

Abstract
Systemic anaplastic large cell lymphoma is a category of T-cell non-Hodgkin’s lymphoma which can be further subdivided into two distinct entities (ALK+ and ALK−) based on the presence or absence of ALK gene rearrangements. Among several pathways triggered by ALK signaling, constitutive activation of STAT3 is strictly required for ALK-mediated transformation and survival. Here we performed genome-wide microRNA profiling and identified 48 microRNA concordantly modulated by the inducible knock-down of ALK and STAT3. To evaluate the functional role of differentially expressed miRNA, we forced their expression in ALK+ anaplastic large cell lymphoma cells, and monitored their influence after STAT3 depletion. We found that the expression of the microRNA-17~92 cluster partially rescues STAT3 knock-down by sustaining proliferation and survival of ALK+ cells. Experiments in a xenograft mouse model indicated that forced expression of microRNA-17~92 interferes with STAT3 knock-down in vivo. High expression levels of the microRNA-17~92 cluster resulted in down-regulation of BIM and TGFβRII proteins, suggesting that their targeting might mediate resistance to STAT3 knock-down in anaplastic large cell lymphoma cells. We speculate that the microRNA-17~92 cluster is involved in lymphomagenesis of STAT3+ ALCL and that its inhibition might represent an alternative avenue to interfere with ALK signaling in anaplastic large cell lymphomas.
Introduction
Anaplastic large cell lymphoma (ALCL) is a subtype of T-cell non-Hodgkin’s lymphoma characterized by marked cellular pleomorphism, a propensity to grow cohesively, and a tendency to colonize lymph node sinuses. ALCL have a unique immunophenotype expressing high levels of CD30, cytotoxic granules (i.e. granzyme and perforin), and often lack T-cell associated/restricted antigens.1 The World Health Organization (WHO) classification of lymphoid malignancies recognizes two systemic forms of ALCL, defined by the presence or the absence of chromosomal translocations involving the anaplastic lymphoma kinase (ALK) gene at the 2p23 locus.2 The most common translocation is the t(2;5)(p23;q35) that codes for the nucleophosmin (NPM)-ALK fusion protein.3 All ALK translocations invariably result in constitutively active ALK chimeric proteins, which lead to the consequent activation of several growth-promoting and anti-apoptotic pathways, including PI3K/AKT/mTOR, JAK/STAT3, RAS/ERK, and others.4 Among them, it is known that the constitutive activation of the transcription factor STAT3 is strictly required for the maintenance of the ALK-mediated phenotype.5–8 Loss of STAT3 signaling in ALK+ ALCL results in cell cycle arrest, followed by the execution of an irreversible program of apoptosis.9,10 This phenomenon is associated with the modulation of a large number of genes, suggesting that the oncogenic activity of ALK+ ALCL cells requires STAT3-mediated transcription.10 Accordingly, a class-prediction analysis of ALCL expression profiles identified a STAT3 signature as a positive predictor for systemic ALK+ ALCL.10
The pleiotropic effects of STAT3 are due to the concomitant activation/repression of multiple sets of genes, which control crucial functions, such as cell cycle, apoptosis, DNA damage, adhesion, motility, G protein signaling, inflammation, immune response, metabolic pathways, and angiogenesis.11 STAT3 mediates the enhanced transcription of both anti-apoptotic factors and cell cycle regulators, such as Bcl-XL, survivin, cyclin D3, C/EBPβ, Mcl-1 and others, providing proliferative and survival signals in ALK+ ALCL cells.5,7,12–14 Moreover, STAT3 controls the expression of molecules that determine T-cell identity and signaling, and is in part responsible for the “null” phenotype of ALK+ ALCL.15 The strict requirement for STAT3 in ALK+ ALCL makes this molecule an ideal therapeutic target, particularly in the perspective of acquired resistance to ALK inhibitors.16,17 Transcription factor have been traditionally considered “undruggable” because the design of small and selective molecules that disrupt protein-protein or protein-DNA interactions bears considerable challenges. However, decoys targeting STAT3 have been successfully employed in vivo,18 and encouraging data are also emerging with small molecules such as S3I-201, niclosamide and pyrimethamine.11,19
An alternative strategy to interfere with key pathways linked to cancer progression was provided by the discovery that non-coding RNA, such as micro-RNA (miRNA, miR), could simultaneously target multiple oncogenes. Consequently, the manipulation of miRNA was intended as a “combination” therapy to impair compensatory mechanisms and feedback loops limiting the effectiveness of standard therapies, or responsible for the development of resistance.20
miRNA are a class of evolutionarily conserved noncoding RNA of 18–22 nucleotides that modulate gene expression through canonical base pairing between the seed sequence of the miRNA and its complementary match sequence present mainly within the 3′-UTR of target mRNA.21 It is known that miRNA can inhibit the expression of target mRNA either by mRNA cleavage or by translational repression.22 Deregulated miRNA have been observed in many cancers, displaying either tumor suppressor (miR-26a, miR-34a) or oncogenic functions (miR-21, miR-17~92 cluster, miR-155).23 Recent studies identified a number of miRNA aberrantly expressed in ALCL patients and suggested the involvement of miR-101,24 miR-29a,25 miR-135-b,26 and miR-1627 in mediating the oncogenic ALK signaling.
Here, we performed genome-wide profiling of STAT3-regulated miRNA in ALK+ ALCL cell lines. Microarray data analysis revealed a series of miRNA concordantly modulated by ALK and STAT3 knock-down (KD). Specifically, we found that the forced expression of miR-17~92 cluster promotes the survival of ALK+ ALCL cells in low serum conditions and partially rescues the STAT3 KD phenotype in vitro and in vivo. These data suggest that the miR-17~92 cluster could sustain oncogenic properties of STAT3 in T-cell lymphoma and that its inhibition could interfere with ALK downstream signaling.
Methods
Cell lines and culture
Human ALCL cells TS-SUP-M2, JB-6, L82, and Karpas-299 were cultured under standard conditions in RPMI-1640 (Sigma-Aldrich, St Luis, MO, USA) supplemented with 10% fetal calf serum (Lonza, Rockland, ME, USA), 2 mM glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin (Eurobio Biotechnology, Les Ulis, France). TS-SUP-M2-A5, TS-SUP-M2 A5M, TS-SUP-M2 S3S (inducible cell lines derived from TS-SUP-M2), and JB6 S3S were generated, as described elsewhere.10,12 Human HEK-293T cells (ATCC, Manassas, VA, USA) were cultured in DMEM medium with identical supplements and conditions.
Plasmid constructs, transfection and lentiviral production
The empty vector [pCCLsin.cPPT.hEF1a (intron with MCS) deltaLNGFR. Wpre], miR-223 (intronic pri-mir 223), and miR-142 (intronic murine pri-mir 142) expressing plasmids were kindly provided by Dr. Naldini. miR-34a, miR-17~92 and miR-17~92inv were cloned in pCCLsin vector. The pTRIPZ- miR-17~92 sponge construct was kindly provided by Dr Fu.28 Transfection of HEK-293T cells was performed with Effectene (Qiagen, Valencia, CA, USA). High titer lentiviral stocks were produced as previously described.5,7,12–14 Aliquots of virus were used to infect exponentially growing cells (1×105/mL) in the presence of 8 μg/mL of polybrene. The infectivity was determined (after 96 h) by real time quantitative polymerase chain reaction (RT-qPCR) analysis of miRNA.
MicroRNA expression profiling
TS-SUP-M2-A5, TS-SUP-M2-A5M and TS-SUP-M2 S3S were treated with doxycycline (1 μg/mL) to induce short hairpin (sh)RNA expression, and monitored for green fluorescent protein (GFP) expression by FACS analysis. STAT3 and ALK KD were monitored by western blotting. Total RNA was isolated at indicated time points from two independent replicates, using Trizol Reagent (Life Technologies, Inc., Rockville, MD, USA). RNA samples were hybridized on a human miRNA microarray (#G4470B, Agilent Technologies, Palo Alto, CA, USA), as described elsewhere.29 An Agilent scanner and Feature Extraction 10.5 software (Agilent Technologies) were used to obtain the raw microarray data. Microarray results were analyzed using GeneSpring GX 10 software (Agilent Technologies). Data transformation was applied to set all negative raw values at 1.0, followed by a quantile normalization. A filter on low gene expression was used to keep only the probes expressed in at least one sample. Differentially expressed genes were selected to have a 1.5-fold expression difference between late time points after KD (84/96 h for ALK and 120/140 h for STAT3) and controls (untreated cells) and a statistically significant P-value (<0.05) using an unpaired t-test with Benjamini-Hochberg correction. Differentially expressed genes were employed for cluster analysis of samples using the Manhattan correlation as a measure of similarity.
Tumor growth in immunocompromised mice
TS-SUP-M2 S3S cells (5×104) were injected subcutaneously in matrigel into the rear flanks of 24 NOD/SCID/IL2Rγ−/− (NSG) mice (Charles River Laboratories) divided into three groups: UTR, 17~92, and 17~92inv. Tumor growth was monitored over time by determining the diameter of tumor masses. Two weeks after injection of the cells, six mice in each group were treated with 0.1 mg/mL doxycycline in a 0.5% sucrose solution in light-proof bottles, refreshed every 4 days. After 24 days of doxycycline treatment, mice were sacrificed and tumors were excised. Animals were housed in the animal facility of the University of Turin and treated in accordance with guidelines approved by the local Ethical Animal Committee.
Results
STAT3-dependent microRNA signature in ALK-positive anaplastic large cell lymphoma cells
Using inducible KD approaches in ALK+ ALCL cell lines, we recently demonstrated that the ALK expression signature largely depends on the transcriptional activity of STAT3.10,12 To identify oncogenic miRNA involved in ALK+ ALCL, we performed genome-wide miRNA expression profiling in the ALK+ ALCL cell line TS-SUP-M2, following ALK (A5) or STAT3 (S3S) inducible KD. Experiments were carried out at different time points (72, 84, 96, 120, and 144 h), based on the kinetics of the doxycycline-mediated decline of ALK and STAT3 protein levels (Figure 1A,B). As controls, we used untreated cells and TS-SUP-M2 cells expressing a non-functional shRNA construct (A5M).12 A total of 24 samples from two independent replicates were hybridized to Agilent G4470B gene chips, which represent 723 human miRNA. Data analysis revealed detectable expression of 205 miRNA, of which 60 resulted significantly regulated by ALK KD and 27 by STAT3 KD (P<0.05), with 14 overlapping miRNA (Online Supplementary Figure S1A). Differential analysis identified 48 miRNA concordantly modulated in ALK and STAT3 KD samples, as compared to controls. Hierarchical clustering of samples according to the differentially expressed miRNA generated a dendrogram with three major branches sorting out controls, early, and late KD samples, independently of ALK or STAT3 silencing (Figure 1C). A tool for annotations of human miRNA30 recognized enrichment of specific miRNA families, such as miR-17, −106a, −181, −193b, −143, and −221 clusters (Figure 1D). It is noteworthy that miR-223, miR-100, and miR-34a were significantly up-regulated, while the miR-17-92 cluster was down-regulated by STAT3 KD (Figures 1C,D and 2A,B). The ALK/STAT3-dependent miRNA signature was further confirmed by RT-qPCR in TS-SUP-M2 (Figure 2C,D) and in JB-6 cells. Specifically, 24 out of 34 miRNA resulted accordingly modulated by STAT3 KD in JB-6 and SUP-M2 cells (Online Supplementary Table S1). To validate the ALCL miRNA signature obtained by gene silencing, and to exclude shRNA-induced off target effects, we took advantage of a highly potent and selective ALK inhibitor (CEP-28122).31 miRNA expression profiling of TS-SUP-M2 cells treated with control diluent or CEP-28122 (200 nM) for 12 h identified 109 mirRNA with a fold-change >1.5. Among these, a significant number of miRNA overlapped with those identified by the ALK (25 out of 60) or ALK/STAT3 (20 out of 48) signatures (Online Supplementary Figure S1B).
Figure 1.

(A–B) Kinetics of ALK and STAT3 knock-down (KD) in the ALK-positive ALCL cell line TS-SUP-M2. (A) TS-SUP-M2 cells co-transduced with pLV-tTR-KRAB/DsRed and pLVTH-ALK-A5/GFP (A5) or with the mutated pLVTH-ALKA5M/GFP (A5M) lentiviral preparations were cultured in the presence (+ Doxy) or absence (− Doxy) of doxycycline (1 μg/mL) and harvested at the indicated times. Whole-cell lysates were analyzed by western blotting with anti-α-tubulin or ALK antibodies. (B) Two clones (2X and 21) derived from TS-SUP-M2 cells co-transduced with pLV-tTR-KRAB/DsRed and pLVTH-STAT3-S3S/GFP (S3S) lentiviral preparations were cultured in the presence (+ Doxy) or absence (− Doxy) of doxycycline (1 μg/mL) for the indicated time intervals and analyzed by western blotting with anti-α-tubulin or STAT3 antibodies. (C) Heatmap representation of miRNA modulated by ALK or STAT3 inducible KD in TS-SUP-M2 cells. Light blue and yellow lanes designate ALK- and STAT3-silenced samples, respectively. RNA samples from two independent replicates were hybridized on a human miRNA microarray (#G4470B, Agilent Technologies). Differentially expressed miRNA were employed for sample cluster analysis using the Manhattan correlation as a measure of similarity. Expression levels are referred to control samples. Upregulated miRNA are shown in red, downregulated miRNA are shown in green. A5M, A5, and S3S indicate control, ALK, or STAT3 shRNA, respectively. (D) miRNA concordantly modulated by ALK or STAT3 KD. miRNA clusters are highlighted by the same color. FC: fold change after STAT3 KD (late time points vs. controls); P value: P value of an unpaired t-test with Benjamini-Hochberg correction; regulation: direction of miRNA expression modulation after STAT3 KD.
Figure 2.
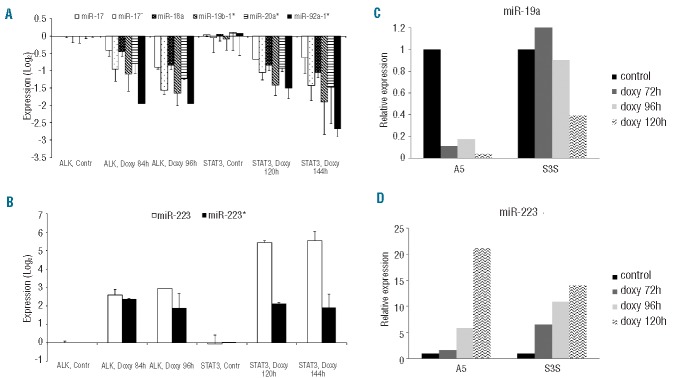
Expression levels of representative miRNA in TS-SUP-M2 cells. Quantification of the miR-17~92 cluster (A) and miR-223 (B), as revealed by genome-wide miRNA profiling following ALK or STAT3 inducible KD. Relative quantities are log2 normalized to control samples. The error bars report standard deviations from duplicates. (C–D) Validation of ALK/STAT3-mediated miR-19a and miR-223 regulation by RT-qPCR. Changes of mature miR-19a and miR-223 levels, normalized to RNU6B expression, were measured at the indicated time points after doxycycline treatment in an independent experiment. Relative quantities are log2 normalized to control samples.
Expression of the miR-17~92 cluster partially rescues STAT3 knock-down in anaplastic large cell lymphoma cells
To establish the role of deregulated miRNA in ALCL pathogenesis, we first sought to examine the effects of stable expression of miR-34a, miR-223, and the miR-17~92 cluster on in vitro cell growth. Empty vector and miR-142, which was not deregulated in our ALCL model system, were used as controls. Lentiviral-mediated expression of miR-34a, miR-223, and the miR-17~92 cluster had no significant effects on cell proliferation or survival of ALK+ ALCL cell lines (TS-SUP-M2 and JB-6), in standard cell culture conditions (Online Supplementary Figure S2). To study whether the indicated miRNA could modify the sensitivity of ALCL cells to stress conditions, we treated miRNA-transduced TS-SUP-M2 cells with increasing concentrations of doxorubicin, ALK inhibitors, or with a suboptimal supplement of fetal calf serum (Online Supplementary Figure S3). In the low serum concentration (2%), TS-SUP-M2 cells expressing miR-34a and miR-223 displayed a progressive increment of apoptotic cells, comparable to controls (data not shown). In contrast, miR-17~92 over-expression significantly decreased the basal level of cell death (Online Supplementary Figure S3C). Accordingly, miR-17~92 transduced cells displayed higher metabolic activity, as measured by ATP production (Online Supplementary Figure S3D). These results prompted us to focus on the miR-17~92 cluster, a known oncogenic miR,32 previously demonstrated to be highly expressed in systemic ALK+ ALCL.24 To confirm STAT3–dependent regulation of the miR-17~92, JB-6, L82, and Karpas-299 ALK+ ALCL cells were treated with increasing concentrations of the STAT3 inhibitor Stattic.33 RT-qPCR of miR-19a and miR-92 indicated that pharmacological inhibition of STAT3 dose-dependently decreased miR-17~92 cluster expression in all cell lines (Online Supplementary Figure S4).
To evaluate the functional role of the miR-17~92 cluster in ALK+ ALCL cells, we transduced TS-SUP-M2 S3S cells with lentiviral particles expressing miR-17~92 cluster (17~92), an antisense construct (17~92inv), or an empty vector (EV) as negative controls, and monitored the changes on proliferation and survival after inducible KD of STAT3 (Figure 3A). miR-17~92 over-expression was first confirmed by RT-qPCR measuring the mature forms of miR-19a and miR-92, at 96 h post-transduction (Figure 3B, and data not shown). Doxycycline treatment of TS-SUP-M2 S3S cells resulted in a significant and reproducible down-regulation of STAT3 protein expression at day 5 (Figure 3C). Kinetics of cell death induced by conditional STAT3 KD revealed that the rates of apoptosis were significantly lower in TS-SUP-M2 S3S cells expressing miR-17~92 cluster than in controls (Figure 3D). These data were further confirmed in a parallel experiment performed in JB-6 S3S cells (Online Supplementary Figure S5). Overall these findings indicate that miR-17~92 partially rescued the STAT3 KD phenotype.
Figure 3.

(A) Experimental design to evaluate the functional role of miR-17~92 cluster expression in the TS-SUP-M2 S3S cell line, after STAT3 KD by doxycycline treatment. (B) miR-19a expression levels in TS-SUP-M2 S3S cells transduced with lentiviral particles expressing the indicated miRNA, as detected by RT-qPCR 4 days after infection. (C) STAT3 and phosho-STAT3 expression in TS-SUP-M2 S3S cells 8 days after doxycycline treatment detected by western blot analysis with the indicated antibodies. (D) Apoptosis analysis in TS-SUP-M2 S3S cells expressing the indicated miRNA at different time points after induction of STAT3 KD by doxycycline. Analysis was performed by TMRM staining-flow cytometry. These findings are representative of three independent experiments. (E) Tumor growth curves of TS-SUP-M2 S3S cells injected into NSG mice in the absence (−) or presence (+) of doxycycline. TS-SUP-M2 S3S cells untransduced (UTR) or transduced with miR-17~92 cluster (17-92) were injected subcutaneously into eight NSG mice. Two weeks after cell injection, six mice of each group were treated with 0.1 mg/mL doxycycline to induce STAT3 KD (+), and two mice remained untreated (−). Tumor growth was monitored over time by determining the volume of tumor masses. Error bars indicate standard deviation. (F) miR-19a expression levels in mice tumors over-expressing the indicated miRNA after sacrifice of the animals, as determined by RT-qPCR. UTR: untransduced cells; EV: empty vector; 17~92: miR-17~92 cluster; 17~92 inv: miR-17~92 cluster antisense.
To corroborate the specificity of the effects of the miR-17~92 cluster, we analyzed the functional changes of reducing miR-17~92 by a doxycycline-inducible sponge construct that sequesters the corresponding miRNA.28 Expression of miR-17~92 sponge lentiviral construct increased cell death of TS-SUP-M2 S3S cells upon doxycycline administration, thus reverting the effects of the miR-17~92 cluster (Online Supplementary Figure S6).
To assess whether forced expression of miR-17~92 interferes with STAT3 KD in vivo, we studied the growth patterns of subcutaneously transplanted TS-SUP-M2 S3S cells, transduced with either the miR-17~92 cluster or the antisense construct as a control. TS-SUP-M2 S3S cells, injected subcutaneously into the flanks of NSG mice, formed visible tumors 2 weeks after injection. In control mice treated with doxycycline (UTR+), tumor masses gradually diminished compared with those in untreated mice (UTR−). In contrast, miR-17~92 over-expressing tumors underwent a minor growth regression upon STAT3 KD, and eventually resumed growth within 2 weeks of doxycycline treatment (Figure 3E). miR-17~92 over-expression was confirmed by quantification of miR-19a after tumor excision (Figure 3F). Overall, these findings support the notion that the miR-17~92 cluster could sustain the oncogenic properties of STAT3 in ALK+ ALCL.
Expression of the microRNA-17~92 cluster sustains proliferation and survival of STAT3-depleted anaplastic large cell lymphoma cells
We have previously demonstrated that conditional STAT3 KD in ALK+ ALCL cells is coupled to a G0/G1 cell-cycle arrest, followed by apoptosis.10 We, therefore, asked whether forced miR-17~92 expression could complement either one or both processes. Cell cycle analysis of STAT3-depleted TS-SUP-M2 S3S cells (5 days after doxycycline treatment) indicated that lentiviral transduction of the miR-17~92 cluster specifically increased the proportion of cells in S-phase and decreased those in G0/G1, as compared to controls cells transduced with empty vector, miR 34a, or 17~92inv (Figure 4A). In contrast, miR-17~92 transduction did not significantly affect the cell cycle or survival of highly-proliferating TS-SUP-M2 S3S cells expressing normal levels of STAT3 (Online Supplementary Figure S7). Consistent with these findings, western blotting analysis revealed that miR-17~92 cluster over-expression attenuated cyclin A, cyclin B1 and cyclin D3 down-regulation induced by STAT3 KD (Figure 4B). Moreover, determination of the DNA content confirmed that expression of the miR-17~92 cluster significantly protected STAT3-depleted ALCL cells from apoptosis, as shown by the reduced fraction of hypodiploid cells (~20%) compared to controls (~50%), 9 days after doxycycline treatment (Figure 4C). Lower levels of apoptosis were associated with reduced activation of known apoptotic effectors, such as caspase 3 and caspase 7, as well as decreased cleavage of the caspases substrate poly (ADP ribose) polymerase 1 (PARP1). Since the induction of apoptosis relies on a fine balance between pro- and anti-apoptotic proteins, we evaluated the effects of miR-17~92 on the level of several survival regulating proteins. Among these, cellular inhibitor-of-apoptosis protein 1 (IAP1) was considerably down-regulated by STAT3 KD. In contrast, IAP2 and XIAP, protein levels remained unchanged (Figure 4D, and data not shown). According to the above-described anti-apoptotic activity of the miR-17~92 cluster, its forced expression attenuated IAP1 down-regulation induced by STAT3 KD. However, IAP1 modulation could not be explained via a direct effect of miR-17~92. We, therefore, searched among validated miR-17~92 cluster targets,34 those potentially implicated in cell death. Western blotting analysis of p21, PTEN, pRB2, E2F1, and BIM proteins indicated that the forced expression of the miR-17~92 cluster induced the exclusive down-modulation of BIM, both in basal and STAT3 KD conditions (Figure 4D, and data not shown). These findings suggest that the miR-17~92 cluster might mediate resistance to STAT3 KD by targeting BIM.
Figure 4.

(A) Cell cycle analysis of TS-SUP-M2 S3S cells expressing the indicated miRNA 5 days after doxycycline treatment. Cells expressing miR-17~92 cluster displayed reduced G0/G1 arrest and increased S-phase as compared to control cells and cells transduced with the indicated lentiviral particles. Cell cycle was analyzed by propidium iodide staining-flow cytometry. These findings are representative of three independent experiments. (B) Western blot analysis of the experiment described above revealed that cells expressing the miR-17~92 cluster showed increased levels of cyclin A, B1 and D3 following STAT3 KD as compared to control cells. (C) TS-SUP-M2 S3S cells expressing the miR-17~92 cluster undergo reduced apoptosis as compared to control cells. Propidium iodide staining analysis was performed at day 9 after induction with doxycycline. The sub-G0/G1 fraction was used to quantify apoptotic cells. These findings are representative of three independent experiments. (D) Western blot analysis of the experiment descibed above (C) revealed that miR-17~92 over-expressing cells displayed lower levels of activated caspase 3 and caspase 7, processed PARP, BIM and higher levels of IAP1 protein. Asterisk (*) indicates unspecific band for IAP1.
Genes modulated by forced expression of the miR-17~92 cluster in anaplastic large cell lymphoma cells
The oncogenic nature of miR-17~92 is supported by the identification of several targets with key roles both in cell cycle control and cell death.35 In this scenario, we asked whether miR-17~92-mediated rescue of STAT3 KD in ALCL cells could occur through mechanisms additional to BIM regulation. To identify potential miR-17~92 targets in ALCL cells, we performed gene expression profiling experiments in the ALK+ ALCL cell line TS-SUP-M2, following inducible STAT3 KD. Samples from two independent replicates were processed and hybridized to HT-12 v4 bead chips (Illumina), which target more than 47,000 human transcripts. Experiments were carried out 96 h after doxycycline or mock treatment. As controls, we used TS-SUP-M2 cells not transduced (UTR) or transduced with the antisense construct (17~92inv). Using as selection criteria fold-change >2, P<0.001, detection >0.99, microarray data analysis identified 87 genes modulated by miR-17~92 in STAT3 KD conditions (Online Supplementary Table S2). Among these, the expression of several known miR-17~92 targets,36–38 such as hypoxia-inducible factor 1α (HIF1α), retinoblastoma-like 2 (RBL2/p130), and the transforming growth factor β receptor II (TGFβR2) genes, was modified as expected (Figure 5). Validations performed by RT-qPCR analyses in independent experiments indicated that TGFβR2 expression was significantly increased by STAT3 KD, and that the miR-17~92 cluster down-regulates TGFβR2 transcripts in both the presence and absence of STAT3 expression (Figure 5C and Online Supplementary Figure S8).
Figure 5.
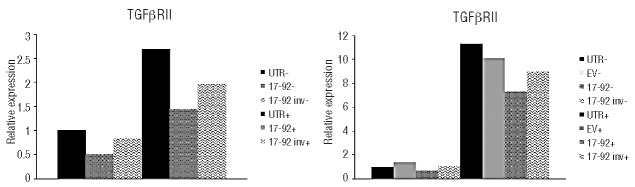
Expression of TGFβRII as detected by gene expression profiling at 96 h (left panel), and by RT-qPCR after 8 days of doxycycline treatment (right panel). TS-SUP-M2 S3S cells were transduced with the indicated lentivectors and cultured in the presence (+) or absence (−) of doxycycline (1 μg/mL). Experiments were performed in biological duplicates.
Discussion
It is known that target therapies will inevitably boost the development of acquired resistance. With no exception, the treatment of ALK+ ALCL with ALK inhibitor(s) also results in the selection of tumor cells unresponsive to the drug(s).17 The lesson learned from treatments with several tyrosine kinase inhibitors and, specifically, from ALK inhibitors in non-small cell lung carcinomas is that resistance could come from either secondary mutations in the ALK tyrosine kinase domain,39 ALK gene amplification, aberrant activation of alternative receptors by amplification,16,40 or by paracrine stimulation.41 To design foresighted therapeutic approaches for ALK+ ALCL patients, we believe that it may be critical to unravel ALK signaling in specific pathological contexts. Since ALK oncogenic properties rely on the constitutive activation of multiple pathways, it is reasonable to envisage combination therapies associating conventional drugs with compounds targeting multiple effectors of ALK signaling. Toward this end, members of the RAS/ERK, PI3K/mTOR, and JAK/STAT3 pathways are the most promising targets for innovative therapeutic strategies in ALK+ ALCL patients.6,42,43 Among these, the vital role of STAT3 demonstrated in ALK+ ALCL models is well established.9,10 The STAT3 KD phenotype of ALK+ ALCL cells is associated with reproducible modulation of a large number of genes (~1500), suggesting that STAT3 signaling leads to concomitant activation of multiple targets.10 Preliminary data from a functional screening of STAT3-regulated genes in ALK+ cell lines indicate that, with rare exceptions, modulation of single genes is not able to determine a remarkable phenotype (R.P. et al., manuscript in preparation). We, therefore, reasoned that an alternative strategy to interfere with multiple ALK key pathways was to target critical miRNA. Accordingly, we performed genome-wide miRNA profiling of ALK+ ALCL cell lines and identified a series of miRNA modulated by the activity of STAT3. Among these, we found that forced expression of the miR-17~92 cluster promotes survival of ALK+ ALCL cells and partially overcomes STAT3 addiction in cell lines as well as in a xenograft model. Specifically, we demonstrated that high expression of the miR-17~92 cluster sustains proliferation and inhibits apoptosis of STAT3-depleted ALCL cells and found that miR-17~92 over-expression is correlated with the down-regulation of known targets such as BIM and TGFβRII. The polycistronic miRNA cluster 17~92, originally designated as oncomiR-1, is one of best-characterized oncogenic miRNA.44 Human miR-17~92 is located at 13q31.3, a region amplified in several hematopoietic malignancies and solid tumors, including diffuse B-cell lymphoma, follicular lymphoma, Burkitt’s lymphoma, and lung carcinoma.45 The first functional evidence to support the oncogenic activity of miR-17~92 came from an in vivo mouse B-cell lymphoma model, in which enforced expression of miR-17-19b collaborated with the c-myc oncogene to accelerate B-cell lymphomagenesis.46 Since this initial observation, the effects of miR-17~92 overexpression have been examined in multiple animal models, human cancers, and cell culture systems for its ability to regulate a number of cellular processes that favor malignant transformation.
The significance of the miR-17~92 cluster in STAT3+ ALCL cell lines is confirmed by its high expression in systemic ALK+ ALCL tissue lesions.24,47 In view of the fact that the miR-17~92 cluster is modulated by interleukin-6/STAT3 signaling in human endothelial cells through a highly conserved STAT3-binding site in its promoter,48 we speculate that an ALK-STAT3-mir17~92 pathway would also play a critical role in ALK+ ALCL cells. This axis could be particularly relevant in light of the indications that STAT3 mediates the resistance of lung cancer cells to the MEK inhibitor AZD6244 through the up-regulation of the miR-17~92 cluster, and consequent block of BIM expression.49 Moreover, the miR-17~92 cluster has been shown to mediate chemoresistance and enhance tumor growth through PI3K/AKT pathway activation also in non-Hodgkin’s lymphomas, such as mantle cell lymphoma.28
We, therefore, suggest that the combination of a small molecule–based inhibitor of ALK with a STAT3 inhibitor or a miR-17~92 inhibitor may be useful to prevent drug resistance in patients with ALK+ ALCL. Moreover, since many human cancers rely on constitutive activation of STAT3, this strategy could be extended to other non-Hodgkin’s lymphomas and many other human tumors.50 Overall, these data suggest that the miR17~92 cluster could sustain the oncogenic properties of STAT3 in T-cell lymphoma, and that its inhibition might represent an alternative avenue to interfere with ALK signaling in ALCL.
Supplementary Material
Acknowledgments
Supported by: Associazione Italiana per la Ricerca sul Cancro (AIRC) grants IG-8675 and AIRC Special Program in Clinical Molecular Oncology 5 × 1000, N. 10007; Ministero dell’Università e Ricerca Scientifica. Regione Piemonte; Università di Torino, Rete Oncologica Piemonte e Valle d’Aosta, and Compagnia di San Paolo, Torino (Progetto Oncologia). ES is recipient of a research fellowship from the Fondazione Internazionale di Ricerca in Medicina Sperimentale, Torino, Italy. We thank Dr. Luigi Naldini for providing the lentiviral cassettes.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Fornari A, Piva R, Chiarle R, Novero D, Inghirami G. Anaplastic large cell lymphoma: one or more entities among T-cell lymphoma¿ Hematol Oncol. 2009;27(4):161–70 [DOI] [PubMed] [Google Scholar]
- 2.Vose J, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–30 [DOI] [PubMed] [Google Scholar]
- 3.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–4 [DOI] [PubMed] [Google Scholar]
- 4.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8(1):11–23 [DOI] [PubMed] [Google Scholar]
- 5.Amin HM, Medeiros LJ, Ma Y, Feretzaki M, Das P, Leventaki V, et al. Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene. 2003;22(35):5399–407 [DOI] [PubMed] [Google Scholar]
- 6.Chiarle R, Simmons WJ, Cai H, Dhall G, Zamo A, Raz R, et al. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11(6):623–9 [DOI] [PubMed] [Google Scholar]
- 7.Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002;21(7):1038–47 [DOI] [PubMed] [Google Scholar]
- 8.Zhang Q, Raghunath PN, Xue L, Majewski M, Carpentieri DF, Odum N, et al. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol. 2002;168(1):466–74 [DOI] [PubMed] [Google Scholar]
- 9.Amin HM, McDonnell TJ, Ma Y, Lin Q, Fujio Y, Kunisada K, et al. Selective inhibition of STAT3 induces apoptosis and G(1) cell cycle arrest in ALK-positive anaplastic large cell lymphoma. Oncogene. 2004;23(32):5426–34 [DOI] [PubMed] [Google Scholar]
- 10.Piva R, Agnelli L, Pellegrino E, Todoerti K, Grosso V, Tamagno I, et al. Gene expression profiling uncovers molecular classifiers for the recognition of anaplastic large-cell lymphoma within peripheral T-cell neoplasms. J Clin Oncol. 2010;28(9):1583–90 [DOI] [PubMed] [Google Scholar]
- 11.Tabbo F, Barreca A, Piva R, Inghirami G. ALK signaling and target therapy in anaplastic large cell lymphoma. Front Oncol. 2012;2:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piva R, Pellegrino E, Mattioli M, Agnelli L, Lombardi L, Boccalatte F, et al. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J Clin Invest. 2006;116(12):3171–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlette EJ, Medeiros LJ, Goy A, Lai R, Rassidakis GZ. Survivin expression predicts poorer prognosis in anaplastic large-cell lymphoma. J Clin Oncol. 2004;22(9):1682–8 [DOI] [PubMed] [Google Scholar]
- 14.Wasik MA, Zhang Q, Marzec M, Kasprzycka M, Wang HY, Liu X. Anaplastic lymphoma kinase (ALK)-induced malignancies: novel mechanisms of cell transformation and potential therapeutic approaches. Semin Oncol. 2009;36(2 Suppl 1):S27–35 [DOI] [PubMed] [Google Scholar]
- 15.Ambrogio C, Martinengo C, Voena C, Tondat F, Riera L, di Celle PF, et al. NPM-ALK oncogenic tyrosine kinase controls T-cell identity by transcriptional regulation and epigenetic silencing in lymphoma cells. Cancer Res. 2009;69(22):8611–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lovly CM, Pao W. Escaping ALK inhibition: mechanisms of and strategies to overcome resistance. Sci Transl Med. 2012;4(120):120ps2. [DOI] [PubMed] [Google Scholar]
- 17.Ceccon M, Mologni L, Bisson W, Scapozza L, Gambacorti-Passerini C. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol Cancer Res. 2013;11(2):122–32 [DOI] [PubMed] [Google Scholar]
- 18.Sen M, Thomas SM, Kim S, Yeh JI, Ferris RL, Johnson JT, et al. First-in-human trial of a STAT3 decoy oligonucleotide abrogates target gene expression in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;2(8):694–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mangolini M, de Boer J, Walf-Vorderwulbecke V, Pieters R, den Boer ML, Williams O. STAT3 mediates oncogenic addiction to TEL-AML1 in t(12;21) acute lymphoblastic leukemia. Blood 2013;122(4):542–9 [DOI] [PubMed] [Google Scholar]
- 20.Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012;482(7385):347–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2011;11(4):252–63 [DOI] [PubMed] [Google Scholar]
- 23.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10(10):704–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merkel O, Hamacher F, Laimer D, Sifft E, Trajanoski Z, Scheideler M, et al. Identification of differential and functionally active miRNAs in both anaplastic lymphoma kinase (ALK)+ and ALK- anaplastic large-cell lymphoma. Proc Natl Acad Sci USA. 2010;107(37):16228–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desjobert C, Renalier MH, Bergalet J, Dejean E, Joseph N, Kruczynski A, et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood. 2011;117(24):6627–37 [DOI] [PubMed] [Google Scholar]
- 26.Matsuyama H, Suzuki HI, Nishimori H, Noguchi M, Yao T, Komatsu N, et al. miR-135b mediates NPM-ALK-driven oncogenicity and renders IL-17-producing immunophenotype to anaplastic large cell lymphoma. Blood. 2011;118(26):6881–92 [DOI] [PubMed] [Google Scholar]
- 27.Dejean E, Renalier MH, Foisseau M, Agirre X, Joseph N, de Paiva GR, et al. Hypoxia-microRNA-16 downregulation induces VEGF expression in anaplastic lymphoma kinase (ALK)-positive anaplastic large-cell lymphomas. Leukemia. 2011;25(12):1882–90 [DOI] [PubMed] [Google Scholar]
- 28.Rao E, Jiang C, Ji M, Huang X, Iqbal J, Lenz G, et al. The miRNA-17 approximately 92 cluster mediates chemoresistance and enhances tumor growth in mantle cell lymphoma via PI3K/AKT pathway activation. Leukemia. 2012;26(5):1064–72 [DOI] [PubMed] [Google Scholar]
- 29.Ferracin M, Zagatti B, Rizzotto L, Cavazzini F, Veronese A, Ciccone M, et al. MicroRNAs involvement in fludarabine refractory chronic lymphocytic leukemia. Mol Cancer. 2010;9:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu M, Shi B, Wang J, Cao Q, Cui Q. TAM: a method for enrichment and depletion analysis of a microRNA category in a list of microRNAs. BMC Bioinformatics. 2010;11:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng M, Quail MR, Gingrich DE, Ott GR, Lu L, Wan W, et al. CEP-28122, a highly potent and selective orally active inhibitor of anaplastic lymphoma kinase with antitumor activity in experimental models of human cancers. Mol Cancer Ther. 2012;11(3):670–9 [DOI] [PubMed] [Google Scholar]
- 32.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132(5):875–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13(11):1235–42 [DOI] [PubMed] [Google Scholar]
- 34.Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, et al. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol. 2007;9(7):775–87 [DOI] [PubMed] [Google Scholar]
- 35.Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, Forloni M, et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One. 2008;3(5):e2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taguchi A, Yanagisawa K, Tanaka M, Cao K, Matsuyama Y, Goto H, et al. Identification of hypoxia-inducible factor-1 alpha as a novel target for miR-17-92 microRNA cluster. Cancer Res. 2008;68(14):5540–5 [DOI] [PubMed] [Google Scholar]
- 37.Wang Q, Li YC, Wang J, Kong J, Qi Y, Quigg RJ, et al. miR-17-92 cluster accelerates adipocyte differentiation by negatively regulating tumor-suppressor Rb2/p130. Proc Natl Acad Sci USA. 2008;105(8):2889–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103(7):2257–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–9 [DOI] [PubMed] [Google Scholar]
- 40.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4(120):120ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S, et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin Cancer Res. 2012;18(13):3592–602 [DOI] [PubMed] [Google Scholar]
- 42.Marzec M, Kasprzycka M, Liu X, El-Salem M, Halasa K, Raghunath PN, et al. Oncogenic tyrosine kinase NPM/ALK induces activation of the rapamycin-sensitive mTOR signaling pathway. Oncogene. 2007;26(38):5606–14 [DOI] [PubMed] [Google Scholar]
- 43.Vega F, Medeiros LJ, Leventaki V, Atwell C, Cho-Vega JH, Tian L, et al. Activation of mammalian target of rapamycin signaling pathway contributes to tumor cell survival in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Cancer Res. 2006;66(13):6589–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olive V, Li Q, He L. mir-17-92: a polycistronic oncomir with pleiotropic functions. Immunol Rev. 2013;253(1):158–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64(9):3087–95 [DOI] [PubMed] [Google Scholar]
- 46.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435(7043):828–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu C, Iqbal J, Teruya-Feldstein J, Shen Y, Dabrowska MJ, Dybkaer K, et al. MicroRNA expression profiling identifies molecular signatures associated with anaplastic large cell lymphoma. Blood. 2013;122(12):2083–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, et al. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res. 2009;104(10):1184–91 [DOI] [PubMed] [Google Scholar]
- 49.Dai B, Meng J, Peyton M, Girard L, Bornmann WG, Ji L, et al. STAT3 mediates resistance to MEK inhibitor through microRNA miR-17. Cancer Res. 2011;71(10):3658–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cabanillas F. Non-Hodgkin’s lymphoma: the old and the new. Clin Lymphoma Myeloma Leuk. 2011;11 (Suppl 1):S87–90 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.