

Abstract
β-thalassemia and sickle cell disease are widespread fatal genetic diseases. None of the existing clinical treatments provides a solution for all patients. Two main strategies for treatment are currently being investigated: (i) gene transfer of a normal β-globin gene; (ii) reactivation of the endogenous γ-globin gene. To date, neither approach has led to a satisfactory, commonly accepted standard of care. The δ-globin gene produces the δ-globin of hemoglobin A2. Although expressed at a low level, hemoglobin A2 is fully functional and could be a valid substitute of hemoglobin A in β-thalassemia, as well as an anti-sickling agent in sickle cell disease. Previous in vitro results suggested the feasibility of transcriptional activation of the human δ-globin gene promoter by inserting a Kruppel-like factor 1 binding site. We evaluated the activation of the Kruppel-like factor 1 containing δ-globin gene in vivo in transgenic mice. To evaluate the therapeutic potential we crossed the transgenic mice carrying a single copy activated δ-globin gene with a mouse model of β-thalassemia intermedia. We show that the human δ-globin gene can be activated in vivo in a stage- and tissue-specific fashion simply by the insertion of a Kruppel-like factor 1 binding site into the promoter. In addition the activated δ-globin gene gives rise to a robust increase of the hemoglobin level in β-thalassemic mice, effectively improving the thalassemia phenotype. These results demonstrate, for the first time, the therapeutic potential of the δ-globin gene for treating severe hemoglobin disorders which could lead to novel approaches, not involving gene addition or reactivation, to the cure of β-hemoglobinopathies.
Introduction
β-thalassemia and sickle cell disease (SCD) are severe hereditary anemias and are among the most common monogenic diseases worldwide.1,2 Both are chronic diseases with considerable morbidity and high mortality. β-thalassemia is generally fatal if not treated in the earliest years of life. Heterologous bone marrow transplantation is the only definitive cure but is currently available only for patients with a matched donor, optimally a sibling. For the majority of patients there is currently no hope for a permanent cure. It is, therefore, necessary to find alternative ways to treat the disease. Two main alternative strategies for the treatment of β-hemoglobinopathies are currently being investigated: (i) gene transfer of a normal β-globin gene, mostly through the use of retroviral vectors;3,4 and (ii) reactivation of the endogenous γ-globin gene that is normally expressed only during fetal life.5 To date, neither approach has led to a satisfactory, commonly accepted standard of care for hemoglobinopathies.1,2
Previous studies by our group,6 as well as by others,7,9 led us to believe that human δ-globin gene activation could represent an alternative approach to the treatment of β-thalassemia and SCD. Relatively few studies have addressed the regulation of the δ-globin gene, which is part of hemoglobin A2 (HbA2).10 HbA2 (δ2α2) is a minor adult hemoglobin usually expressed at levels less than 3% in normal individuals. The δ and β human globin genes derive from a common gene ancestor and have maintained a strong sequence homology by gene conversion events. HbA2 has functional properties that are nearly identical to those of HbA (α2β2).10 Moreover HbA2 inhibits HbS polymerization by acting as an anti-sickling agent.11
Treatment of β-thalassemia and SCD by stimulation of the adult δ-globin gene has, in principle, some advantages over reactivation of the fetal γ-globin gene. HbF and HbA2 are minor components of adult blood which are equally effective at inhibiting HbS polymerization.11 However, in adult blood, HbF is expressed only in a small fraction of cells (less than 2%), called F cells, whereas the expression of the δ-globin chain is always pancellular. The γ-globin gene is therefore in an inactive chromatin configuration in most adult erythroid cells, whereas the δ-globin gene is in an active chromatin configuration and is transcribed in all erythroid cells, although at a low efficiency. The low expression of δ-globin in adult blood is mostly due to a mutation in the Kruppel-like factor 1 (Klf1) binding site (CACCC box) within the δ-globin proximal promoter region.6–9 Some instability of the δ-globin mRNA may also contribute.10
Several research groups, including ours, have demonstrated in transient cell culture expression assays6–8 as well as in primary erythroid cells,9 the feasibility of transcriptionally activating the human δ-globin gene promoter by inserting the β-globin proximal CACCC box, which has high affinity for the transcription factor Klf1.6–9 Klf1 is an erythroid tissue-restricted transcription factor belonging to the zinc-finger family of DNA binding proteins.12 Its pivotal importance in the expression of the adult β-globin gene has been highlighted by gene ablation (knock-out) experiments in mice.13,14 Klf1 stimulation of the β-globin gene promoter depends on binding to the CACCC box.15 The activation of the δ-globin gene promoter obtained with the insertion of the β-CACCC is theoretically sufficient to compensate the imbalance of globin chains caused by β-thalassemia.6–9
Based on these results, we have undertaken studies in vivo by creating transgenic mice containing the same DNA constructs as those tested in cell cultures. Here we show in vivo that it is possible to activate the promoter of the δ-globin gene to a level similar to that of the wild-type (wt) β-globin gene promoter confirming in an animal model the efficacy demonstrated in previous in vitro studies.6–9 Moreover, a single copy transgenic mouse line carrying the full length δ-globin gene coding sequence under the control of the Klf1 activated δ-globin gene promoter expresses the δ-globin gene to a high level.
To better highlight the therapeutic potential of the δ-globin chain for the treatment of β-hemoglobinopathies, we crossed transgenic mice carrying the activated δ-globin gene with a mouse model of β-thalassemia intermedia (Hbbth3/+). The resulting animals have substantially increased hemoglobin levels, which are maintained in adult life over time, and a dramatic improvement of hematologic parameters.
These results are the first proof of principle validation of the therapeutic potential of the δ-globin gene in β-hemoglobinopathies and provide the basis for the development of approaches based on δ-globin gene expression for the therapy of β-thalassemia and SCD.
Methods
Constructs
In vivo reporter analysis was carried out using HS2δFLβRL constructs.6 A DNA construct containing the mini-LCR (HS1–HS4) and the δ-globin gene driven by the CACCC-containing δ-globin gene promoter (CACCCδ-LCR) was produced using the GSE 1417 vector.16 A similar DNA construct containing the δ-globin gene driven by the wt δ-globin promoter (δ-LCR) was also produced.
Generation of transgenic mice
Transgenic mice were generated by standard techniques. Genotype was determined by polymerase chain reaction. All animal protocols used in this study were approved by national and institutional policies on laboratory animal care.
Luciferase assay
Crude proteins extracted were assayed for luciferase versus renilla activity by the Dual-Luciferase Reporter Assay System (Promega) on a Lumat LB9501 luminometer (Bertold and Wallac).
Copy number analysis
The average transgene copy number was determined by Southern blot assay as well as real-time quantitative PCR (RT-qPCR).
Gene expression analysis
RNA extraction and retrotranscription were carried out using standard techniques. Gene expression was assessed by an S1 protection assay, carried out as described by Stroubulis J et al.17 and Poddie et al.,18 as well as RT-qPCR.
Hematologic analyses
Blood samples from adult mice were obtained by retro-orbital puncture under anesthesia. Hematologic parameters were determined using an Automated Hematology Cell Counter MS4 (Melet Schloesing Lab) and the HemoCue Hemoglobin System.
Red cell morphology
Blood smears were obtained from wild-type, Hbbth3/+ and Hbbth3/+CACCCδ-LCR (homozygous and heterozygous) adult mice and stained with May-Grünwald Giemsa (Sigma) for morphological analysis of red blood cells. The analysis was performed using a double-blind procedure.
High-performance liquid chromatography
Globin chains were separated by high-performance liquid chromatography (HPLC) using analytical Beckman System Gold equipment.
Flow cytometry
Fluorescence-activated cell sorting (FACS) analysis was carried out by classical CD71 and Ter119 co-staining.
Measurement of tissue iron content
The iron content of the liver, heart, and spleen was determined by non-heme iron analysis, as described by Torrance and Bothwell.19
Statistical analysis
Statistical differences between means were calculated with Student’s t-test (Microsoft Excel).
Results
Expression analysis of the δ-globin promoter in vivo using reporter constructs
To evaluate the activation of the CACCC-containing δ-globin gene promoter in competition with the β-globin gene promoter in vivo we created transgenic mice constructs in which the wt β-globin gene promoter and the δ-globin gene promoter containing the proximal CACCC (or the wt δ-globin gene promoter in a control construct) were linked in cis to a single enhancer (HS2δFLβRL in Figure 1A).6
Figure 1.
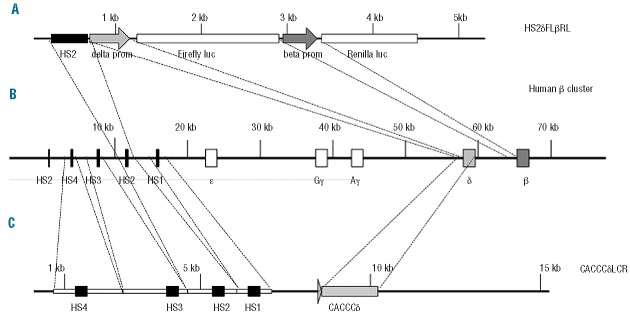
Schematic representation of the constructs used in the present study. (A) Schematic representation of the HS2δFLβRL construct used for in vivo reporter analysis. The construct is composed of a single HS2 enhancer, the δ-globin gene promoter wt (HS2δwtLβRL) or the CACCC-containing δ-globin gene promoter (HS2CACCCδFLβRL) driving firefly luciferase and the β-globin gene promoter driving renilla luciferase. (B) Schematic representation of the human β-globin cluster showing the relative location of the five hypersensitive sites (HS) of the LCR, ε, Gγ, Aγ, δ and β-genes. (C) Schematic representation of the δ-LCR construct used for in vivo studies. The construct is composed of mini-LCR containing HS4-HS3-HS2-HS1 sites and the full length CACCC-containing δ-globin gene (CACCCδ-LCR).
In this construct the wt β-globin gene promoter drives the renilla luciferase cDNA and the δ-globin gene promoter drives the firefly luciferase cDNA. We produced and analyzed three independent transgenic mice lines bearing the CACCC-containing δ-globin gene promoter (HS2δCACCCFLβRL) and three transgenic mice lines bearing the wt δ-globin gene promoter (HS2δFLβRL). Transgenic copy number was determined by Southern blotting. All transgenic lines were multicopy (data not shown).
The level of expression of the two reporter genes was assessed in the yolk sac at days 10.5 post-coitum (pc), in the fetal liver at days 12.5, 14.5 and 16.5 pc and in the peripheral blood in adult mice. For each transgenic line we analyzed three different pregnancies. In each experiment the luciferase activity of the β-globin promoter was considered as 100% and all other values were calculated as percentages of the β-globin promoter activity (relative luciferase activity, RLA). In the yolk sac at 10.5 pc the CACCC-containing δ-globin gene promoter did not show any increase in RLA compared to that of the wt δ-globin gene promoter (Figure 2).
Figure 2.
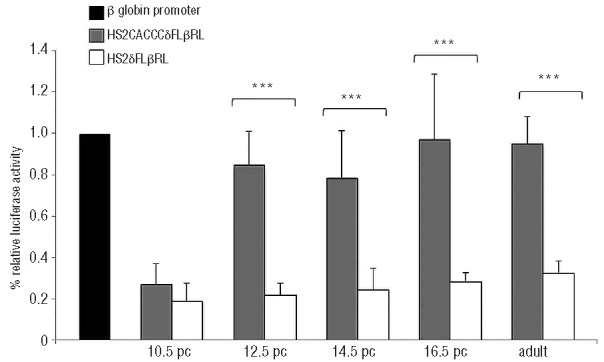
The histogram represents the relative luciferase activity (RLA), expressed as a percentage of activity of the wt β-globin gene promoter. Black indicates wt β-globin promoter, gray the CACCC-containing δ-globin promoter (HS2CACCCδFLβRL) and white the wt δ-globin promoter (HS2δFLβRL). The level of promoter activity was evaluated at 10.5 pc, 12.5 pc, 14.5 pc, 16.5 pc and in adulthood by measurement of firefly luciferase activity relative to the internal control renilla luciferase activity. Fold increase was calculated compared with expression in wt δ-globin gene promoter. 10.5 pc P=0.23; 12.5 pc P=1.57×10−6, 14.5 pc P=0,000232; 16.5 pc P=0.000605 and adult P=0.0056. Error bars indicate SD of the mean of at least three independent experiments.
This result indicates that the insertion of the CACCC box (Klf1 consensus sequence) does not alter the stage specificity of the δ-globin gene promoter, which is not expressed in the yolk sac (embryonic erythropoiesis). On the other hand, the same construct displayed increased δ-globin promoter activity in the fetal liver at the stage of definitive erythropoiesis (Figure 2). In comparison to the activity of the wt β-globin gene promoter in cis, the RLA rose to 82±17% (t-test, P=1.57×10−6) in the 12.5 pc fetal liver, 75±22% (t-test, P=0.0002) in the 14.5 pc fetal liver and 97±29% (t-test, P=0.0006) in the 16.5 pc fetal liver. In adult peripheral blood, the RLA of the δ-globin promoter increased up to 95%±13% (t-test, P=0.00058).
These data are in agreement with previous results6 and indicate that the creation of a single Klf1 consensus sequence inside an otherwise wt δ-globin gene promoter is sufficient to enhance the expression in erythroid tissue in vivo to an extent comparable to the wt β-globin gene promoter.
Expression of the activated δ-globin gene promoter was confined to hematopoietic tissues as ectopic expression was undetectable (data not shown).
In summary our results show that the activation of the CACCC-modified human δ-globin gene promoter in definitive erythropoiesis is comparable to the wt β-globin gene promoter and is tissue- and stage-specific.
Expression analysis of single copy activated δ-globin gene transgenic mice
To evaluate the ability of the activated δ-globin gene promoter to drive the expression to the high level of a full length δ-globin gene we produced three single-copy transgenic mouse lines carrying the mini LCR (HS1–HS4) and a full length δ-gene driven by the CACCC containing δ promoter (CACCCδ-LCR) (Figure 1C). The mini-LCR confers erythroid-specific, site-of-integration-independent expression of the transgene.16
Single copy transgenic lines were produced by microinjection of diluted DNA constructs. The presence of a single copy transgene was assessed by qPCR by comparison to an established single copy transgene that contains the full β-globin cluster and therefore a single-copy δ-globin gene (line 72 described by Stroboulis et al.17) Of the three lines produced, two expressed comparable levels of δ-globin mRNA whereas the third did not express any δ-globin mRNA as determined by qPCR. Southern blotting analysis of the third transgenic line revealed that the mini-LCR had unwanted deletions that impaired functionality and was not, therefore, further investigated (data not shown). One of the two expressing lines was further investigated to establish the full therapeutic potential of the δ-globin gene.
We also produced single copy wt δ-globin transgenic lines. None of the three single copy wt δ-globin gene transgenic lines produced detectable levels of expressed transgene. Southern blotting analysis revealed that the transgene was rearranged in two lines (data not shown). The third line was intact but did not express detectable levels of the transgene. Therefore, as a control for single copy δ-globin expression level we preferred to use the transgenic line 72, which expresses detectable, although low, levels of δ-globins.
The expression of the δ-globin transgene was first analyzed in adult blood of heterozygous CACCCδ-LCR mice by S1 nuclease protection assay in comparison to the endogenous α-globin murine gene and to line 72 (Figure 3A).
Figure 3.
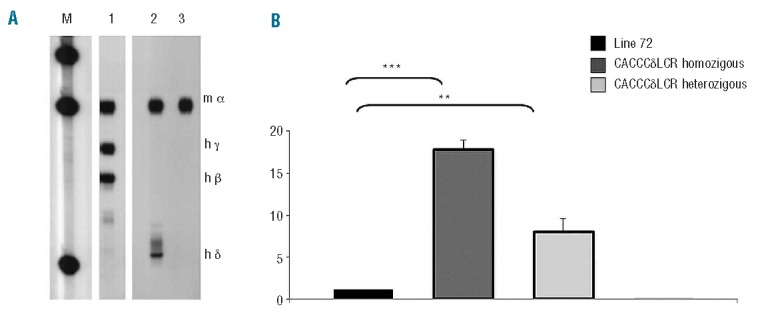
Determination of δ-globin gene expression level. (A) S1 nuclease protection analysis of 12.5 day pc fetal liver RNA from transgenic line 72 as a positive control for δ-globin gene expression (lane 1), transgenic line CACCCδ-LCR (lane 2) and wt whole blood mouse as a negative control for δ-globin gene expression (lane 3). M is the DNA molecular weight marker plasmid ΦX174 HaeIII-digested. (B) Relative expression levels of δ-globin gene by RT-qPCR. The histogram represents the expression level of δ-globin gene analyzed in blood samples from the transgenic line bearing the entire human β-globin cluster line 72 (black), the transgenic line bearing the δ-globin gene (CACCCδ-LCR) in homozygosity (dark gray) or heterozygosity (light gray). mRNA levels of reference genes were used for normalization in all cases. ***P<0.001 or **P<0.01 according to a t-test.
S1 probes were a mixture of mouse α and human δ, β and γ globins. Blood RNA from wt mice was included in the experiment as a negative control for δ-globin gene expression. In S1 analysis, the correct and expected protected fragments for mouse α-globin mRNA were present in all the lines. In addition the expected human δ-globin S1 protected fragment was present in the CACCCδ-LCR sample (Figure 3A, lane 2). The relative expression level was 20% compared to that of the endogenous α-globin mouse gene, as determined by phosphoimager with a correction for probe-specific activity. As shown in Figure 3A (lane 1) the δ-globin gene expression signal of the control line 72 was barely visible making it difficult to compare the expression level and to evaluate the extent of activation of the CACCCδ-LCR transgene versus the wt δ-globin gene. On the other hand, in this transgenic line the δ-globin mRNA is detectable by RT-qPCR. Therefore, to analyze the level of expression of the activated δ-globin transgene in comparison to the wt δ-globin gene further, we performed RT-qPCR analysis on our single copy transgenic line (CACCCδ-LCR) in comparison to line 72. The expression level of the δ globin gene was approximately 18-fold (±1.59) more in the homozygous mice and 8.9-fold (±1.21) more in the heterozygous animals with respect to the single copy transgenic line bearing the entire human β globin cluster.
Rescuing the thalassemic phenotype of the Hbbth3/+ mice model
To evaluate whether the production of δ-globin in our transgenic model was able to ameliorate a β-thalassemic phenotype, we crossed the homozygous transgenic line CACCCδ-LCR with the heterozygous mouse model of β° thalassemia (Hbbth3/+).20
Phenotype rescue was evaluated by comparison of hematologic parameters, total hemoglobin, red blood cell (RBC) count, hematocrit, mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean cell hemoglobin concentration (MCHC), and red cell distribution width (RDW), in wt, Hbbth3/+ and Hbbth3/+ CACCCδ-LCR adult mice either heterozygous or homozygous for the transgene.
Correction of anemia was inferred mainly from the hemoglobin levels that increased from 8.75±0.97 g/dL in Hbbth3/+ mice to 11.32±0.97 g/dL in Hbbth3/+ CACCCδ-LCR heterozygous mice and to 12.96±0.59 g/dL in Hbbth3/+ CACCCδ-LCR homozygous mice, which was very close to the normal value of 14.49±1.28 g/dL in the wt control mice (Figure 4).
Figure 4.
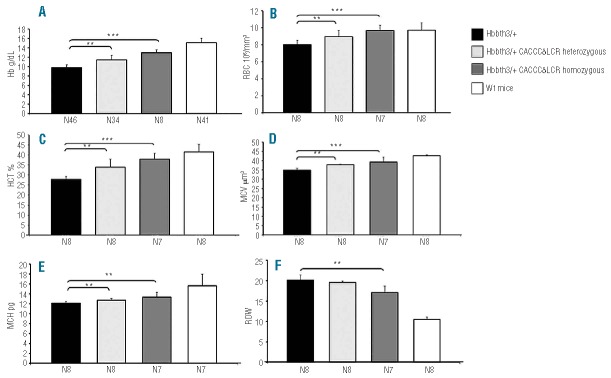
Hematologic parameters. (A) Hb: hemoglobin levels. (B) RBC: red blood cell counts. (C) HCT: hematocrit percentage. (D) MCV: mean corpuscular volume. (E) MCH: mean corpuscular hemoglobin. (F) RDW: red cell distribution width. N indicates the number of mice.
Relative to Hbbth3/+ mice, the Hbbth3/+ CACCCδ-LCR homozygous adult mice show not only an increase in hemoglobin, but also a significant improvement in all other hematologic parameters whether measured (hematocrit, RBC count) or derived (RDW, MCV, MCH, MCHC) (Figure 4).
High-performance liquid chromatography
HPLC elution analysis was performed on Hbbth3/+ and Hbbth3/+CACCCδ-LCR homozygous blood samples from the single Hbb genotype mice. This elution analysis showed the characteristic globin chain profiles with the mouse α and β-chain peaks in mice with the Hbbth3/+ genotype (Figure 5A) and the presence of an additional peak formed by human δ-chain in mice with the Hbbth3/+CACCCδ-LCR genotype (Figure 5B).
Figure 5.
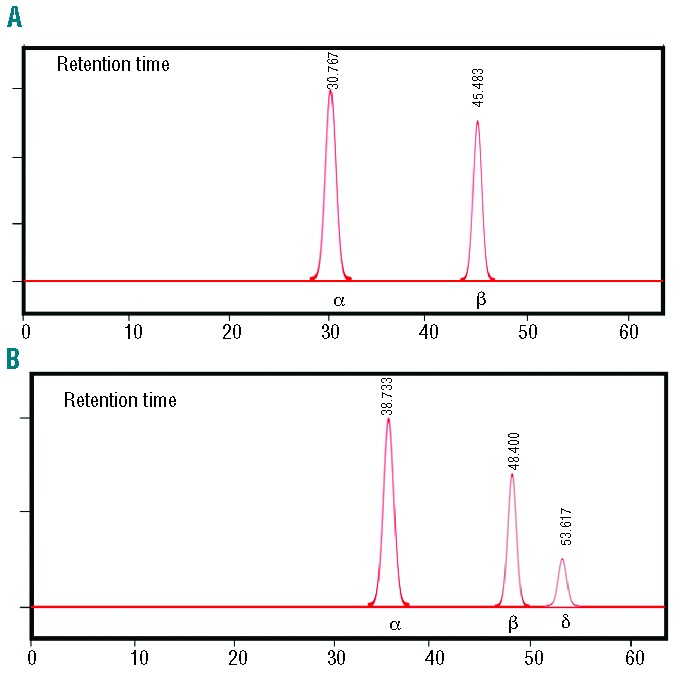
Representative HPLC elution profiles of globin chains from whole blood samples. (A) Transgenic line Hbbth3/+. (B) Transgenic line Hbbth3/+CACCCδ-LCR. The spectra of the transgenic line Hbbth3/+CACCCδ-LCR show the presence on δ-globin chain beside the normal globin chains (α and β).
Red cell morphology
Blood smears were obtained from wt (Figure 6A), Hbbth3/+ (Figure 6B) and Hbbth3/+ CACCCδ-LCR homozygous (Figure 6C) adult mice. Blind microscopic evaluation of Hbbth3/+ mice blood smears showed characteristics typical of those seen in patients with severe thalassemia, including marked anisocytosis and poikilocytosis, microcytes and frequent target cells. Red cells were generally hypochromic with variable numbers of teardrop, oval forms and fragmented cells (Figure 6B). In comparison to blood smears from Hbbth3/+ mice, those from Hbbth3/+CACCCδ-LCR homozygous mice showed a much lesser degree of anisocytosis and poikilocytosis, fewer target and fragmented cells and the RBC were normochromic rather than hypochromic (Figure 6C).
Figure 6.
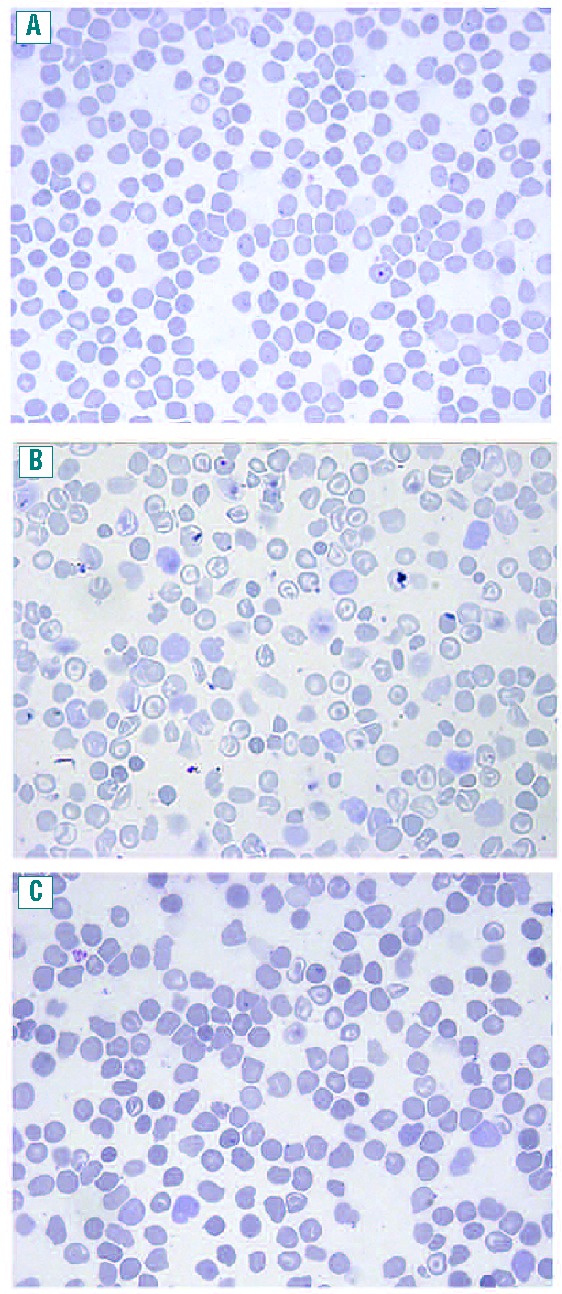
Peripheral blood smears. (A) Adult C57BL/6 mouse, (×63, May-Grunwald stain) (B) Adult Hbbth3/+ mouse, (×63, May-Grunwald stain) (C) Adult Hbbth3/+CACCCδ-LCR homozygous mouse, (×63, May-Grunwald stain) Blood smears from Hbbth3/+CACCCδ-LCR homozygous mouse show less anisocytosis and poikilocytosis, fewer target cells and fragmented cells compared to Hbbth3/+ mouse blood smears and normochromic instead of hypochromic red cells.
Activation of δ-gene improves erythropoiesis in Hbbth3/+ mice
To estimate the extent of ineffective erythropoiesis we evaluated the fraction of spleen and bone marrow cells in discrete erythroid populations by flow cytometry using co-staining for the transferrin-receptor CD71 and erythroid-specific TER119. The analysis was performed on wt, Hbbth3/+CACCCδ-LCR homozygous, Hbbth3/+CACCCδ-LCR heterozygous and Hbbth3/+ mice lines at 2.5 months of age (Figure 7A,B). The CD71+/Ter119+ population corresponds to early erythroid cells (basophilic erythroblasts, late basophilic and chromatophilic erythroblasts), whereas the CD71−/Ter119+ population corresponds to orthochromatophilic erythroblasts and, mostly, to enucleated erythroid cells. FACS analysis performed on spleen and bone marrow cells showed that the percentage of early erythroid cells (CD71+/Ter119+) is significantly lower in Hbbth3/+CACCCδ-LCR heterozygous and Hbbth3/+CACCCδ-LCR homozygous mice lines than in Hbbth3/+ mice, whereas the relative number of more differentiated erythroid cells (CD71−/Ter119+) was higher (representative FACS profiles of splenic erythroid cells are shown in Figure 7A).
Figure 7.

Effect of the activated δ-globin gene on ineffective erythropoiesis and organ iron content in animals with a β-thalassemia intermedia phenotype (Hbbth3/+ mice) at 2.5 months of age. (A) Erythroid populations in splenic cell suspensions measured by classical CD71 and Ter119 co-staining showing the percentage of double-positive cells and single-positive Ter119 cells. FACS profiles from four representative mice: wt mice, Hbbth3/+ CACCCδ-LCR homozygous, Hbbth3/+ CACCCδ-LCR heterozygous and Hbbth3/+ transgenic lines. (B) Amount of ineffective erythropoiesis estimated in the spleen and in the bone marrow based on the ratio between early precursor (Ter119+/CD71+) and more mature erythroid cells (Ter119+/CD71−) in wt mice, Hbbth3/+CACCCδ-LCR homozygous, Hbbth3/+CACCCδ-LCR heterozygous and Hbbth3/+ transgenic lines. (C) Spleen weights normalized to body weight of wt, Hbbth3/+CACCCδ-LCR homozygous, Hbbth3/+CACCCδ-LCR heterozygous and Hbbth3/+ mice lines. (D) Spleen size observed in three representative mice: δ1) wt mouse; 2) Hbbth3/+CACCCδ-LCR homozygous and 3) Hbbth3/+ transgenic lines. (E) Total iron (mg) content of the spleen, liver and heart as measured by non-heme iron analysis in wt mice, Hbbth3/+CACCCδ-LCR homozygous, Hbbth3/+CACCCδ-LCR heterozygous and Hbbth3/+ mice transgenic lines. *P<0.05; **P<0.01; ***P<0.001 according to the t-test. Data are presented as mean ± SD. N indicates the number of mice.
The amount of ineffective erythropoiesis in the spleen and bone marrow was estimated based on the ratio between early precursor (Ter119+/CD71+) and more mature erythroid cells (Ter119+/CD71−) (Figure 7B).
Ineffective erythropoiesis was significantly improved in Hbbth3/+CACCCδ-LCR heterozygous mice (spleen: P=0.0022; bone marrow: P=0.042) and Hbbth3/+CACCCδ-LCR homozygous mice (spleen: P=0.0089; bone marrow: P=8.4×10−4) compared to that in Hbbth3/+ mice (Figure 7B).
The improvement of ineffective erythropoiesis in our mice model was further evaluated by measuring the spleen size in the different animals. We observed a marked reduction of splenomegaly in Hbbth3/+CACCCδ-LCR homozygous mice (P=3.1×10−5) and a slight reduction in Hbbth3/+CACCCδ-LCR heterozygous mice (P=0.048), both compared to Hbbth3/+ mice (Figure 7C,D).
Organ iron content
We evaluated the iron content of organs involved in RBC production and iron homeostasis. The iron content of the spleen, liver, and heart was analyzed by non-heme iron quantification19 in all the different animals. At 2.5 months the iron content was significantly lower in the spleen (P=0.0012), liver (P=1×10−4), and heart (P=6.9×10−4) of Hbbth3/+CACCCδ-LCR homozygous mice compared to that in the corresponding organs of Hbbth3/+ mice (Figure 7E).
In the Hbbth3/+CACCCδ-LCR heterozygous transgenic mice only the heart showed a significantly lower iron content compared to that of the Hbbth3/+ mice (P=0.006) (Figure 7E).
However, the assessment of iron content at 5 months revealed that Hbbth3/+CACCCδ-LCR heterozygous mice had a significant decrease of iron content compared to the Hbbth3/+ mice (data not shown) also in the spleen (P=2.73×10−5) and in the liver (P=0.0037).
Discussion
β-thalassemia and SCD have a pronounced impact on both socio-economic structure and national health care systems. Gene therapy of β-thalassemia has been a goal pursued for decades. Encouraged by preclinical studies,21–23 the first clinical trial using a lentiviral construct to treat patients with β-thalassemia was started in 2007.24,25 In this trial, a single patient with βE-thalassemia major (βE/βT) established 10% engraftment and an increase of hemoglobin of 3 g/dL compared to the pre-treatment level. However, a detailed analysis showed that in a partially dominant hematopoietic clone, the viral vector had integrated into a proto-oncogene, which could be a harbinger of a leukemic transformation as seen previously in gene therapy protocols with retroviruses.24,25
Another important line of research for the treatment of β-thalassemia and SCD is the activation of the γ-globin gene, not normally expressed in adults, although success with this strategy had been limited.5
Although promising none of the treatments so far explored is resolutive for all patients and it is, therefore, important to develop alternative and complementary approaches such as boosting the expression of the δ-globin gene that we propose here.
The validation of the δ-globin as a possible therapeutic target for the treatment of β-hemoglobinopathies is novel and could open a promising field of translational research.
In this report we present in vivo results demonstrating that the creation of a single Klf1 consensus sequence (proximal β-globin CACCC box) in the context of a normal δ-globin gene promoter increases the level of promoter activity almost to the level of that of the normal β-globin gene promoter in both fetal liver and adult peripheral blood. Hence, the activation of the δ-globin promoter occurs, as predicted, in the sites of adult erythropoiesis, but not in the embryonic yolk sac indicating that the expression is tissue- and developmental stage-specific.
In our assay, the wt δ-globin gene promoter exhibited about 20% residual activity compared to the β-globin gene promoter. This ratio of expression in our reporter constructs is higher than expected and could be explained by the absence of the second intervening sequence (IVS2), which contains an enhancer in the β-globin gene,26 and to the lack of the enhancer located 3′ to the β-globin gene.27
Next, we produced transgenic mice lines carrying a single copy of a full length δ-globin gene driven by its own promoter, wt or modified by the insertion of the proximal CACCC box, and by a mini-LCR. However, the lack of δ-globin expression in the wt construct forced us to use, as a reference for expression, a transgenic line that contains the full β-globin gene cluster (line 72), which expresses low but detectable levels of δ-globin RNA. In this assay, the CACCC-activated δ-globin gene was expressed at high levels in vivo, nine times more than the expression observed in line 72.
The extent of activation of the human δ-globin gene was evaluated in a transgenic line in which the δ-globin gene was under the control of the mini-LCR in comparison to a transgenic line containing the full β-globin locus. Hence, our reference construct might have had lower δ-globin expression because of the potential competition between the δ and β-globin gene for the LCR. Nevertheless previous results6–9 as well as those reported here, on when the δ and β-globin gene promoters were allowed to compete freely for the HS2, strongly support the autonomous activation of the CACCC-modified δ-globin gene. Further support comes from the knowledge that the Lepore β-thalassemic gene, which is a δ/β hybrid single gene and thus not exposed to competition, is poorly expressed in both humans28 and BAC transgenic mice.29 Finally, the observation that human β-thalassemic carriers (including those with β-globin gene promoter deletions) have a much lower increase in δ-expression than that observed here, is in agreement with the boosting of the δ-globin gene specifically due to the presence of the Klf1 binding site and not to the lack of competition with the wt β-globin promoter.
To further validate the δ-globin gene as a therapeutic target for β-hemoglobinopathies, we crossed the activated δ-globin transgenic line with the Hbbth3/+ β-thalassemia intermedia mouse model in an attempt to rescue the thalassemic phenotype. Resulting animals showed an average hemoglobin increase of 4.21 g/dL. This increase in hemoglobin concentration persisted lifelong and was the likely reason for the laboratory and clinical improvement of the β-thalassemia phenotype. These data are the first experimental evidence showing that the δ-globin chain can efficiently compensate the unbalanced globin chain synthesis that characterizes β-thalassemia.
Interestingly, the observed increase in hemoglobin concentration was greater than that obtained in previous studies with lentivirus-based gene therapy in two murine models of β-thalassemia.21–23 The hemoglobin gain was also greater than the 3 g/dL observed in the unique β-thalassemia patient recently cured by gene transfer with a lentivirus-encoded β-globin construct.24,25
Here we have strongly improved the β-thalassemia intermedia phenotype of Hbbth3/+ mice. The improvement of hematologic parameters and the decrease in the number of immature erythroid cells indicate that expression of the δ-gene in Hbbth3/+ mice had a positive effect on ineffective erythropoiesis and was correlated with decreased spleen, liver and heart iron load and a reduction of splenomegaly.
We also attempted to rescue the homozygous Hbbth3 β-thalassemia major phenotype. However, no rescued Hbbth3 homozygous mice were detected among 72 live-born pups (data not shown). Hence the 4.21 g/dL increase in hemoglobin concentration is very likely not sufficient to rescue the Hbbth3 homozygous β-thalassemic phenotype in mice, which is lethal in utero20 and thus far more severe than human β0-thalassemia. A murine model of Cooley’s anemia, more similar to the human phenotype, based on fetal liver hematopoietic transplantation, has been proposed by Rivella et al.23 The effect of the activated δ-globin gene in this β-thalassemia major mice model will be investigated in the near future. Nevertheless, the observed increase in hemoglobin concentration could represent the difference between transfusion-dependence and transfusion-inependence in most β-thalassemia patients and is predicted to be curative in SCD.
The 30% increase in hemoglobin concentration obtained by the full length activated δ-globin gene is significantly lower than the 82–95% RLA of the isolated promoter seen with the reporter constructs. This result is best explained by the difference among β and δ second intervening sequences (IVS2). β-IVS2 is in fact critical for high mRNA expression in erythroid cells.26 Some instability of the δ-globin mRNA compared to the β-globin mRNA may also contribute to this difference.10 Another reason for the difference in the expression level between reporter and gene constructs might be the use of only the HS2 in the reporter constructs, in place of the mini-LCR used in the full gene construct, which could act differently on the δ and β globin gene promoters.
A previous preliminary report suggested severe red cell abnormalities in transgenic mice expressing high levels of normal human δ-chain.30 In our transgenic mice, we did not observe any abnormality due to the expression of the δ-globin gene in spite of the comparable level of expression of the transgene (30% in homozygous mice). This difference is most likely due to the fact that those abnormalities were observed in transgenic mice homozygous for the β-major deletion and expressing high levels of unassembled human α-globin chains.30
In practice, δ globin gene activation could be obtained with the use of molecules selected through high-throughput screening, by transcription factors engineered to boost δ globin gene expression and by gene therapy approaches.
Zinc finger (ZF) proteins engineered to target specific DNA sequences are easily produced and commercially available.31 Recently up-regulation of the human δ-globin gene (as well as the γ-globin gene) by recombinant Klf1 factors fused to GATA1 has been reported.32 Using this approach, δ-globin gene was up-regulated 2.7-fold in CD34+ cells. The modest level of increase of the δ-globin gene expression was most likely due to the fact that the authors used a chimeric transcription factor harboring the GATA1 DNA binding domain. This artificial protein binds to a GATA1 binding site at position −74bp from the cap site of the δ-globin gene promoter. This interaction is likely to hamper the binding of the endogenous wt GATA1 transcription factor, which is of pivotal importance for the function of the δ-globin gene promoter.33
Activation could most likely be obtained by designing an artificial chimeric transcription factor made of ZF DNA binding domains designed to interact with the δ-globin gene promoter in the site where we created the Klf1 binding site, fused to the Klf1 activation domain. This artificial transcription factor should be able to activate the δ-globin gene promoter specifically without interfering with other transcription binding sites crucial for the δ-globin promoter architecture.
Another strategy could rely on genome editing through homologous recombination promoted by ZF nucleases targeting the δ-globin promoter to create a Klf1 binding site. Homologous recombination promoted by ZF nuclease appears to be highly efficient and has been successfully used against various human genes.31,34 A single couple of ZF nucleases could be designed to target the δ-globin gene promoter and endorse homologous recombination to create a Klf1 binding site. This approach would avoid the need to design different couples of ZF nucleases for each of the more than 200 mutations causing β-thalassemia.
In summary, here we have shown for the first time that it is possible to activate in vivo the human δ-globin gene to a considerable extent by the creation of a single Klf1 binding site in the promoter and that δ-globin activation is tissue- and stage-specific. The expression of the activated δ-globin gene in a β-thalassemia mice model greatly improves the phenotype, validating the δ-globin chain as a therapeutic target for β-hemoglobinopathies.
Acknowledgments
The authors would like to thank Michael Whalen for proofreading, and Liliana Maccioni for technical support. This work is dedicated in loving memory to Professor Antonio Cao and Professor Renzo Galanello.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
MFMan. was supported by Regione Autonoma della Sardegna (L.R. 08/07/2007 N 7, Promozione della Ricerca Scientifica e dell’Innovazione Tecnologica in Sardegna). This work was supported by AMGEN (through a grant to MSR).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;2(2):61–76 [DOI] [PubMed] [Google Scholar]
- 3.Persons DA. The challenge of obtaining therapeutic levels of genetically modified hematopoietic stem cells in beta-thalassemia patients. Ann NY Acad Sci. 2010; 1202:69–74 [DOI] [PubMed] [Google Scholar]
- 4.Rivière I, Dunbar CE, Sadelain M. Hematopoietic stem cell engineering at a crossroads. Blood. 2012;119(5):1107–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bauer DE, Kamran SC, Orkin SH. Reawakening fetal hemoglobin: prospects for new therapies for the β-globin disorders. Blood. 2012;120(15):2945–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ristaldi MS, Casula S, Porcu S, Pirastu M, Cao A. Activation of the delta globin gene by the beta-CACCC motif. Blood Cells Mol. Dis. 1999;25(3–4):193–209 [DOI] [PubMed] [Google Scholar]
- 7.Donze D, Jeancake PH, Townes TM. Activation of delta-globin gene expression by erythroid Krupple-like factor: a potential approach for gene therapy of sickle cell disease. Blood. 1996;88(10):4051–7 [PubMed] [Google Scholar]
- 8.Tang DC, Ebb D, Hardison RC, Rodgers GP. Restoration of the CCAAT box or insertion of the CACCC motif activates [corrected] delta-globin gene expression. Blood. 1997;90(1):421–7 [PubMed] [Google Scholar]
- 9.Tang DC, Rodgers GP. Activation of the human delta-globin gene promoter in primary adult erythroid cells. Br J Haematol. 1998;103(3):835–8 [DOI] [PubMed] [Google Scholar]
- 10.Steinberg MH, Adams JG., 3rd Hemoglobin A2: origin, evolution, and aftermath. Blood. 1991;78(9):2165–77 [PubMed] [Google Scholar]
- 11.Poillon WN, Kim BC, Rodgers GP, Noguchi CT, Schechter AN. Sparing effect of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S at physiologic ligand saturations. Proc Natl Acad Sci USA. 1993;90(11):5039–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siatecka M, Bieker JJ. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood. 2011;118(8):2044–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nuez B, Michalovich D, Bygrave A, Ploemacher R, Grosveld F. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature. 1995;375(6529):316–8 [DOI] [PubMed] [Google Scholar]
- 14.Perkins AC, Sharpe AH, Orkin SH. Lethal beta-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. 1995;375(6529):318–22 [DOI] [PubMed] [Google Scholar]
- 15.Lee JS, Ngo H, Kim D, Chung JH. Erythroid Kruppel-like factor is recruited to the CACCC box in the beta-globin promoter but not to the CACCC box in the gamma-globin promoter: the role of the neighboring promoter elements. Proc Natl Acad Sci. USA. 2000;97(6):2468–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collis P, Antoniou M, Grosveld F. Definition of the minimal requirements within the human beta-globin gene and the dominant control region for high level expression. EMBO J. 1990;9(1):233–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stroboulis J, Dillon N, Grosveld F. Developmental regulation of a complete 70-kb human β-globin locus in transgenic mice. Genes Dev. 1993;6(10):1857–64 [DOI] [PubMed] [Google Scholar]
- 18.Poddie D, Marongiu MF, Ferrari SC, Porcu S, Ristaldi MS. Delta-globin gene structure and expression in the K562 cell line. Hemoglobin. 2003;27(4):219–28 [DOI] [PubMed] [Google Scholar]
- 19.Torrance JD, Bothwell TH. Tissue iron stores. In: Iron Methods in Hematology. Vol. 1 New York, NY; Churchill Livingstone: 1980; p. 90–115 [Google Scholar]
- 20.Yang B, Kirby S, Lewis J, Detloff PJ, Maeda N, Smithies O. A mouse model for beta 0-thalassemia. Proc Natl Acad Sci USA. 1995;92(25):11608–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.May C, Rivella S, Callegari J, Heller G, Gaensler KM, Luzzatto L, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406(6791):82–6 [DOI] [PubMed] [Google Scholar]
- 22.May C, Rivella S, Chadburn A, Sadelain M. Successful treatment of murine beta-thalassemia intermedia by transfer of the human beta-globin gene. Blood. 2002;99(6):1902–8 [DOI] [PubMed] [Google Scholar]
- 23.Rivella S, May C, Chadburn A, Rivière I, Sadelain M. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood. 2003;101(8):2932–9 [DOI] [PubMed] [Google Scholar]
- 24.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467(7313):318–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Persons DA. Gene therapy: targeting β-thalassaemia. Nature. 2010;467(7313):277–8 [DOI] [PubMed] [Google Scholar]
- 26.Kosche KA, Dobkin C, Bank A. DNA sequences regulating human beta globin gene expression. Nucleic Acids Res. 1985;13(21):7781–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kollias G, Hurst J, deBoer E, Grosveld F. The human beta-globin gene contains a downstream developmental specific enhancer. Nucleic Acids Res. 1987;15(14):5739–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weatherall DJ, Clegg JB. The Thalassemia Syndromes, 4th edn: Blackwell Science, Oxford [Google Scholar]
- 29.Sloane-Stanley J, Roberts NA, Olivieri N, Weatherall DJ, Wood WG. Globin gene expression in Hb Lepore-BAC transgenic mice. Br J Haematol. 2006;135(5):735–7 [DOI] [PubMed] [Google Scholar]
- 30.Nagel RL, Sharma A, Kumar R, Fabry ME. Severe red cell abnormalities in transgenic mice expressing high levels of normal human delta chains. Blood. 1995;86(Suppl 1):251a [Google Scholar]
- 31.Klug A. The discovery of zinc fingers and their development for pratical applications in gene regulation and genome manipulation. Q Rev Biophys. 2010;43(1):1–21 [DOI] [PubMed] [Google Scholar]
- 32.Zhu J, Chin K, Aerbajinai W, Trainor C, Gao P, Rodgers GP. Recombinant erythroid Kruppel-like factor fused to GATA1 up-regulates delta- and gamma-globin expression in erythroid cells. Blood. 2011;117(11):3045–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuda M, Sakamoto N, Fukumaki Y. Delta-thalassemia caused by disruption of the site for an erythroid-specific transcription factor, GATA-1, in the delta-globin gene promoter. Blood. 1992;80(5):1347–51 [PubMed] [Google Scholar]
- 34.Cheng LT, Sun LT, Tada T. Genome editing in induced pluripotent stem cells. Genes Cells. 2012;17(6):431–8 [DOI] [PubMed] [Google Scholar]