

Abstract
A series of triazolylpurine analogues show interesting and unintuitive structure−activity relationships against the A2A adenosine receptor. As the 2-substituted aliphatic group is initially increased to methyl and isopropyl, there is a decrease in potency; however, extending the substituent to n-butyl and n-pentyl results in a significant gain in potency. This trend cannot be readily explained by ligand−receptor interactions, steric effects, or differences in ligand desolvation. Here, we show that a novel method for characterizing solvent thermodynamics in protein binding sites correctly predicts the trend in binding affinity for this series based on the differential water displacement patterns. In brief, small unfavorable substituents occupy a region in the A2A adenosine receptor binding site predicted to contain stable waters, while the longer favorable substituents extend to a region that contains several unstable waters. The predicted binding energies associated with displacing water within these hydration sites correlate well with the experimental activities.
Keywords: A2A adenosine receptor, antagonist, GPCR, WaterMap, binding energy, desolvation
G-protein coupled receptors (GPCRs) constitute the largest protein family in the human genome and represent one of the most important target classes in pharmaceutical research. Recent estimates suggest that over 30% of currently approved therapeutic agents1 and more than 25 of the top 100 selling drugs target members of this protein family.2 Historically, computational approaches to GPCR research have mainly relied on ligand-based techniques due to the lack of 3D structural information. In the past decade, there have been an increasing number of successful applications of structure-based drug design approaches3 that used homology models based on the rhodopsin GPCR structure.4 However, because of the intrinsic inaccuracy of homology models of GPCRs built with currently available methodologies5 and the fact that only a single template was available for model building, widespread application of structure-based drug design for GPCRs has remained elusive. Fortunately, in recent years, a number of novel GPCR structures6,7 have become available that have made structure-based approaches more tractable.
Among the GPCR family, the adenosine receptors are promising candidate targets for therapeutic intervention due to the cytoprotective functions of the endogenous ligand adenosine during instances of hypoxia, ischemia, and seizure activity.8 The adenosine receptor subfamily includes four different members, namely, the A1, A2A, A2B, and A3 subtypes, each with distinct pharmacology, tissue distribution, and second messenger coupling.
The A2A adenosine receptor subfamily is responsible for regulating myocardial oxygen consumption and coronary blood flow by modulating vasodilation of the coronary arteries. The A2A receptor is also expressed in the brain, where it regulates glutamate and dopamine release, making it an attractive target for the treatment of pain, depression, and Parkinson's disease. Several high affinity and in some cases selective A2A ligands have been discovered.9 2-substituted adenosine derivatives have been shown to be selective agonists,10 while a number of nitrogen polyheterocyclic compounds and styrylxanthines derivatives11 as well as the triazolotriazine ZM24138512 are moderately selective high-affinity A2A receptor antagonists. The selective agonist BIIB014 (Biogen/Vernalis) has progressed to phase II clinical trials for the treatment of Parkinson's disease.13
The recent publication of the A2A receptor structure cocrystallized with the antagonist, ZM241385, represents a major breakthrough in the understanding of the A2A receptor's structure and function.7 The structure reveals key interactions between the heterocylic rings of the ligand and several residues, including Phe168, Glu169 (located in extracellular loop 2, EL2), and Asn253 (6.55, numbering according to the Ballesteros and Weinstein scheme14). Despite the availability of a crystal structure, many aspects of ligand structure−activity relationships (SAR) cannot be accounted for by only considering steric and electrostatic factors, and additional computational analysis is required for a comprehensive explanation of the available SAR.
In this work, we present the results of molecular dynamics simulations coupled with a statistical thermodynamic analysis of water molecules to explain the SAR within a series of substituted triazolylpurine A2A adenosine ligands.15 The method, called WaterMap, computes the free energy of water occupying hydration sites of a protein active site and is well-suited to the analysis of congeneric molecules where small structural modifications can result in substantial changes in activity.16 The method has been successfully applied in other works, such as peptides binding to PDZ domains,17 kinase selectivity,18 and ranking of congeneric molecules binding to factor Xa and CDK2.19 Here, we show that the method is able to describe the activity profile of the triazolylpurine A2A adenosine ligands, which is otherwise difficult to explain using traditional techniques. WaterMap was run on the A2A adenosine receptor crystal structure (PDB ID: 3EML) as described in the Experimental Procedures.
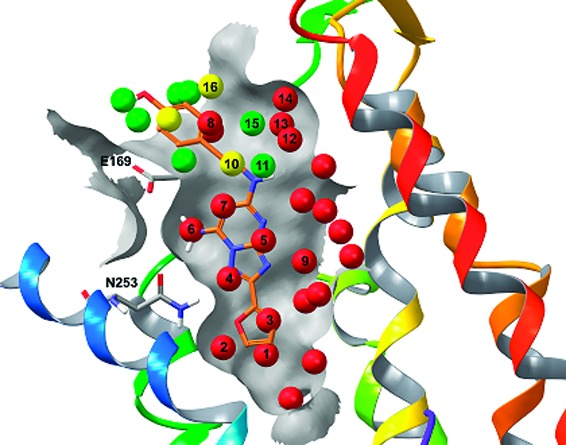
The hydration sites in the proximity of the crystal structure antagonist ZMA241385 can be seen in Figure 1. Stable hydration sites (ΔG < 0.0 kcal/mol) are shown in green, unstable hydration sites (ΔG > 1.0 kcal/mol) are shown in red, and those that are moderately unstable (0.0 < ΔG < 1.0 kcal/mol) are shown in yellow. The core of the ZMA241385 ligand overlaps with seven hydration sites, all of which are unfavorable with free energies ranging from 1.4 to 7.0 kcal/mol. There are two particularly unstable hydration sites that correspond to locations where hydrogen bonds are made between the ligand and the receptor. One of these high-energy sites (hydration site 6), with a ΔG of 4.5 kcal/mol, overlaps with the amine group of the ligand which the amine group makes one hydrogen bond to the side chain of Glu169 (EL2) and another to the side chain of Asn253 (6.55). The other high-energy site (hydration site 4) is the most unstable in the binding site with a ΔG of 7.0 kcal/mol and overlaps with a nitrogen atom in the triazolotriazine core, which also makes a hydrogen bond with the side chain of Asn253. The phenethyl tail extends beyond a shell of stable water molecules and overlaps with a high-energy water molecule (hydration site 8) with a ΔG of 3.3 kcal/mol.
Figure 1.
WaterMap results for the A2A adenosine receptor. TM7 has been omitted for clarity. The cocrystallized ligand, ZMA241385, is shown in orange. Residues Glu169 (EL2) and Asn253 (6.55) are shown explicitly. Stable hydration sites (ΔG < 0 kcal/mol) are shown in green, significantly unstable hydration sites (ΔG > 1 kcal/mol) are shown in red, and moderately unstable sites (0.0 < ΔG < 1.0 kcal/mol) are shown in yellow. Key hydration sites important for the ligand SAR are labeled 1−16.
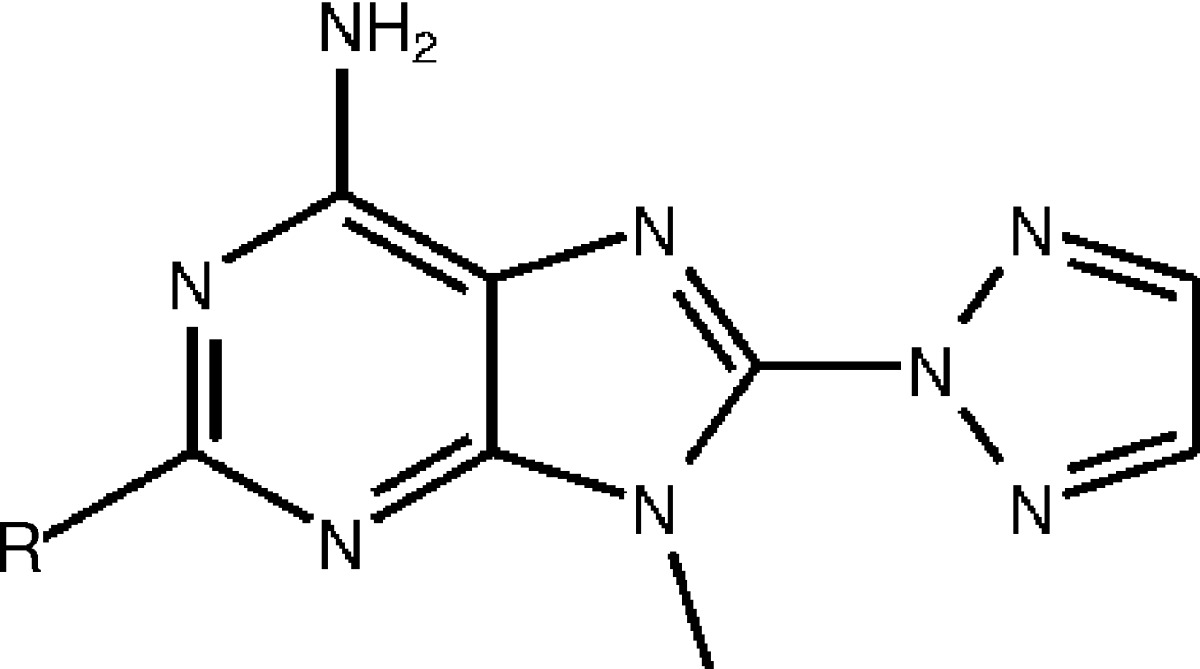
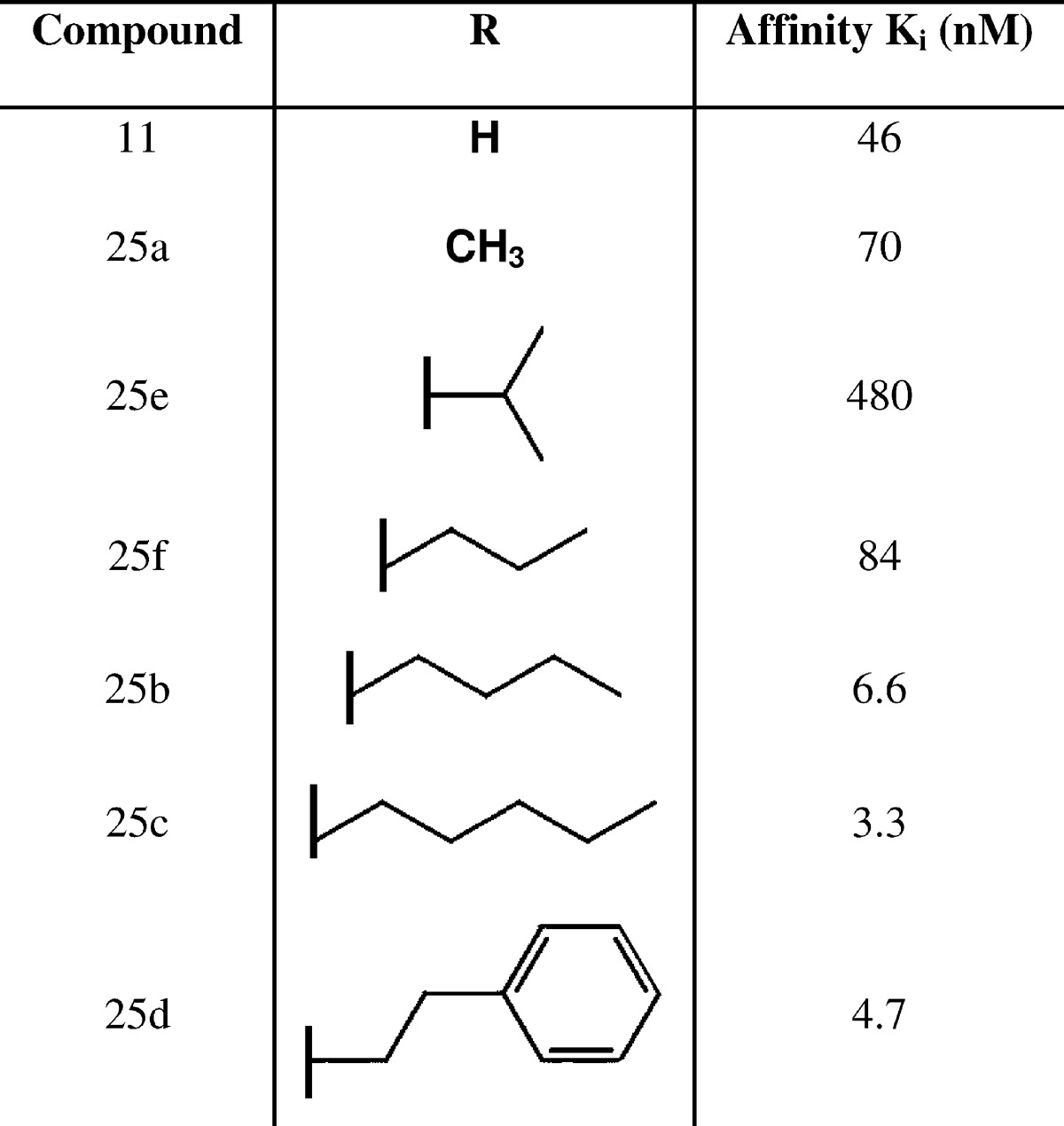
Next, we investigated whether the A2A WaterMap results can explain the SAR within the congeneric series from Minetti et al.15 shown in Table 1. The unsubstituted compound (11, numbering according to Minetti et al.) has an activity of 46 nM. Interestingly, the addition of a methyl group (25a) results in diminished activity to 70 nM. Extending further, the isopropyl derivative (25e) is the least potent compound in the series with an activity of 480 nM. As the alkyl chain is extended to propyl (25f), butyl (25b), and pentyl (25c), the activity is improved to 84, 6.6, and 3.3 nM, respectively. The reduced affinity of the smaller methyl and isopropyl derivatives cannot be explained by steric effects, since the larger compounds are highly active and therefore must fit in the binding site. Below, we explain the pose of each compound in Table 1 and show that the displacement energies from the WaterMap hydration sites accurately explain the observed experimental activity in the series. The binding free energies are computed relative to compound 11 (i.e. only the variable part of the molecule is different and therefore only the WaterMap energy associated with the substituents is shown).
Table 1. Binding Affinity of A2A Adenosine Receptor Antagonists Identified by Minetti et al.,15 Ordered by Increasing Substituent Size.
 |
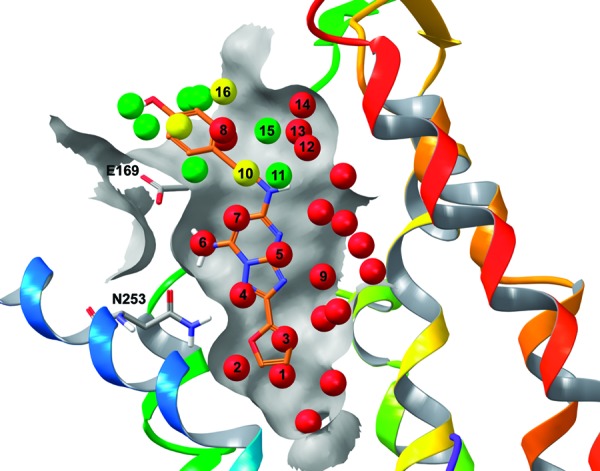
The methyl derivative (25a) is predicted to be slightly less potent than the lead compound as a result of partially displacing water within hydration site 11. This hydration site is favorable by −0.9 kcal/mol in the binding site relative to bulk water; therefore, the ligand is penalized for displacing water within this hydration site into bulk. The methyl group also partially overlaps with hydration site 10, which is roughly neutral energetically (+0.2 kcal/mol). The total change in binding energy computed by WaterMap is +0.2 kcal/mol relative to the lead compound based on the amount of overlap of sites 10 and 11 (Figure 2A). The isopropyl variant, 25e, overlaps more with the stable hydration site 11 (Figure 2B) and therefore has a worse computed binding energy (ΔΔG of +0.5 kcal/mol). This is the worst predicted binder in the series, consistent with the experimental data.
Figure 2.
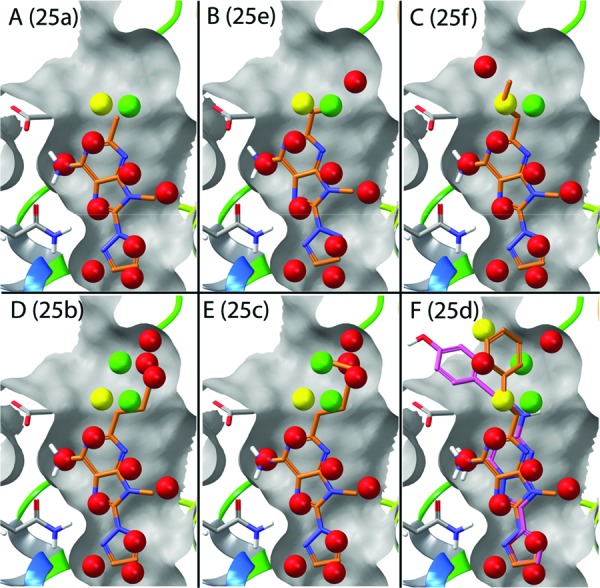
The predicted binding pose and overlapping hydration sites for the triazolylpurine analogues in Table 1 taken from Minetti et al.15 (A) The methyl derivative 25a, (B) isopropyl derivative 25e, (C) n-propyl derivative 25f, (D) n-butyl derivative 25b, (E) n-pentyl derivative 25c, and (F) phenethyl derivative 25d, shown in orange, along with the crystal structure pose of ZMA241385 shown in pink.
The n-propyl derivative, 25f, partially overlaps with hydration sites 10 and 11, as does the methyl variant, before extending toward hydration site 8 (ΔG of +3.3 kcal/mol). The partial overlap with hydration site 8 compensates for the unfavorable overlap with hydration site 11 and results in a total computed ΔΔG of −0.7 kcal/mol (Figure 2C). As described below, the overprediction of compound 25f affinity can be improved by adding a simple entropy correction based on the number of rotatable bonds. As the alkyl chain is extended in the n-butyl variant (25b), three additional unstable hydration sites show either complete or partial overlap (site 12, ΔG = 2.5 kcal/mol; site 13, ΔG = 2.0 kcal/mol; and site 14, ΔG = 2.2 kcal/mol). Additionally, there is overlap with one roughly neutral site (site 15, ΔG = −0.1 kcal/mol), which does not significantly impact the computed binding energy (Figure 2D). The computed WaterMap ΔΔG for the n-butyl variant is −3.5 kcal/mol, placing it as the third most potent compound in the series, in agreement with the experimental trend. The n-pentyl variant (25c) overlaps with the same hydration sites as the n-butyl derivative (Figure 2E), although the overlap is more complete and results in a more favorable computed WaterMap energy of −5.0 kcal/mol. This is the most favorable binder in the series as computed by WaterMap, consistent with the experimental results.
Finally, the phenethyl substituent (25d) is predicted to occupy a different part of the binding site from the rest of the ligand substitutions. Figure 2F shows that the docked pose prediction for 25d is similar to the cocrystallized ligand ZMA241385. The computed WaterMap energy for this pose is −3.8 kcal/mol, which is in agreement with the experimental trend, placing it as the second most potent compound in the series. Rather than overlapping with the unstable hydration sites 12 and 13, the phenethyl variant is predicted to completely displace water within hydration site 8 (ΔG = 3.3 kcal/mol) and partially displace water within hydration sites 14 (ΔG = 2.2 kcal/mol) and 16 (ΔG = 0.5 kcal/mol), resulting in a WaterMap ΔΔG of −3.8 kcal/mol.
Overall, compounds 25b, 25c, and 25d are predicted to be significantly more favorable than the other compounds, while the isopropyl derivative (25e) is computed to be the worst compound in the series. The other two compounds (methyl derivative 25a and n-propyl derivative 25f) are predicted to be slightly worse and slightly better than the lead compound. This general classification of the affinities relative to the lead compound is in close agreement with the experimental results.
A quantitative prediction of the hydration site locations and energetic properties as presented above would be difficult without using a rigorous statistical thermodynamic method, such as WaterMap. However, visual inspection of the structure reveals some interesting characteristics associated with the hydration sites. First, the stable hydration sites 10 and 11 are located at the edge of a hydrophobic pocket near the side chains of Phe168 (located in EL2), Met270 (7.35), and Ile274 (7.39). However, they are not interacting directly with these side chains. The enthalpic component of site 10 is −0.6 kcal/mol and that of site 11 is −2.7 kcal/mol, meaning that the water molecules occupying these hydration sites make more favorable interactions in the binding site than in bulk solvent. These two sites make a large average number of hydrogen bonds with other water molecules (3.7 and 3.8, respectively), resulting in an enthalpically stable environment with minimal loss of entropy (0.8 and 1.8 kcal/mol, respectively).
On the other hand, the hydration sites overlapped by the longer alkyl chains are in a hydrophilic region where they are restricted in movement and do not interact as much with other water molecules, leading to large entropy losses that are not compensated by favorable enthalpic interactions. For example, hydration site 12 has an enthalpy of −0.7 kcal/mol and an entropy (-TdS) of 3.2 kcal/mol. Water molecules in this hydration site can only hydrogen bond with the side chain of Tyr271 (7.36) of the protein, thus locking them into place and resulting in an unfavorable entropy that is not compensated by the average of 2.4 hydrogen bonds with other water molecules. Hydration sites 13 and 14 follow a slightly different pattern. Although both of these waters are located in a polar region and could potentially form hydrogen bonds with the backbone of Cys166 (EL2), Phe168 (EL2), and Ile66 (2.65), the geometry is not ideal. As a result, both the enthalpies (0.8 and 0.4 kcal/mol) and the entropies (1.2 and 1.8 kcal/mol) for these two hydration sites are unfavorable.
We also calculated the binding free energy for these compounds using molecular mechanics generalized Born surface area (MM-GBSA) (as implemented in Prime20) to see if a free energy method based on an implicit solvation model would be able to rationalize the SAR data. As shown in Table 2, the calculated MM-GBSA binding energies for all compounds are more favorable than 11. While 25c (the n-pentyl variant) is correctly predicted to be the most potent, the least potent compounds in the series (isopropyl variant, 25e) is predicted to be more potent than the unsubstituted lead compound (11) and the n-butyl variant (25b). Overall, MM-GBSA has little predictive ability for this series, which is consistent with the hypothesis that explicit water molecules play a key role in the difference in binding of these compounds. The correlation with experimental activity for WaterMap calculations (Figure 3A) is significantly better than that of MM-GBSA (Figure 3B), further highlighting that the activities of the compounds within this series are difficult to rationalize with traditional scoring methods.
Table 2. Predicted Binding Free Energies Using WaterMap and MM-GBSA for the Series of Triazolylpurine A2A Adenosine Antagonistsa.
| Compounds | Substituent | Relative affinity (kcal/mol) | WaterMap ΔΔG (kcal/mol) | WaterMap ΔΔG (kcal/mol)b | MM-GBSA ΔΔG (kcal/mol)b |
|---|---|---|---|---|---|
| 11 | hydrogen | 0.0 | 0.0 | 0.0 | 0.0 |
| 25a | methyl | 0.3 | 0.2 | 0.2 | −2.5 |
| 25e | isopropyl | 1.4 | 0.5 | 1.0 | −1.3 |
| 25f | N-propyl | 0.4 | −0.7 | 0.3 | −3.5 |
| 25b | N-butyl | −1.1 | −3.5 | −2.0 | −1.1 |
| 25c | N-pentyl | −1.5 | −5.0 | −3.0 | −3.7 |
| 25d | phenethyl | −1.3 | −3.8 | −2.3 | −2.0 |
All energies given are relative to compound 11.
Includes a 0.5 kcal/mol per rotatable bond entropy correction penalty.
Figure 3.

Correlation between the experimental activities and the calculated free energies of binding from either WaterMap (A) or MM-GBSA (B).
While the overall correlation between predicted and experimental affinity is very good (R2 = 0.86), the trend in going from 3 to 5 carbons is too steep, and the n-propyl compound is predicted to be more potent than the lead compound. One potential reason for this is the neglect of ligand conformational entropy in the WaterMap scoring. Applying a correction of 0.5 kcal/mol per rotatable bond, which is a simple estimate based on empirical and experimental work (see the Experimental Procedures), results in an improved correlation of R2 = 0.94. Perhaps more importantly, the predicted ordering of the compounds is in perfect agreement with the experimental activity trend. Given the results presented above, it can be inferred that novel compounds with increased rigidity of the substituent that overlaps with the same unstable hydration sites as compounds 25b and 25c could lead to further gains in free energy by displacing high-energy waters while not incurring the ligand conformational entropy loss.
Experimental Procedures
All calculations were performed on the crystal structure of human A2A adenosine receptor, 3EML,7 obtained from the Protein Data Bank.21 The structure was prepared with the Protein Preparation Wizard in Maestro22 using default options, described briefly here. First, hydrogens were added, bond orders were assigned to the ligand, and crystallographic water molecules were removed. The hydrogen-bonding network of the receptor was then optimized by reorienting hydroxyl and thiol groups, amide groups of Asn and Gln, and selecting appropriate states and orientations of the His imidazole group.
Ligands were obtained from Minetti et al.,15 and 3D structures were generated with LigPrep.23 For each compound, only the neutral state of the scaffold was retained. Docking calculations were performed with Glide.24 Energy grids were generated using the prepared structure described above. Each of the ligands was docked using the Extra Precision (XP) mode of Glide25 followed by postdocking minimization. WaterMap was run in the default mode using the crystal structure ligand to define the binding site but with the ligand removed in the simulations.
For each ligand, the top scoring pose based on Emodel26 was selected and scored with WaterMap using the default scoring function, which computes the binding free energy of a ligand as the sum of the free energies associated with the displacement of water from the hydration sites by the ligand upon binding, as described in previous works.16,17 This approximation of free energy is based exclusively on the displacement of water molecules within a hydration site upon binding of the ligand and ignores other terms such as protein−ligand van der Waals contacts, electrostatic interactions, internal strain (ligand and protein), and entropy changes. However, as discussed above, for a congeneric series of molecules with aliphatic substitutions, it can be expected that many of the energetic terms not computed by WaterMap will roughly cancel within a series for modifications that fit in the protein binding site.
MM-GBSA calculations using Prime20 were run to see if a simpler solvation model could account for the binding energetics within the congeneric series. These calculations involve minimization of the ligand within the receptor complex as well as free in solution, all using generalized Born implicit solvent.27 The receptor was kept fixed for all MM-GBSA calculations.
To approximate the loss of ligand conformational entropy upon binding, we added 0.5 kcal/mol per rotatable bond. This value was chosen to be consistent with empirically derived literature values (0.30−0.54 kcal/mol)28 and measured entropy changes per rotor for freezing of liquid alkanes (0.38−0.86 kcal/mol).29 Other components of the binding thermodynamics could in principle be added to the WaterMap score to more completely describe the total binding free energy; however, this is beyond the scope of this work.
Acknowledgments
We thank Robert Abel, Ramy Farid, and Jeremy Greenwood for reviewing the manuscript and providing helpful comments.
Supporting Information Available
Predicted relative binding free energy (ΔΔG), entropy (−TΔΔS), and enthalpy (ΔΔH) for WaterMap calculations on the series of triazolylpurine A2A adenosine receptor antagonists is available in the supporting information along with additional details about the docking and WaterMap scoring calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Hopkins A. L.; Groom C. R. The druggable genome. Nat. Rev. Drug Discovery 2002, 1, 727–730. [DOI] [PubMed] [Google Scholar]
- Klabunde T.; Hessler G. Drug design strategies for targeting G-protein-coupled receptors. ChemBioChem 2002, 3, 928–944. [DOI] [PubMed] [Google Scholar]
- Bissantz C.; Bernard P.; Hibert M.; Rognan D. Protein-based virtual screening of chemical databases. II. Are homology models of G-Protein Coupled Receptors suitable targets?. Proteins 2003, 50, 5–25. [DOI] [PubMed] [Google Scholar]
- Palczewski K.; Kumasaka T.; Hori T.; Behnke C. A.; Motoshima H.; Fox B. A.; Le Trong I.; Teller D. C.; Okada T.; Stenkamp R. E.; Yamamoto M.; Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [DOI] [PubMed] [Google Scholar]
- Mobarec J. C.; Sanchez R.; Filizola M. Modern homology modeling of G-protein coupled receptors: Which structural template to use?. J. Med. Chem. 2009, 52, 5207–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V.; Rosenbaum D. M.; Hanson M. A.; Rasmussen S. G.; Thian F. S.; Kobilka T. S.; Choi H. J.; Kuhn P.; Weis W. I.; Kobilka B. K.; Stevens R. C. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola V. P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y.; Lane J. R.; Ijzerman A. P.; Stevens R. C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Gao Z. G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discovery 2006, 5, 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristalli G.; Muller C. E.; Volpini R. Recent developments in adenosine A2A receptor ligands. Handb. Exp. Pharmacol. 2009, 59–98. [DOI] [PubMed] [Google Scholar]
- Jacobson K. A. Introduction to adenosine receptors as therapeutic targets. Handb. Exp. Pharmacol. 2009, 193, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraldi P. G.; Tabrizi M. A.; Gessi S.; Borea P. A. Adenosine receptor antagonists: Translating medicinal chemistry and pharmacology into clinical utility. Chem. Rev. 2008, 108, 238–263. [DOI] [PubMed] [Google Scholar]
- Poucher S. M.; Keddie J. R.; Singh P.; Stoggall S. M.; Caulkett P. W.; Jones G.; Coll M. G. The in vitro pharmacology of ZM 241385, a potent, non-xanthine A2a selective adenosine receptor antagonist. Br. J. Pharmacol. 1995, 115, 1096–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie R. J.; Bamford S. J.; Botting R.; Comer M.; Denny S.; Gaur S.; Griffin M.; Jordan A. M.; Knight A. R.; Lerpiniere J.; Leonardi S.; Lightowler S.; McAteer S.; Merrett A.; Misra A.; Padfield A.; Reece M.; Saadi M.; Selwood D. L.; Stratton G. C.; Surry D.; Todd R.; Tong X.; Ruston V.; Upton R.; Weiss S. M. Antagonists of the human A(2A) adenosine receptor. 4. Design, synthesis, and preclinical evaluation of 7-aryltriazolo[4,5-d]pyrimidines. J. Med. Chem. 2009, 52, 33–47. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Minetti P.; Tinti M. O.; Carminati P.; Castorina M.; Di Cesare M. A.; Di Serio S.; Gallo G.; Ghirardi O.; Giorgi F.; Giorgi L.; Piersanti G.; Bartoccini F.; Tarzia G. 2-n-butyl-9-methyl-8-[1,2,3]triazol-2-yl-9H-purin-6-ylamine and analogues as A2A adenosine receptor antagonists. Design, synthesis, and pharmacological characterization. J. Med. Chem. 2005, 48, 6887–6896. [DOI] [PubMed] [Google Scholar]
- Abel R.; Young T.; Farid R.; Berne B. J.; Friesner R. A. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuming T.; Farid R.; Sherman W. High-energy water sites determine peptide binding affinity and specificity of PDZ domains. Protein Sci. 2009, 18, 1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D. D.; Sherman W.; Farid R. Understanding kinase selectivity through energetic analysis of binding site waters. ChemMedChem 2010, 5, 618–627. [DOI] [PubMed] [Google Scholar]
- Guimaraes C. R.; Mathiowetz A. M.. Addressing limitations with the MM-GB/SA scoring procedure using the WaterMap method and free energy perturbation calculations. J. Chem. Inf. Model. 2010, DOI: 10.1021/ci900497d. [DOI] [PubMed] [Google Scholar]
- Prime v2.1; Schrödinger, Inc.: Portland, OR. [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestro v9.0; Schrödinger, Inc.: Portland, OR. [Google Scholar]
- LigPrep v2.3; Schrödinger, Inc.: Portland, OR. [Google Scholar]
- Glide v5.5; Schrödinger, Inc.: Portland, OR. [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- a Jacobson M. P.; Kaminski G. A.; Friesner R. A.; Rapp C. S. Force Field Validation Using Protein Side Chain Prediction. J. Phys. Chem. B 2002, 106, 11673–11680. [Google Scholar]; b Yu Z.; Jacobson M. P.; Friesner R. A. What role do surfaces play in GB models? A new-generation of surface-generalized born model based on a novel gaussian surface for biomolecules. J. Comput. Chem. 2006, 27, 72–89. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yu Z.; Jacobson M. P.; Josovitz J.; Rapp C. S.; Friesner R. A. First-Shell Solvation of Ion Pairs: Correction of Systematic Errors in Implicit Solvent Models. J. Phys. Chem. B 2004, 108, 6643–6654. [Google Scholar]
- Lazaridis T.; Masunov A.; Gandolfo F. Contributions to the binding free energy of ligands to avidin and streptavidin. Proteins 2002, 47, 194–208. [DOI] [PubMed] [Google Scholar]
- Searle M. S.; Williams D. H. The cost of conformational order: Entropy changes in molecular associations. J. Am. Chem. Soc. 1992, 114, 10690–10697. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.