

Abstract
The phosphoinositide 3-kinase pathway represents an important anticancer target because it has been implicated in cancer cell growth, survival, and motility. Recent studies show that PI3K may also play a role in the development of resistance to currently available therapies. In a broad range of cancers, various components of the phosphoinositide 3-kinase signaling axis are genetically modified, and the pathway can be activated through many different mechanisms. The frequency of genetic alterations in the phosphoinositide 3-kinase pathway, coupled with the impact in oncogenesis and disease progression, make this signaling axis an attractive target in anticancer therapy. A better understanding of the critical function of the phosphoinositide 3-kinase pathway in leukemias and lymphomas has led to the clinical evaluation of novel rationally designed inhibitors in this setting. Three main categories of phosphoinositide 3-kinase inhibitors have been developed so far: agents that target phosphoinositide 3-kinase and mammalian target of rapamycin (dual inhibitors), pan-phosphoinositide 3-kinase inhibitors that target all class I isoforms, and isoform-specific inhibitors that selectively target the α, -β, -γ, or -δ isoforms. Emerging data highlight the promise of phosphoinositide 3-kinase inhibitors in combination with other therapies for the treatment of patients with hematologic malignancies. Further evaluation of phosphoinositide 3-kinase inhibitors in first-line or subsequent regimens may improve clinical outcomes. This article reviews the role of phosphoinositide 3-kinase signaling in hematologic malignancies and the potential clinical utility of inhibitors that target this pathway.
Introduction
Phosphoinositide 3-kinase (PI3K) signaling plays a key role in protein synthesis, gene transcription, cell growth, and motility. The PI3K pathway is dysregulated in various cancers, and aberrant PI3K signaling is associated with oncogenesis and disease progression in solid tumors and hematologic malignancies.1–3 Oncogenic activation of the PI3K pathway can occur via several mechanisms, including mutations or amplification of the PIK3CA gene, activating mutations in upstream receptor tyrosine kinases (RTKs), activating mutations of the downstream effector protein AKT, or inactivating mutations or loss of the phosphatase and tensin homolog deleted on chromosome 10 (PTEN, a negative regulator of the PI3K pathway).1 Although PIK3CA mutations are rare in hematologic malignancies, constitutive activation of the PI3K pathway is common, and certain PI3K isoforms are expressed mainly in hematopoietic cells.4 Interest in this pathway has led to the development of novel inhibitors that target PI3K itself or downstream effectors such as mammalian target of rapamycin (mTOR) and AKT.5,6 mTOR inhibitors, everolimus and temsirolimus, are approved for the treatment of various cancers. Other mTOR inhibitors, such as OSI-027, AZD 2014, INK-128, and CC-223 are being investigated in patients with solid tumors. Inhibitors of AKT, including perifosine, MK2206, and GSK2110183, are also in clinical development. This article will focus on PI3K inhibitors because mTOR and AKT inhibitors have been extensively reviewed elsewhere.5–7 Herein, we explore the rationale for targeting PI3K in hematologic malignancies and discuss the clinical development of PI3K inhibitors in these diseases.
Overview of the PI3K pathway
The PI3K family of heterodimeric enzymes is grouped into three main categories: classes I, II, and III. Class I enzymes are the most studied and are often implicated in cancer. Class I enzymes are further divided into classes IA and IB, depending on the structure of their regulatory and catalytic subunits. Class IA encompasses five different regulatory subunits (p85α, p85β, p55α, p55γ, or p50α) and three distinct catalytic subunits (p110α, p110β, or p110δ).8,9 Class IB PI3Ks are composed of two different regulatory subunits (p101 or p87) and one catalytic subunit (p110γ).10 Notably p110δ and p110γ are exclusively expressed in hematopoietic cells, whereas the α and β isoforms are ubiquitous.1,11,12 Class IA PI3Ks are activated by direct interaction with RTKs, non-RTKs, G-protein coupled receptors (GPCRs), and Ras, whereas class IB enzymes are activated by GPCRs and Ras.13–16 The regulatory subunits mediate membrane localization, receptor binding, and activation, whereas the catalytic subunit phosphorylates phosphatidylinositol-4,5-bisphosphate (PI-4,5-P2) to yield phosphatidylinositol-3,4,5-trisphosphate (PIP3). The resulting PIP3 initiates downstream signaling by recruiting 3-phosphoinositide-dependent kinase 1 (PDK1) to the membrane, where it activates the AKT serine/threonine kinase by phosphorylating the threonine 308 residue (Figure 1). In turn, AKT phosphorylates many downstream substrates, including forkhead box protein O (FOXO), glycogen synthase kinase-3 (GSK-3), and Bcl-2 associated death promoter (BAD). AKT-mediated phosphorylation also inhibits the proline-rich AKT substrate of 40 kDa (PRAS40) and tuberous sclerosis complex 2 (TSC2), thereby activating mTOR complex 1 (mTORC1). mTORC1 consists of mTOR, mTOR-associated protein LST8 homolog (mLST8), regulatory associated protein of mTOR (RAPTOR), DEP domain TOR-binding protein (DEPTOR), and PRAS40. Activated mTORC1 stimulates protein synthesis by phosphorylating the ribosomal kinase proteins S6K1 (p70S6K/p85S6K) and S6K2 (p54S6K), and the eukaryotic initiation factor 4E-binding proteins (4E-BP1, 4E-BP2, and 4E-BP3).17,18 mTORC2 is physiologically distinct from mTORC1 and consists of mTOR, mLST8, DEPTOR, rapamycin-insensitive companion of mTOR (RICTOR), protein observed with RICTOR (PROTOR), and mammalian stress-activated protein kinase interaction protein 1 (mSIN1). Unlike mTORC1, mTORC2 is considered rapamycin-insensitive because its inhibition requires prolonged drug exposure. While its function is not fully elucidated, mTORC2 is known to phosphorylate AKT at serine 473, which is necessary for maximum activation of AKT.19 Thus, mTOR plays important roles upstream and downstream of AKT.
Figure 1.

Key features of the phosphoinositide 3-kinase (PI3K) signaling pathway. Upon activation, class IA and B PI3Ks initiate signaling by phosphorylating phosphatidylinositol-4,5-bisphosphate (PI-4,5-P2) to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3), which recruits AKT and 3-phosphoinositide-dependent kinase 1 (PDK1) to the plasma membrane. AKT-mediated phosphorylation can activate (arrows) or inhibit (bars) downstream proteins. PI3K signaling affects cell growth, apoptosis, cell-cycle regulation, glucose metabolism, and DNA repair. Some molecular details have been omitted from this diagram for simplicity.
PI3K is negatively regulated by PTEN and Src homology domain-containing inositol phosphatases (SHIP1 and 2). PTEN is ubiquitously expressed and often mutated or epigenetically silenced in many tumor types, which can lead to sustained activation of the PI3K pathway (Figure 1).20,21 SHIP2 is also ubiquitously expressed while SHIP1 is restricted to hematopoietic cells and has been implicated in B-cell malignancies.22 Hydrolysis of PIP3 by PTEN generates PI-4,5-P2, thereby terminating downstream PI3K signaling. In contrast, SHIP modulates but does not terminate PI3K signaling because SHIP yields phosphatidylinositol-3,4-bisphosphate (PI-3,4-P2), which can initiate distinct signaling by binding to proteins such as AKT, Bam32, and tandem PH domain-containing proteins (TAPP1/2).23
Role of the PI3K pathway in hematologic malignancies
Numerous molecular abnormalities that result in constitutive activation of the PI3K pathway in hematologic malignancies have been reported (Table 1), demonstrating the importance of targeting PI3K in leukemias and lymphomas.
Table 1.
Genetic aberrations that activate the PI3K pathway in hematologic malignancies
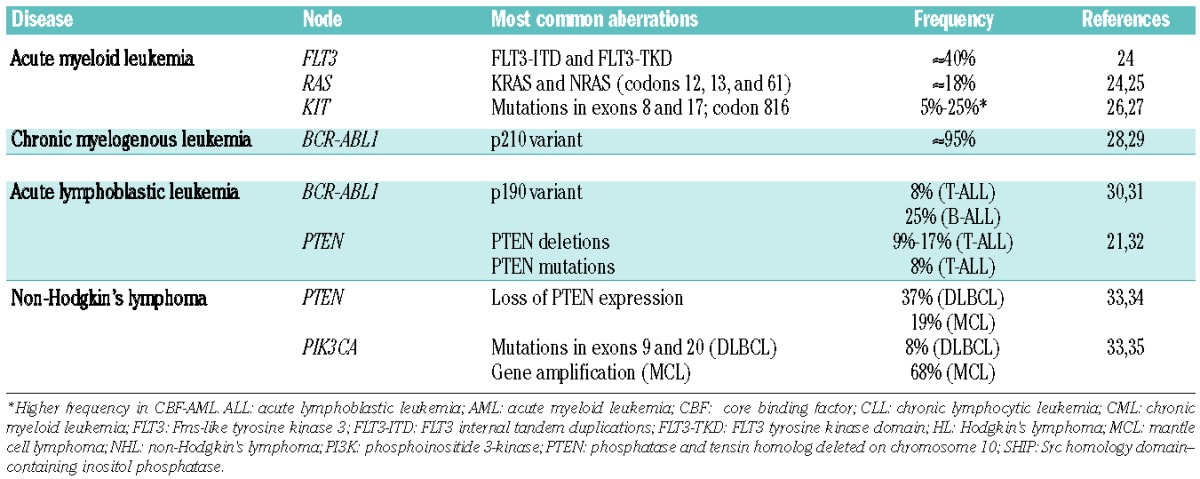
Leukemia
Acute myeloid leukemia
While most patients with acute myeloid leukemia (AML) respond to polychemotherapy regimens, the frequency of relapse is high and patients with relapsed or refractory AML have a poor prognosis.36 A major therapeutic challenge in AML is the presence of leukemic stem cells (LSCs), which are primarily quiescent and resistant to anticancer therapies. Eradication of LSCs is critical because they are associated with metastases, resistance, and relapse.37
Genetic mutations in Fms-like tyrosine kinase 3 (FLT3), KIT receptor tyrosine kinase, N-Ras, and K-Ras, that are associated with oncogenic activation of the PI3K pathway are frequently reported in AML cell lines and patient samples (Table 1).26,38,39 PI3Kδ is ubiquitously expressed in AML blast cells, and the pathway is constitutively activated in 50–80% of AML cases.3,38,40 Constitutive activation of AKT and FOXO1 is also associated with shorter overall survival (OS) in patients with AML.40 Pre-clinical evidence shows that PI3K inhibitors block downstream signaling and suppress growth of AML cells.3 Exploiting the PI3K pathway may help identify new agents and/or innovative regimens to improve outcomes in AML.
Chronic myelogenous leukemia
Tyrosine kinase inhibitors (TKIs) induce remissions in most patients with chronic myelogenous leukemia (CML); however, development of resistance or suboptimal response to therapy can result in relapse and progression to accelerated or blastic phases of the disease.41 The constitutively activated BCR-ABL1 tyrosine kinase is the main oncogenic driver in the pathogenesis of CML, leading to downstream activation of the PI3K signaling pathway. Expression of p85α is critical for the proliferation and survival of BCR-ABL1–expressing CML cells. Furthermore, p85δ has been detected in BCR-ABL1 immunoprecipitates from CML cell lysates.13 BCR-ABL1 also mediates specific upregulation of p110γ in CML.28 Treatment with the TKI imatinib leads to compensatory activation of the PI3K pathway, which precedes the development of imatinib resistance and could be an escape mechanism that results in BCR-ABL1–positive minimal residual disease (MRD) in patients with CML.42 Furthermore, quiescent LSCs that are not effectively targeted by TKIs can persist in the bone marrow.41 Klejman et al. showed that PI3K inhibition enhanced the activity of imatinib against CML cell lines.43 Notably, BCR-ABL1–independent activation of the PI3K signaling pathway has been reported in TKI-resistant CML cell lines.44 Therefore, combination of TKIs and PI3K inhibitors may be an effective therapeutic strategy to eliminate LSCs and target residual CML cells via BCR-ABL1–dependent or –independent mechanisms. Although there are no ongoing studies of PI3K inhibitors specifically targeting CML, clinical evaluation of novel combinations could improve long-term clinical outcomes.
Acute lymphoblastic leukemia
Although recent therapeutic advances in polychemotherapy protocols and hematopoietic stem cell transplantation have improved survival rates, both adult and pediatric patients with relapsed or refractory disease have a very poor prognosis.45,46 Persistence of drug-resistant LSCs can also contribute to subsequent relapses in ALL.47 Research efforts are, therefore, focused on new therapeutic approaches for these patients.
Approximately 25% of adult ALL cases are positive for the Philadelphia (Ph) chromosome and express BCR-ABL1, which (in analogy to CML) activates the PI3K pathway.30,48 Inhibition of PI3K blocked BCR-ABL–induced leukemogenesis in a murine model of Ph+ pre–B-cell ALL.48 Hyperactivation of the PI3K pathway is reported in up to 88% of T-cell ALL (T-ALL) cases.49 Concomitant inactivation of PI3Kγ and PI3Kδ suppressed tumor formation, and the activity of either isoform alone supported malignant transformation of T cells in PTEN-deficient mice.50 PTEN inactivation has been reported in T-ALL cell lines and patient samples.21 Inhibition of the PI3K/AKT/mTOR pathway induced apoptosis in T-ALL cell lines and patient samples, including those enriched for LSCs.51 These results indicate that PI3K inhibition could be beneficial therapy for patients with T- or B-ALL.
Chronic lymphocytic leukemia
Except for the few patients who are eligible for allogeneic stem cell transplantation, chronic lymphocytic leukemia (CLL) remains an incurable disease, and all patients eventually relapse after cytotoxic or biological therapy.52 Therapeutic decisions are more challenging in patients with high-risk chromosomal abnormalities including 17p deletion or TP53 mutation, which confer drug resistance and are associated with short survival rates.53 The poor prognosis for CLL patients calls for the development of new treatment options to improve clinical outcomes.
The mechanisms by which the PI3K pathway is activated in CLL is not well understood. Although PI3K is not often mutated in CLL cells, it mediates antiapoptotic signaling downstream of the B-cell receptor.54 Sustained PI3K activation in CLL cells is associated with increased expression of antiapoptotic proteins such as Bcl-xL, Mcl-1, and X-linked inhibitor of apoptosis (XIAP). PI3K activity correlates with nuclear factor kappa B (NFκB) activation and prevents caspase-3–based apoptotic signaling in CLL cells.54 Constitutive activation of PI3K in CLL is dependent on the p110δ isoform.2 Inhibition of p110δ decreased secretion of chemokines and blocked molecular interactions between CLL and bone marrow stromal cells that are critical for tumor growth and survival.55 Additionally, CLL cells over-express casein kinase 2 (CK2), which inactivates PTEN leading to dysregulated PI3K signaling.56 Inhibition of CK2 is associated with PTEN activation and increased phosphorylation of Protein Kinase Cδ, thereby inducing apoptosis of CLL cells.56
Non-Hodgkin’s lymphoma (NHL)
Follicular lymphoma
The PI3K pathway has emerged as an important target in follicular lymphoma (FL) because of the constitutive phosphorylation of AKT and the mTOR substrates p70S6K and 4E-BP1 in FL cells and patient samples.57 Protein microarray studies showed that serine 473 phosphorylation of AKT signified a clinically relevant molecular event in FL, differentiating FL from follicular hyperplasia.58 There was no significant difference in expression of total AKT or AKT phosphorylated at threonine 308, suggesting that selective phosphorylation of AKT at serine 473, which usually occurs via mTORC2, is of biological importance in FL.
Diffuse large B-cell lymphoma
The PI3K pathway is activated in diffuse large B-cell lymphoma (DLBCL) cell lines and primary tumor samples.33,59 Downstream molecules AKT, FOXO1, and GSK-3 are constitutively phosphorylated in DLBCL cell lines and PI3K inhibition blocks phosphorylation of these proteins and induces apoptosis.59 Clinical data indicate that PTEN deletions and mutations result in activation of the PI3K pathway in DLBCL. Mutational analysis of 215 DLBCL cases showed that 37% of samples had reduced or loss of PTEN expression.33 Another study confirmed that expression of PTEN and SHIP was lowest in patients with DLBCL in comparison with FL or normal B cells.22 Loss of PTEN expression in DLBCL is associated with shorter survival rates.60
Mantle cell lymphoma
Studies using cell lines and primary tumor samples indicate that PI3K inhibition could be an effective therapeutic strategy in mantle cell lymphoma (MCL). Constitutive activation of the PI3K pathway via the B-cell receptor leads to deregulation of cell-cycle progression in MCL cells.61 PTEN phosphorylation is also enhanced in MCL cells; this phosphorylation renders PTEN inactive, leading to activated PI3K signaling.61 While p110δ is widely expressed in MCL, p110δ inhibitors have shown modest activity. Evidence suggests that p110α may be a more important target in MCL because PIK3CA gene amplification is a frequent genetic alteration. Analysis of MCL cell lines and primary samples suggest that PIK3CA amplification may be a mechanism of resistance to p110δ inhibition.35
Hodgkin’s lymphoma
PI3K is constitutively activated in Hodgkin’s and ReedSternberg (HRS) cells and is associated with prolonged cell survival.62 In Hodgkin’s lymphoma (HL) cell lines, PI3Kδ was detected at higher levels than other class I PI3K isoforms.62 AKT was also found to be constitutively phosphorylated in HL cell lines and tumor samples, particularly in response to signals from the bone marrow stromal cells.62 Specific inhibition of PI3Kδ blocked stroma-induced phosphorylation of AKT and increased apoptosis of HL cells.62 The tumor suppressor FOXO1, which is inactivated by AKT, is repressed in HRS cells; only one of 32 cases analyzed by Xie et al. was FOXO1 positive.63 Inhibition of AKT caused a dose-dependent increase in FOXO1 expression in classic HL cell lines.63
Together, these findings emphasize the important role of PI3K in hematologic malignancies and suggest that inhibition of this pathway may be a viable therapeutic approach. Further investigation of PI3K inhibitors in hematologic malignancies may help to establish the role of this drug class in the treatment armamentarium and improve patient outcomes.
Evaluating PI3K inhibitors in hematologic malignancies
One of the earliest known PI3K inhibitors was wortmannin, a natural product that binds irreversibly to PI3K.64 The synthetic compound LY294002 was found to reversibly inhibit PI3K at micromolar concentrations.65 Both wortmannin and LY290042 blocked PI3K signaling and induced apoptosis in cancer cells. Despite their demonstrated antitumor activity, these agents were associated with significant toxicity in animal models. It is unclear whether the observed toxicity is related to the fact that both wortmannin and LY294002 are broad-spectrum inhibitors whose targets include multiple PI3K isoforms, phosphoinositide 4-kinase, mTOR, and DNA-PK.66 Given their lack of selectivity, the toxicity associated with both compounds, and the poor solubility of LY294002, these agents were not considered good drug candidates. However, the promising activity led to the development of newer and more specific PI3K inhibitors with improved pharmacokinetic properties, efficacy, and tolerability. The three categories of PI3K inhibitors include dual PI3K/mTOR inhibitors, pan-PI3K inhibitors, and isoform-specific nhibitors (Figure 2). These agents have demonstrated anticancer activity and are in various stages of clinical development.
Figure 2.

Chemical structures of PI3K inhibitors in development. 1Pre-clinical or clinical activity in hematologic malignancies.
Dual PI3K/mTOR inhibitors
Given the structural similarity in the catalytic domains of p110 and mTOR, the rationale for developing dual inhibitors is to use a single agent to target the pathway at multiple levels and block downstream signaling of PI3K, mTORC1 and mTORC2. This strategy is theoretically advantageous because it provides maximal signaling shutdown, circumvents feedback activation of the pathway via mTORC2, and blocks potential tumor escape mechanisms. Dual inhibition may also be useful in clinical contexts such as AML and FL, where PI3K-independent activation of mTOR has been demonstrated.57,67 Dual inhibitors being investigated in the clinical setting include NVP-BEZ235, SF1126, XL765 (SAR245409), PF-04691502, PF-05212384, GDC-0980, GSK2126458, and DS-7423.
The imidazoquinazoline derivative, NVP-BEZ235 has demonstrated activity against hematologic malignancies in pre-clinical models. NVP-BEZ235 blocked PI3K signaling and induced apoptosis in AML cells but not in normal hematopoietic cells.3 As a single agent or in combination with cytotoxic agents, NVP-BEZ235 induced cell-cycle arrest and apoptosis in T-ALL cell lines, including a drug-resistant cell line (CEM-R).68 NVP-BEZ235 also suppressed cell growth and induced apoptosis in Ph+ and Ph− B-ALL cells.69 In bortezomib-resistant MCL cell lines, NVP-BEZ235 suppressed PI3K activation and cell growth.70 Treatment with NVP-BEZ235 inhibited survival of FL cells in vitro, restored sensitivity to bortezomib in a bortezomib-resistant FL cell line (SUDHL4), and significantly suppressed tumor growth in xenograft models.71 One ongoing phase I study is evaluating NVP-BEZ235 as a single agent in patients with relapsed or refractory acute leukemia (NCT01756118). Early data showed one complete response (CR) and one hematologic improvement in patients with Ph− B-ALL, and stable disease (SD) in one AML patient.72 The estimated study completion date is May 2014 (www.clinicaltrials.gov).
SF1126 is a peptide-linked derivative of LY294002 with improved solubility and tolerability compared with the parent compound. In pre-clinical studies, SF1126 blocked proliferation and induced apoptosis of MCL cells more potently than the isoform-specific inhibitor idelalisib (GS1101, CAL-101).73 A phase I study of SF1126 in patients with solid tumors or B-cell malignancies reported SD in 58% of patients (including 2 with B-cell malignancies). One patient with CLL who had progressed on rituximab therapy achieved SD after two months of SF1126 monotherapy. Notably, pharmacodynamic analyses did not provide conclusive evidence regarding target inhibition and response to SF1126. One grade 3 diarrhea was reported.74 The observed safety profile of SF1126 is noteworthy, given the non-specificity of the parent compound LY294002. Further investigation is needed to determine the clinical utility of LY294002 derivatives.
XL765 (SAR245409), a quinoxaline derivative, demonstrated anticancer activity in a phase I trial involving 34 patients with advanced solid tumors. Five patients (15%) had SD lasting over three months, and dose-limiting toxicities (DLTs) included elevated liver enzymes, rash, nausea, and vomiting. Evidence of PI3K inhibition (defined as decrease in pAKT and p4EBP1 levels from baseline to Day 27) was observed in patient peripheral blood mononuclear cells (PBMCs), hair follicles, skin, and tumors.75 Ongoing clinical studies are evaluating the efficacy and safety of XL765 (SAR245409) as a single-agent or in combination with other agents in patients with hematologic malignancies (Table 2).
Table 2.
Ongoing clinical evaluation of PI3K inhibitors in hematologic malignancies
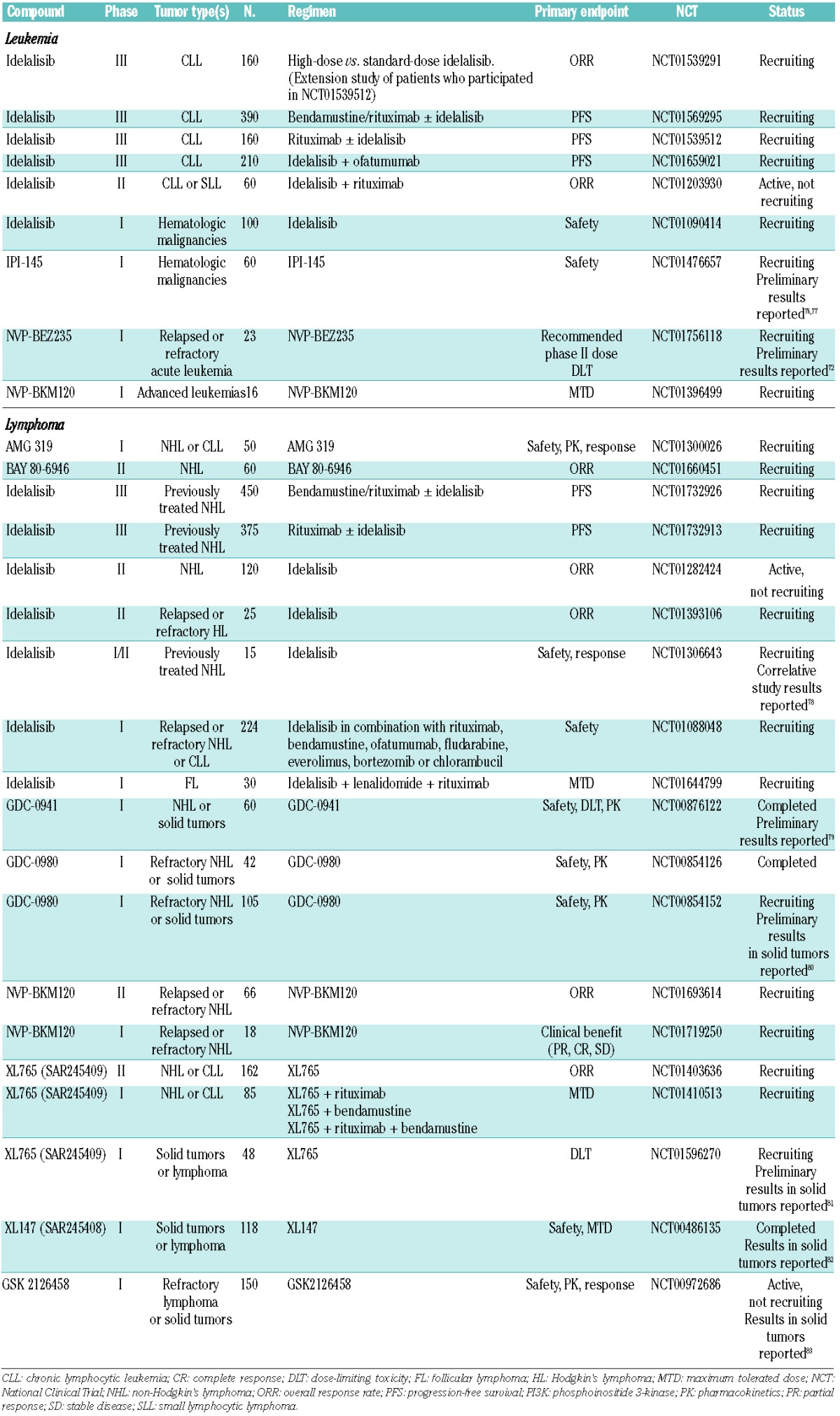
Other dual PI3K/mTOR inhibitors in development include GDC-0980, PF-04691502, PF-05212384, DS-7423, and the pyridylsulfonamide GSK2126458. These agents are currently being investigated in phase I and II trials in patients with advanced solid tumors.
With the potential to block compensatory signaling or feedback loops, dual PI3K/mTOR inhibitors represent an interesting new drug class. Preliminary data shows that simultaneous inhibition of all PI3K isoforms, mTORC1, and mTORC2 is feasible in the clinical setting. The most frequently reported adverse events include diarrhea, nausea, and vomiting. The demonstrated efficacy of dual PI3K/mTOR inhibitors in drug-resistant cell lines highlights the possibility of overcoming limitations associated with conventional therapies. Robust clinical trials are needed to determine whether dual PI3K/mTOR inhibitors have a therapeutic advantage over selective PI3K or mTOR inhibitors.
Pan-PI3K inhibitors
Agents in this category block oncogenic signaling by selectively targeting all class I PI3K isoforms (α, β, γ, and δ). It has been hypothesized that inhibiting all 4 isoforms of class I PI3K may be advantageous over inhibiting specific isoforms. Some pre-clinical studies suggest that functional redundancy may lead to compensatory signaling after genetic or pharmacological inactivation of specific isoforms.84 However, the concept of functional redundancy is controversial, and the clinical relevance of redundant roles remains unclear. Pan-PI3K inhibitors such as NVP-BKM120, XL147 (SAR245408), BAY 80-6946, PX-866, and GDC-0941 have demonstrated potent anticancer activity.
NVP-BKM120 is a dimorpholinopyrimidine derivative that blocks AKT phosphorylation and inhibits tumor growth as a single agent or in combination with other therapies such as docetaxel and temozolomide.85 A phase I study in 35 patients with previously treated solid tumors showed that NVP-BKM120 treatment led to a partial response (PR) in one patient with triple-negative breast cancer and SD in 16 patients. BKM120 demonstrated the ability to cross the blood-brain barrier resulting in 28% shrinkage of a metastatic brain tumor in one patient. Target inhibition was assessed using pre- and post-treatment levels of pS6K in skin samples; 80% of patients had 40–85% decrease in pS6K levels after exposure to NVP-BKM120. Frequently reported grade 3/4 adverse events included rash (11 %), hyperglycemia (9%), and mood alterations (6%). The mood alterations were attributed to the activity of BKM120 in the central nervous system.86 NVP-BKM120 also has activity in hematologic malignancies and was shown to induce apoptosis in Ph+ and Ph− B-ALL.69 Three studies are investigating NVP-BKM120 monotherapy in patients with advanced leukemias and lymphomas (Table 2). The data from these ongoing trials are eagerly anticipated.
Another quinoxaline derivative, XL147 (SAR245408), is currently in clinical development. Early safety data from a phase I trial of XL147 in patients with refractory CLL (n=5) and relapsed lymphoma (n=10) showed that common grade 3/4 adverse events were neutropenia (27%), thrombocytopenia (7%), and hyperglycemia (7%). Pharmacodynamic analyses demonstrated evidence of target inhibition in patient PBMCs, hair follicles, skin, and tumors.82,87 Efficacy data from this study have not yet been reported.
BAY 80-6946 is a reversible pan-PI3K inhibitor with broad anticancer activity in pre-clinical models. BAY 80-6946 was more potent than the p110δ-specific inhibitor idelalisib against B-cell lymphomas with median IC50s of 0.49 μM and 37 μM, respectively.88 A phase I study of BAY 80-6946 in patients with relapsed NHL reported 5 PRs in 6 evaluable patients (83%). Significant lymphoma shrinkage was seen within 48 h of the first dose on 18F-fluorodeoxyglucose positron emission tomography and computed tomography (18FDG-PET/CT). The most frequently reported adverse events included hyperglycemia, nausea, diarrhea, and fatigue.89 An ongoing phase II study is evaluating BAY 80-6946 as a single agent in patients with relapsed, indolent, or aggressive NHL (clinicaltrials.gov identifier:01660451). Results from this study have not been reported.
PX-866 is a wortmannin derivative that binds irreversibly to PI3K and is active in PIK3CA mutant or PTEN null xenografts. In a phase I study, 28.6% evaluable patients with solid tumors had SD as best response to PX-866. Within 4 h of treatment, pharmacodynamic assays in 7 patients showed that PX-866 treatment resulted in from 13% to 94% reduction in pAKT levels.90 There are no ongoing studies in patients with hematologic malignancies.
The thieno [3,2,-d] pyrimidine, GDC-0941, blocked PI3K signaling in a mouse model of FL.91 As a single agent or in combination with other compounds, GDC-0941 induced apoptosis in AML cells under hypoxic conditions, which suggests efficacy in targeting residual AML cells sequestered in a hypoxic bone marrow microenvironment.92 In MCL tumor samples, GDC-0941 blocked proliferation and induced apoptosis more effectively than the p1 10δ-specific inhibitor, idelalisib. The superior activity of GDC-0941 was attributed to the change in isoform expression in relapsed MCL. While PI3Kδ was the most consistently expressed, PI3Kα expression was significantly greater in patients with MCL who had relapsed multiple times and PIK3CA amplification was associated with resistance to p110δ inhibition.35 Several ongoing trials are evaluating GDC-0941, including a phase I dose-escalation study in patients with NHL or solid tumors (clinicaltrials.gov identifier:00876122). Preliminary results in patients with solid tumors have been reported.79
Overall, pan-PI3K inhibitors have shown encouraging anticancer activity as single agents or in combination with other agents, and results from ongoing trials are eagerly awaited. Combination regimens may have better efficacy because single-agent activity of pan-PI3K inhibitors could theoretically be limited by PI3K-independent activation of mTOR. Pan-PI3K inhibition with BAY 80-6946 was superior to isoform-specific inhibition in pre-clinical studies,88 but it is unknown whether this will translate to superior clinical efficacy. Further investigation is needed to determine whether complete inhibition of all isoforms is necessary, and whether the doses required for total target inhibition will be tolerable in patients. Consistent with other inhibitors of the PI3K pathway, the most common adverse events are hyperglycemia, diarrhea, and rash.
Isoform-specific inhibitors
Selective PI3K inhibitors may have benefit in tumors in which a particular isoform is predominantly expressed or more critical for survival. PI3Kα is expressed and frequently mutated in many tumor types, whereas the β isoform has been shown to regulate the growth of PTEN-null tumors.1,93 PI3Kδ and PI3Kγ are mainly expressed in hematopoietic cells.11 Blocking the activity of specific isoforms that are crucial for survival could theoretically provide complete target inhibition at tolerable doses and maximize the therapeutic index of this drug class.
PI3Kα-specific inhibitors are expected to be highly effective in tumors harboring PIK3CA mutations. Notably, PIK3CA mutations are rare in hematologic malignancies. NVP-BYL719, INK1117, and GDC-0032 are being evaluated in patients with solid tumors.
KIN-193 and GSK2636771 specifically target the PI3K-β isoform. First identified in a kinase inhibitor screen, KIN-193 demonstrated pre-clinical activity but has not yet entered clinical trials.93 GSK2636771 is being evaluated in a phase I trial in patients with PTEN-deficient solid tumors (clinicaltrials.gov identifier:01458067).
IPI-145 selectively inhibits both PI3Kδ and PI3Kγ isoforms. A phase I trial is evaluating IPI-145 as a single agent in patients with hematologic malignancies (clinicaltrials.gov identifier:01476657). Preliminary data from 34 CLL patients reported partial response in 12 of 22 evaluable patients. Nodal response (defined as ≥50% reduction in tumor mass without PR) was observed in 7 patients. The most common grade 3 or over adverse event was neutropenia (25%). Pharmacodynamic analysis in 26 patients showed a significant reduction in pAKT levels within 1 h of treatment. There was also a marked decrease in cytokines and chemokines.76
The restricted expression of PI3Kδ, and its important role in B-cell receptor signaling, make it a key anticancer target in hematologic malignancies. PI3Kδ-specific inhibitors include AMG 319 and idelalisib (GS-1101, CAL-101), both of which are in clinical trials for the treatment of patients with hematologic malignancies. AMG 319 monotherapy is being investigated in a phase I trial in patients with relapsed or refractory lymphoid malignancies with an expansion cohort in patients with CLL (clinicaltrials.gov identifier:01300026). Results have not been reported.
Of the PI3K inhibitors in development, idelalisib is the most studied in hematologic malignancies and is being evaluated in several phase III studies (Table 2). Pre-clinical data suggested that idelalisib activity may not be solely dependent on isoform expression; despite the ubiquitous expression of PIK3δ in AML, idelalisib was not very effective against AML samples from patients but had a greater therapeutic potential against lymphoid malignancies, such as CLL.2 A phase I trial in 55 patients with relapsed/refractory CLL showed that idelalisib monotherapy led to an objective response rate (ORR) of 26%. The most common grade 3/4 adverse events were pneumonia (24%), neutropenia (24%), thrombocytopenia (7%), and febrile neutropenia (7%). Idelalisib therapy also led to a transient increase (≥50%) in absolute lymphocyte count in 58% of patients.94 In another phase I study involving patients with relapsed or refractory CLL, idelalisib was administered in combination with rituximab (n=19), bendamustine (n=17), or both (n=15). ORRs were 78%, 82%, and 87%, respectively. Approximately 85% of patients had 50% or more reduction in lymphadenopathy. The 2-year progression-free survival (PFS) was 63%, and 2-year OS rate was 84%. Neutropenia was the most common grade 3 or over adverse event reported in 32%, 76%, and 67% of patients receiving idelalisib with rituximab, bendamustine, or both, respectively. Treatment with idelalisib was associated with a significant reduction in disease-associated chemokines/cytokines.95 Idelalisib has also demonstrated activity in combination with ofatumumab (anti-CD20), resulting in rapid reduction in lymphadenopathy in 9 of 11 patients (82%) with previously treated CLL.96
Correlative analyses of a phase I/II study showed that idelalisib blocked downstream PI3K signaling in primary samples from patients with indolent NHL.78 Idelalisib was evaluated in combination with rituximab (n=31), bendamustine (n=34), or both (n=14) in patients with relapsed or refractory NHL. Nearly all patients in this study had received prior rituximab or bendamustine. ORRs were 71%, 88%, and 79% in the respective treatment arms and 20-month PFS was 72% among responders. Complete responses were observed in 19%, 27%, and 43%, respectively. The most commonly reported grade 3 or over adverse events were pneumonia (15%), rash (8%) and diarrhea (8%). ALT/AST elevations (grade ≥3) were reported in 18% of patients.97 Notably, idelalisib-based therapy led to a significant reduction in chemokines/cytokines compared with base-line levels.98 Interim analysis of a phase II study in 125 patients with indolent NHL refractory to both rituximab and an alkylating agent, showed that idelalisib treatment led to an ORR of 50% (CR 4%, PR 46%) and median PFS of 11.4 months. Frequently reported grade 3 or over adverse events included neutropenia (26%), diarrhea (10%) and dyspnea (3%). Grade 3 or over ALT/AST elevations occurred in 13% of patients.99
As a single agent or in combination with other agents, idelalisib has demonstrated impressive results in clinical trials, with durable responses, rapid reductions in lymphadenopathy and cytokine levels. Idelalisib is currently being evaluated in 6 phase III trials involving patients with CLL and NHL (Table 2). Results from these trials are eagerly awaited.
Conclusions and outlook
The few effective treatment options available for patients with relapsed or refractory hematologic malignancies highlight the opportunities to improve first-line or subsequent therapy. Aberrant activation of the PI3K signaling pathway plays a critical role in the pathogenesis of hematologic malignancies; thus, there is considerable interest in the development of PI3K inhibitors. Agents in the three categories of PI3K inhibitors have demonstrated anticancer activity. It remains uncertain whether functional redundancy among isoforms, or activation of feedback loops, will have significant impact on the clinical activity of these agents.
With the exception of idelalisib, most of the PI3K inhibitors have been studied mainly in solid tumors; however, there is promising indication of efficacy in patients with hematologic malignancies. For example, idelalisib and BAY 80-6946 resulted in durable responses in patients with hematologic malignancies.89,95,98 Idelalisib also induced rapid reductions in lymphadenopathy and baseline cytokine levels.95,98 As impressive as the idelalisib results are, other agents in development may have even better efficacy. BAY 80-6946 and SF1126 showed superior in vitro activity to idelalisib,73,88 but it remains to be seen whether this will translate into superior clinical efficacy.
Despite the demonstrated single-agent activity, PI3K inhibitors may work best in combination with other therapies. For example, targeting PI3K in combination with other therapies such as TKIs could potentially eradicate MRD in diseases such as CML or Ph+ ALL.47 Therefore, investigators are seeking to identify the ideal combination with targeted or cytotoxic agents that will improve patient outcomes. On the basis of pre-clinical studies showing activity of PI3K inhibitors in drug-resistant cell lines,85,100 there is the potential to overcome limitations of conventional therapies and restore sensitivity to prior treatment. Restoring sensitivity to therapy could be particularly important for patients with relapsed or refractory disease. Overall toxicity is another key consideration for combination regimens. Thus far, PI3K inhibitors have demonstrated a predictable and manageable safety profile; the most common toxicities are on-target effects, including hyperglycemia, rash, and diarrhea. Nevertheless, careful consideration of overall toxicity is needed to ensure that the agents do not have overlapping toxicities or exacerbate adverse events.
Overall, PI3K is a viable therapeutic target in hematologic malignancies, and preliminary findings warrant further investigation in robust clinical trials. Results from ongoing hypothesis-driven trials are eagerly anticipated, because they may further define the role of PI3K inhibitors in therapeutic strategies for patients with hematologic malignancies. Emerging opportunities involve the validation of pathway biomarkers to monitor response and determination of PI3K expression or mutation status to assist in selecting appropriate therapy for individual patients.
Acknowledgments
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals Corporation. The authors would like to thank Nene Anadu, PhD, for medical editorial assistance with this manuscript.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3(10):1221–4 [DOI] [PubMed] [Google Scholar]
- 2.Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapuis N, Tamburini J, Green AS, Vignon C, Bardet V, Neyret A, et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin Cancer Res. 2010;16(22):5424–35 [DOI] [PubMed] [Google Scholar]
- 4.Bousquet M, Recher C, Queleen C, Demur C, Payrastre B, Brousset P. Assessment of somatic mutations in phosphatidylinositol 3-kinase gene in human lymphoma and acute leukaemia. Br J Haematol. 2005;131(3):411–3 [DOI] [PubMed] [Google Scholar]
- 5.Polak R, Buitenhuis M. The PI3K/PKB signaling module as key regulator of hematopoiesis: Implications for therapeutic strategies in leukemia. Blood. 2012;119(4):911–23 [DOI] [PubMed] [Google Scholar]
- 6.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10(3):143–53 [DOI] [PubMed] [Google Scholar]
- 7.Younes A, Samad N. Utility of mTOR inhibition in hematologic malignancies. Oncologist. 2011;16(6):730–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inukai K, Funaki M, Ogihara T, Katagiri H, Kanda A, Anai M, et al. p85alpha gene generates three isoforms of regulatory subunit for phosphatidylinositol 3-kinase (PI 3-kinase), p50alpha, p55alpha, and p85alpha, with different PI 3-kinase activity elevating responses to insulin. J Biol Chem. 1997;272(12):7873–82 [DOI] [PubMed] [Google Scholar]
- 9.Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: Stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18(3):1379–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krugmann S, Hawkins PT, Pryer N, Braselmann S. Characterizing the interactions between the two subunits of the p101/p110gamma phosphoinositide 3-kinase and their role in the activation of this enzyme by G beta gamma subunits. J Biol Chem. 1999;274(24):17152–8 [DOI] [PubMed] [Google Scholar]
- 11.Saudemont A, Garcon F, Yadi H, Roche-Molina M, Kim N, Segonds-Pichon A, et al. P110gamma and P110delta isoforms of phosphoinositide 3-kinase differentially regulate natural killer cell migration in health and disease. Proc Natl Acad Sci USA. 2009;106(14):5795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454(7205):776–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16(20):6151–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jimenez C, Hernandez C, Pimentel B, Carrera AC. The p85 regulatory subunit controls sequential activation of phosphoinositide 3-kinase by tyr kinases and ras. J Biol Chem. 2002;277(44):41556–62 [DOI] [PubMed] [Google Scholar]
- 15.Murga C, Laguinge L, Wetzker R, Cuadrado A, Gutkind JS. Activation of Akt/protein kinase B by G protein-coupled receptors. A role for alpha and beta gamma subunits of heterotrimeric G proteins acting through phosphatidylinositol-3-OH kinasegamma. J Biol Chem. 1998;273(30):19080–5 [DOI] [PubMed] [Google Scholar]
- 16.Kurig B, Shymanets A, Bohnacker T, Prajwal, Brock C, Ahmadian MR, et al. Ras is an indispensable coregulator of the class IB phosphoinositide 3-kinase p87/p110gamma. Proc Natl Acad Sci USA. 2009;106(48):20312–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee-Fruman KK, Kuo CJ, Lippincott J, Terada N, Blenis J. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene. 1999;18(36):5108–14 [DOI] [PubMed] [Google Scholar]
- 18.Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17(3):249–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–68 [DOI] [PubMed] [Google Scholar]
- 20.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–7 [DOI] [PubMed] [Google Scholar]
- 21.Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114(3):647–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miletic AV, Anzelon-Mills AN, Mills DM, Omori SA, Pedersen IM, Shin DM, et al. Coordinate suppression of B cell lymphoma by PTEN and SHIP phosphatases. J Exp Med. 2010;207(11):2407–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krahn AK, Ma K, Hou S, Duronio V, Marshall AJ. Two distinct waves of membrane-proximal B cell antigen receptor signaling differentially regulated by src homology 2-containing inositol polyphosphate 5-phosphatase. J Immunol. 2004;172(1):331–9 [DOI] [PubMed] [Google Scholar]
- 24.Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1909–18 [DOI] [PubMed] [Google Scholar]
- 25.Neubauer A, Maharry K, Mrozek K, Thiede C, Marcucci G, Paschka P, et al. Patients with acute myeloid leukemia and RAS mutations benefit most from postremission high-dose cytarabine: A cancer and leukemia group B study. J Clin Oncol. 2008;26(28):4603–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen Y, Zhu YM, Fan X, Shi JY, Wang QR, Yan XJ, et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood. 2011;118(20):5593–603 [DOI] [PubMed] [Google Scholar]
- 27.Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, et al. Incidence and prognostic impact of c-kit, FLT3, and ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia. 2006;20(6):965–70 [DOI] [PubMed] [Google Scholar]
- 28.Hickey FB, Cotter TG. BCR-ABL regulates phosphatidylinositol 3-kinase-p110gamma transcription and activation and is required for proliferation and drug resistance. J Biol Chem. 2006;281(5):2441–50 [DOI] [PubMed] [Google Scholar]
- 29.Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36(1):93–9 [DOI] [PubMed] [Google Scholar]
- 30.Moorman AV, Harrison CJ, Buck GA, Richards SM, Secker-Walker LM, Martineau M, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): Analysis of cytogenetic data from patients treated on the medical research council (MRC) UKALLXII/Eastern cooperative oncology group (ECOG) 2993 trial. Blood. 2007;109(8):3189–97 [DOI] [PubMed] [Google Scholar]
- 31.Hagemeijer A, Graux C. ABL1 rearrangements in T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2010;49(4):299–308 [DOI] [PubMed] [Google Scholar]
- 32.Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13(10):1203–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abubaker J, Bavi PP, Al-Harbi S, Siraj AK, Al-Dayel F, Uddin S, Al-Kuraya K. PIK3CA mutations are mutually exclusive with PTEN loss in diffuse large B-cell lymphoma. Leukemia. 2007;21(11):2368–70 [DOI] [PubMed] [Google Scholar]
- 34.Rudelius M, Pittaluga S, Nishizuka S, Pham TH, Fend F, Jaffe ES, et al. Constitutive activation of akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood. 2006;108(5):1668–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iyengar S, Clear A, Bodor C, Maharaj L, Lee A, Calaminici M, et al. P110alpha-mediated constitutive PI3K signaling limits the efficacy of p110delta-selective inhibition in mantle cell lymphoma, particularly with multiple relapse. Blood. 2013;121(12):2274–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hengeveld M, Suciu S, Karrasch M, Specchia G, Marie JP, Muus P, et al. Intensive consolidation therapy compared with standard consolidation and maintenance therapy for adults with acute myeloid leukaemia aged between 46 and 60 years: Final results of the randomized phase III study (AML 8B) of the European Organization for Research and Treatment of Cancer (EORTC) and the Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto (GIMEMA) Leukemia Cooperative Groups. Ann Hematol. 2012;91(6):825–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(-) fraction. Blood. 2010;115(10):1976–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muranyi AL, Dedhar S, Hogge DE. Combined inhibition of integrin linked kinase and FMS-like tyrosine kinase 3 is cytotoxic to acute myeloid leukemia progenitor cells. Exp Hematol. 2009;37(4):450–60 [DOI] [PubMed] [Google Scholar]
- 39.Ahmad EI, Gawish HH, Al Azizi NM, Elhefni AM. The prognostic impact of K-RAS mutations in adult acute myeloid leukemia patients treated with high-dose cytarabine. Onco Targets Ther. 2011;4:115–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Min YH, Eom JI, Cheong JW, Maeng HO, Kim JY, Jeung HK, et al. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: Its significance as a prognostic variable. Leukemia. 2003;17(5):995–7 [DOI] [PubMed] [Google Scholar]
- 41.Kumari A, Brendel C, Hochhaus A, Neubauer A, Burchert A. Low BCR-ABL expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood. 2012;119(2):530–9 [DOI] [PubMed] [Google Scholar]
- 42.Burchert A, Wang Y, Cai D, von Bubnoff N, Paschka P, Muller-Brusselbach S, et al. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia. 2005;19(10):1774–82 [DOI] [PubMed] [Google Scholar]
- 43.Klejman A, Rushen L, Morrione A, Slupianek A, Skorski T. Phosphatidylinositol-3 kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene. 2002;21(38):5868–76 [DOI] [PubMed] [Google Scholar]
- 44.Quentmeier H, Eberth S, Romani J, Zaborski M, Drexler HG. BCR-ABL1-independent PI3Kinase activation causing imatinib-resistance. J Hematol Oncol. 2011;4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kantarjian HM, Thomas D, Ravandi F, Faderl S, Jabbour E, Garcia-Manero G, et al. Defining the course and prognosis of adults with acute lymphocytic leukemia in first salvage after induction failure or short first remission duration. Cancer. 2010;116(24):5568–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ko RH, Ji L, Barnette P, Bostrom B, Hutchinson R, Raetz E, et al. Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: A therapeutic advances in childhood leukemia consortium study. J Clin Oncol. 2010;28(4):648–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cox CV, Diamanti P, Evely RS, Kearns PR, Blair A. Expression of CD133 on leukemia-initiating cells in childhood ALL. Blood. 2009;113(14):3287–96 [DOI] [PubMed] [Google Scholar]
- 48.Kharas MG, Janes MR, Scarfone VM, Lilly MB, Knight ZA, Shokat KM, Fruman DA. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J Clin Invest. 2008;118(9):3038–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008;118(11):3762–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Subramaniam PS, Whye DW, Efimenko E, Chen J, Tosello V, De Keersmaecker K, et al. Targeting nonclassical oncogenes for therapy in T-ALL. Cancer Cell. 2012;21(4):459–72 [DOI] [PubMed] [Google Scholar]
- 51.Bressanin D, Evangelisti C, Ricci F, Tabellini G, Chiarini F, Tazzari PL, et al. Harnessing the PI3K/Akt/mTOR pathway in T-cell acute lymphoblastic leukemia: Eliminating activity by targeting at different levels. Oncotarget. 2012;3(8):811–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia Updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–6 [DOI] [PubMed] [Google Scholar]
- 54.Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating antiapoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood. 2008;111(2):846–55 [DOI] [PubMed] [Google Scholar]
- 55.Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shehata M, Schnabl S, Demirtas D, Hilgarth M, Hubmann R, Ponath E, et al. Reconstitution of PTEN activity by CK2 inhibitors and interference with the PI3-K/Akt cascade counteract the antiapoptotic effect of human stromal cells in chronic lymphocytic leukemia. Blood. 2010;116(14):2513–21 [DOI] [PubMed] [Google Scholar]
- 57.Leseux L, Hamdi SM, Al Saati T, Capilla F, Recher C, Laurent G, Bezombes C. Syk-dependent mTOR activation in follicular lymphoma cells. Blood. 2006;108(13):4156–62 [DOI] [PubMed] [Google Scholar]
- 58.Gulmann C, Espina V, Petricoin E, 3rd, Longo DL, Santi M, Knutsen T, et al. Proteomic analysis of apoptotic pathways reveals prognostic factors in follicular lymphoma. Clin Cancer Res. 2005;11(16):5847–55 [DOI] [PubMed] [Google Scholar]
- 59.Uddin S, Hussain AR, Siraj AK, Manogaran PS, Al-Jomah NA, Moorji A, et al. Role of phosphatidylinositol 3′-kinase/AKT pathway in diffuse large B-cell lymphoma survival. Blood. 2006;108(13):4178–86 [DOI] [PubMed] [Google Scholar]
- 60.Robledo C, Garcia JL, Caballero D, Conde E, Arranz R, Flores T, et al. Array comparative genomic hybridization identifies genetic regions associated with outcome in aggressive diffuse large B-cell lymphomas. Cancer. 2009;115(16):3728–37 [DOI] [PubMed] [Google Scholar]
- 61.Dal Col J, Zancai P, Terrin L, Guidoboni M, Ponzoni M, Pavan A, et al. Distinct functional significance of akt and mTOR constitutive activation in mantle cell lymphoma. Blood. 2008;111(10):5142–51 [DOI] [PubMed] [Google Scholar]
- 62.Meadows SA, Vega F, Kashishian A, Johnson D, Diehl V, Miller LL, et al. PI3Kdelta inhibitor, GS-1101 (CAL-101), attenuates pathway signaling, induces apoptosis, and overcomes signals from the microenvironment in cellular models of Hodgkin lymphoma. Blood. 2012;119(8):1897–900 [DOI] [PubMed] [Google Scholar]
- 63.Xie L, Ushmorov A, Leithauser F, Guan H, Steidl C, Farbinger J, et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood. 2012;119(15):3503–11 [DOI] [PubMed] [Google Scholar]
- 64.Arcaro A, Wymann MP. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: The role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem J. 1993;296(Pt 2):297–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem. 1994;269(7):5241–8 [PubMed] [Google Scholar]
- 66.Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, Panayotou G. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol. 1996;16(4):1722–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tamburini J, Chapuis N, Bardet V, Park S, Sujobert P, Willems L, et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: Rationale for therapeutic inhibition of both pathways. Blood. 2008;111(1):379–82 [DOI] [PubMed] [Google Scholar]
- 68.Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A, et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res. 2010;70(20):8097–107 [DOI] [PubMed] [Google Scholar]
- 69.Badura S, Tesanovic T, Pfeifer H, Hohnloser C, Liebermann M, Serve H, et al. Differential suppressive effects of selective PI3K and mTOR and dual PI3K/mTORC1/C2 inhibition on long-term cultured primary human acute lymphoblastic leukemia (ALL) cells implicate a distinct role of mTORC2. ASH Annual Meeting Abstracts. 2010;116(21):1032 [Google Scholar]
- 70.Kim A, Park S, Lee JE, Jang WS, Lee SJ, Kang HJ, Lee SS. The dual PI3K and mTOR inhibitor NVP-BEZ235 exhibits anti-proliferative activity and overcomes bortezomib resistance in mantle cell lymphoma cells. Leuk Res. 2012;36(7):912–20 [DOI] [PubMed] [Google Scholar]
- 71.Bhende PM, Park SI, Lim MS, Dittmer DP, Damania B. The dual PI3K/mTOR inhibitor, NVP-BEZ235, is efficacious against follicular lymphoma. Leukemia. 2010;24(10):1781–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wunderle L, Lang F, Badura B, Wolf A, Serve HN, Goekbuket N, et al. A phase I, dose-finding study of the oral, dual PI3-kinase/mTOR inhibitor BEZ235 in adult patients with relapsed or refractory acute leukemia. Haematologica. 2013;98(Suppl 1). Abstract P620 [Google Scholar]
- 73.Mahadevan D, Qi W, Stejskal A, Cooke L, Garlich JR. SF1126, a pan-PI3K inhibitor has superior preclinical activity to CAL-101 a PI3K delta-specific inhibitor in aggressive B-cell non-hodgkin’s lymphoma. Blood. 2011;118(21). Abstract 2720 [Google Scholar]
- 74.Mahadevan D, Chiorean EG, Harris WB, Von Hoff DD, Stejskal-Barnett A, Qi W, et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur J Cancer. 2012;48(18):3319–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.LoRusso P, Markman B, Tabernero J, Shazer R, Nguyen L, Heath E, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL-765, a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced solid tumors. J Clin Oncol. 2009;27(15s). Abstract 3502 [Google Scholar]
- 76.Patel MR, Kahl BS, Horwitz SM, Younes A, Foss FM, Oki Y, et al. Preliminary safety and efficacy of IPI-145, a potent inhibitor of phosphoinositide-3-kinase-δ,γ, in patients with relapsed/refractory CLL. J Clin Oncol. 2013;31(suppl). Abstract 7070 [Google Scholar]
- 77.Kahl B, Patel M, Younes A, Horwitz S, Foss FM, Oki Y, et al. Preliminary safety and efficacy of IPI-145, a potent inhibitor of phosphoinositide-3-kinase-d-g in patients with relapsed/refractory B-cell lymphoma. Hematol Oncol. 2013;31(suppl 1). Abstract 066 [Google Scholar]
- 78.Arita A, Hanlon K, Chkourko H, Lannutti BJ, Johnson DM, Gabrilove JL, et al. Effect of phosphotidylinositol 3-kinase-delta inhibitor idelalisib (GS-1101) on signaling in primary non-hodgkin lymphoma cells: Correlative studies from NCT01306643. J Clin Oncol. 2013;31(suppl). Abstract 8579 [Google Scholar]
- 79.Banerjee S, Baird R, Basu B, Shah K, Tunariu N, Moreno Garcia V, et al. A phase I study evaluating GDC-0941, a pan-phosphoinositide-3 kinase (PI3K) inhibitor, in patients (pts) with advanced solid tumours, multiple myeloma, and PIK3Ca mutant (mt) tumours. Eur J Cancer. 2011;47(Suppl 1). Abstract 1248P [Google Scholar]
- 80.Wagner AJ, Bendell JC, Dolly S, Morgan JA, Ware JA, Fredrickson J, et al. A first-inhuman phase I study to evaluate GDC-0980, an oral PI3K/mTOR inhibitor, administered QD in patients with advanced solid tumors. J Clin Oncol. 2011;29(suppl). Abstract 3020 [Google Scholar]
- 81.Brana I, LoRusso P, Baselga J, Heath EI, Patnaik A, Gendreau S, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765 (SAR245409), a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced malignancies. J Clin Oncol. 2010;28(suppl). Abstract 3030 [Google Scholar]
- 82.Edelman G, Bedell C, Shapiro G, Pandya SS, Kwak EL, Scheffold C, et al. A phase I dose-escalation study of XL147 (SAR245408), a PI3K inhibitor administered orally to patients (pts) with advanced malignancies. J Clin Oncol. 2010;28 Abstract 3004 [Google Scholar]
- 83.Munster P, Specht J, Werner TL, Dees EC, Tan A, Schellens JHM, et al. PI3K kinase inhibitor GSK2126458 (GSK458): Clinical activity in select patient (PT) populations defined by predictive markers (study P3K112826). Ann Oncol. 2013;23(suppl 10). Abstract 442O [Google Scholar]
- 84.Foukas LC, Berenjeno IM, Gray A, Khwaja A, Vanhaesebroeck B. Activity of any class IA PI3K isoform can sustain cell proliferation and survival. Proc Natl Acad Sci USA. 2010;107(25):11381–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–28 [DOI] [PubMed] [Google Scholar]
- 86.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, et al. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282–90 [DOI] [PubMed] [Google Scholar]
- 87.Brown JR, Davids MS, Rodon J, Abrisqueta P, DeCillis AP, Rockich K, et al. Phase I trial of SAR245408 (S08), a pan-phosphatidylinositol 3 kinase (PI3K) inhibitor, in patients with chronic lymphocytic leukemia (CLL) and lymphoma. Blood. 2011;118(21). Abstract 2683 [Google Scholar]
- 88.Jeffers M, Dubowy RL, Lathia CD, Mallon R, Appleman LJ, Ramanathan RK, Patnaik A. Evaluation of the PI3K inhibitor BAY 80-6946 in hematologic malignancies. J Clin Oncol. 2012;30(suppl). Abstract e13576 [Google Scholar]
- 89.Patnaik A, Ramanathan RK, Appleman LJ, Tolcher AW, Mountz JM, Beerham M, et al. Phase I study of intravenous PI3K inhibitor BAY 80-6946: Preliminary activity in patients with relapsed non-hodgkin lymphoma (NHL) treated in an MTD expansion cohort. Blood. 2012;120(21). Abstract 3704 [Google Scholar]
- 90.Hong DS, Bowles DW, Falchook GS, Messersmith WA, George GC, O’Bryant CL, et al. A multicenter phase I trial of PX-866, an oral irreversible phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(15):4173–82 [DOI] [PubMed] [Google Scholar]
- 91.Garcia-Martinez JM, Wullschleger S, Preston G, Guichard S, Fleming S, Alessi DR, Duce SL. Effect of PI3K- and mTOR-specific inhibitors on spontaneous B-cell follicular lymphomas in PTEN/LKB1-deficient mice. Br J Cancer. 2011;104(7):1116–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jin L, Tabe Y, Zhou Y, Miida T, Andreeff M, Konopleva M. Efficacy and mechanisms of apoptosis induction by simultaneous inhibition of PI3K with GDC-0941 and blockade of bcl-2 (ABT-737) or FLT3 (sorafenib) in AML cells in the hypoxic bone marrow microenvironment. Blood. 2010;116(21). Abstract 777 [Google Scholar]
- 93.Ni J, Liu Q, Xie S, Carlson C, Von T, Vogel K, et al. Functional characterization of an isoform-selective inhibitor or PI3K-p110beta as a potential anticancer agent. Cancer Discovery. 2012;2:425–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coutre SE, Byrd JC, Furman RR, Brown JR, Benson DM, Wagner-Johnston ND, et al. Phase I study of CAL-101, an isoform-selective inhibitor of phosphatidylinositol 3-kinase P110d, in patients with previously treated chronic lymphocytic leukemia. J Clin Oncol. 2011;29(suppl). Abstract 6631 [Google Scholar]
- 95.Coutre SE, Leonard JP, Furman RR, Barrientos JC, De Vos S, Flinn IW, et al. Combinations of the selective phosphatidylinositol 3-kinased-delta (PI3Kdelta) inhibitor GS-1101 (CAL-101) with rituximab and/or bendamustine are tolerable and highly active in patients with relapsed or refractory chronic lymphocytic leukemia (CLL): Results from a phase I study. Blood 2012;120(21). Abstract 191 [Google Scholar]
- 96.Furman RR, Barrientos JC, Sharman JP, De Vos S, Leonard J, Coutre SE, et al. A phase I/II study of the selective phosphatidylinositol 3-kinase-delta (PI3K) inhibitor, GS-1101 (CAL-101), with ofatumumab in patients with previously treated chronic lymphocytic leukemia (CLL). J Clin Oncol. 2012;30(suppl). Abstract 6518 [Google Scholar]
- 97.Leonard JP, Wagner-Johnston ND, Coutre SE, Flinn IW, Schreeder MT, Fowler NH, et al. Combinations of the PI3Kd inhibitor idelalisib (GS-1101) with rituximab and/or bendamustine are tolerable and highly active in patients with previously treated, indolent non-hodgkin lymphoma (INHL): Updated results from a phase I study. Hematol Oncol. 2013;31(suppl 1). Abstract 068 [Google Scholar]
- 98.Fowler NH, De Vos S, Schreeder MT, Leonard JP, Flinn IW, Coutre SE, et al. Combinations of the phosphatidylinositol 3-kinase-delta (PI3Kdelta) inhibitor GS-1101 (CAL-101) with rituximab and/or bendamustine are tolerable and highly active in previously treated, indolent non-hodgkin lymphoma: Results from a phase I study. Blood. 2012;120(21). Abstract 3645 [Google Scholar]
- 99.Salles GA, Kahl BS, De Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, et al. Interim results from a phase 2 study of PI3Kd inhibitor idelalisib in patients with relapsed indolent non-hodgkin lymphoma (INHL) refractory to both rituximab and an alkylating agent. Hematol Oncol. 2013;31(suppl 1). Abstract 064 bis [Google Scholar]
- 100.Leung E, Kim JE, Rewcastle GW, Finlay GJ, Baguley BC. Comparison of the effects of the PI3K/mTOR inhibitors NVP-BEZ235 and GSK2126458 on tamoxifen-resistant breast cancer cells. Cancer Biol Ther. 2011;11(11):938–46 [DOI] [PMC free article] [PubMed] [Google Scholar]