [PubMed] [Google Scholar]
- 6.Nishi M, Nishimura R, Suzuki N, Sawada A, Okamura T, Fujita N, et al. Reduced-intensity conditioning in unrelated donor cord blood transplantation for familial hemophagocytic lymphohistiocytosis. Am J Hematol. 2012;87(6):637–9 [DOI] [PubMed] [Google Scholar]
- 7.Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. 2012;119(12):2754–63 [DOI] [PubMed] [Google Scholar]
- 8.Greystoke B, Bonanomi S, Carr T F, Gharib M, Khalid T, Coussons M, et al. Treosulfan-containing regimens achieve high rates of engraftment associated with low transplant morbidity and mortality in children with non-malignant disease and significant comorbidities. Br J Haematol. 2008;142(2):257–62 [DOI] [PubMed] [Google Scholar]
- 9.Horne A, Trottestam H, Arico M, Egeler RM, Filipovich AH, Gadner H, et al. Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol. 2008;140(3):327–35 [DOI] [PubMed] [Google Scholar]
- 10.Marsh RA, Rao K, Satwani P, Lehmberg K, Muller I, Li D, et al. Allogeneic hematopoietic cell transplantation for XIAP deficiency: an international survey reveals poor outcomes. Blood. 2012;121(6):877–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pagel J, Beutel K, Lehmberg K, Koch F, Maul-Pavicic A, Rohlfs AK, et al. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5). Blood. 2012;119(25):6016–24 [DOI] [PubMed] [Google Scholar]
- 12.Corbacioglu S, Cesaro S, Faraci M, Valteau-Couanet D, Gruhn B, Rovelli A, et al. Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet. 2012;379(9823):1301–9 [DOI] [PubMed] [Google Scholar]
- 13.Bernardo ME, Piras E, Vacca A, Giorgiani G, Zecca M, Bertaina A, et al. Allogeneic hematopoietic stem cell transplantation in thalassemia major: results of a reduced-toxicity conditioning regimen based on the use of treosulfan. Blood. 2012;120(2):473–6 [DOI] [PubMed] [Google Scholar]
- 14.Wachowiak J, Sykora KW, Cornish J, Chybicka A, Kowalczyk JR, Gorczynska E, et al. Treosulfan-based preparative regimens for allo-HSCT in childhood hematological malignancies: a retrospective study on behalf of the EBMT pediatric diseases working party. Bone Marrow Transplant. 2011;46(12):1510–8 [DOI] [PubMed] [Google Scholar]
- 15.Slatter MA, Rao K, Amrolia P, Flood T, Abinun M, Hambleton S, et al. Treosulfan-based conditioning regimens for hematopoietic stem cell transplantation in children with primary immunodeficiency: United Kingdom experience. Blood. 2011;117(16):4367–75 [DOI] [PubMed] [Google Scholar]
- 16.Beier R, Schulz A, Honig M, Eyrich M, Schlegel PG, Holter W, et al. Long-term follow-up of children conditioned with Treosulfan: German and Austrian experience. Bone Marrow Transplant. 2013;48(4):491–501 [DOI] [PubMed] [Google Scholar]
- 17.Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2012;60(1):101–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakrabarti S, Mackinnon S, Chopra R, Kottaridis PD, Peggs K, O’Gorman P, et al. High incidence of cytomegalovirus infection after nonmyeloablative stem cell transplantation: potential role of Campath-1H in delaying immune reconstitution. Blood. 2002;99(12):4357–63 [DOI] [PubMed] [Google Scholar]
- 19.Morris EC, Rebello P, Thomson KJ, Peggs KS, Kyriakou C, Goldstone AH, et al. Pharmacokinetics of alemtuzumab used for in vivo and in vitro T-cell depletion in allogeneic transplantations: relevance for early adoptive immunotherapy and infectious complications. Blood. 2003;102(1):404–6 [DOI] [PubMed] [Google Scholar]
- 20.Ouachee-Chardin M, Elie C, de Saint Basile G, Le Deist F, Mahlaoui N, Picard C, et al. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. 2006;117(4):e743–50 [DOI] [PubMed] [Google Scholar]
- 21.Rohr J, Beutel K, Maul-Pavicic A, Vraetz T, Thiel J, Warnatz K, et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica. 2010;95(12):2080–7 [DOI] [PMC free article] [PubMed] [Google Scholar]