

Abstract

The discovery of novel and selective inhibitors of human 5-lipoxygenase (5-LO) is described. These compounds are potent, orally bioavailable, and active at inhibiting leukotriene biosynthesis in vivo in a dog PK/PD model. A major focus of the optimization process was to reduce affinity for the human ether-a-go-go gene potassium channel while preserving inhibitory potency on 5-LO. These efforts led to the identification of inhibitor (S)-16 (MK-0633, setileuton), a compound selected for clinical development for the treatment of respiratory diseases.
Keywords: Human 5-lipoxygenase, leukotriene biosynthesis, MK-0633, setileuton, respiratory diseases
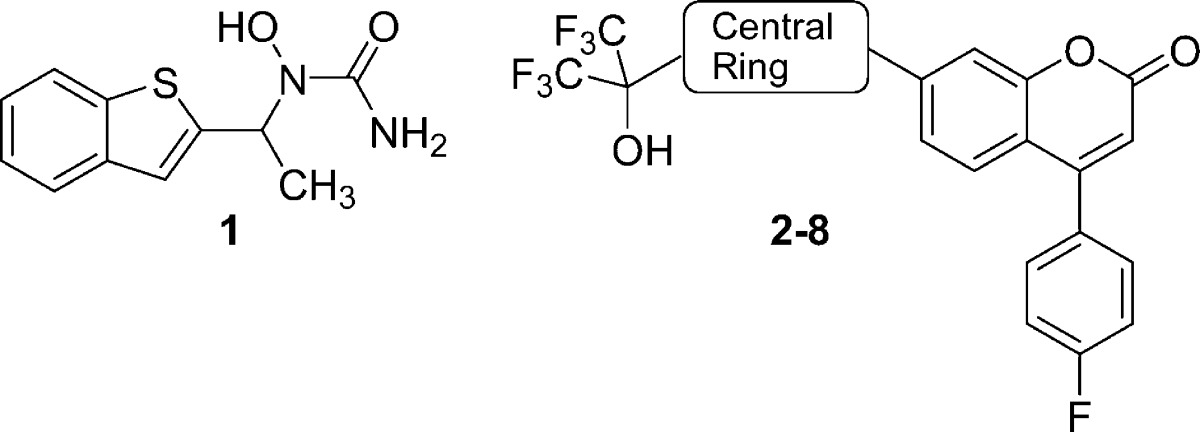
Leukotrienes (LTs) are potent lipid inflammatory mediators derived from arachidonic acid metabolism and released from cells involved in the inflammatory process.1 The synthesis of all LTs requires the action of the enzyme 5-lipoxygenase2 (5-LO) and its accessory protein, 5-LO-activating protein3 (FLAP). Inhibition of 5-LO reduces the production of both LTB4 and cysteinyl LTs (CysLTs) LTC4, LTD4, and LTE4. CysLTs increase vascular permeability, contract smooth muscle cells, and are involved in increased mucus production. LTB4 is a potent chemoattractant for neutrophils, macrophages, and other inflammatory cells and induces chemokinesis and adhesion of these cells to the vascular endothelium. Therefore, 5-LO inhibitors have potential therapeutic utility for the treatment of inflammatory disorders.4 Asthma, an inflammatory disorder of the lower airway, is characterized by variable and reversible bronchoconstriction, airway edema, mucus secretion, and airway remodeling, which leads to recurrent episodes of wheezing, breathlessness, chest tightness, and coughing.5 The clinical importance of the LTs in asthma has been well-demonstrated both by drugs that inhibit the biosynthesis of these mediators such as zileuton6 (1, Table 1; the only marketed 5-LO inhibitor) and MK-05917 (a FLAP inhibitor) and by potent CysLT1 receptor antagonists such as montelukast.8 Theoretically, 5-LO inhibition could provide added benefit over CysLT1 receptor antagonists by blocking LTB4-mediated inflammation in addition to reducing CysLTs production. LTs are also thought to play an important role in the pathogenesis of chronic obstructive pulmonary disease (COPD). COPD is characterized by an inflammatory process in the airways dominated by neutrophils and effector T-cells. LTB4 may be important in contributing to the inflammatory process via both cell types and has been detected in both the sputum and the lungs of patients with COPD. Currently available chronic medications for COPD have limited effectiveness. Thus, COPD could be another indication for inhibitors of 5-LO.9 Accumulating evidence suggests that 5-LO inhibitors could also be useful for the treatment of atherosclerosis,10 pain,11 and cancer.12 Herein, we describe the design of a novel series of coumarin analogues demonstrating high potency at inhibiting 5-LO, leading to the identification of (S)-16 (MK-0633, setileuton) as a development candidate.
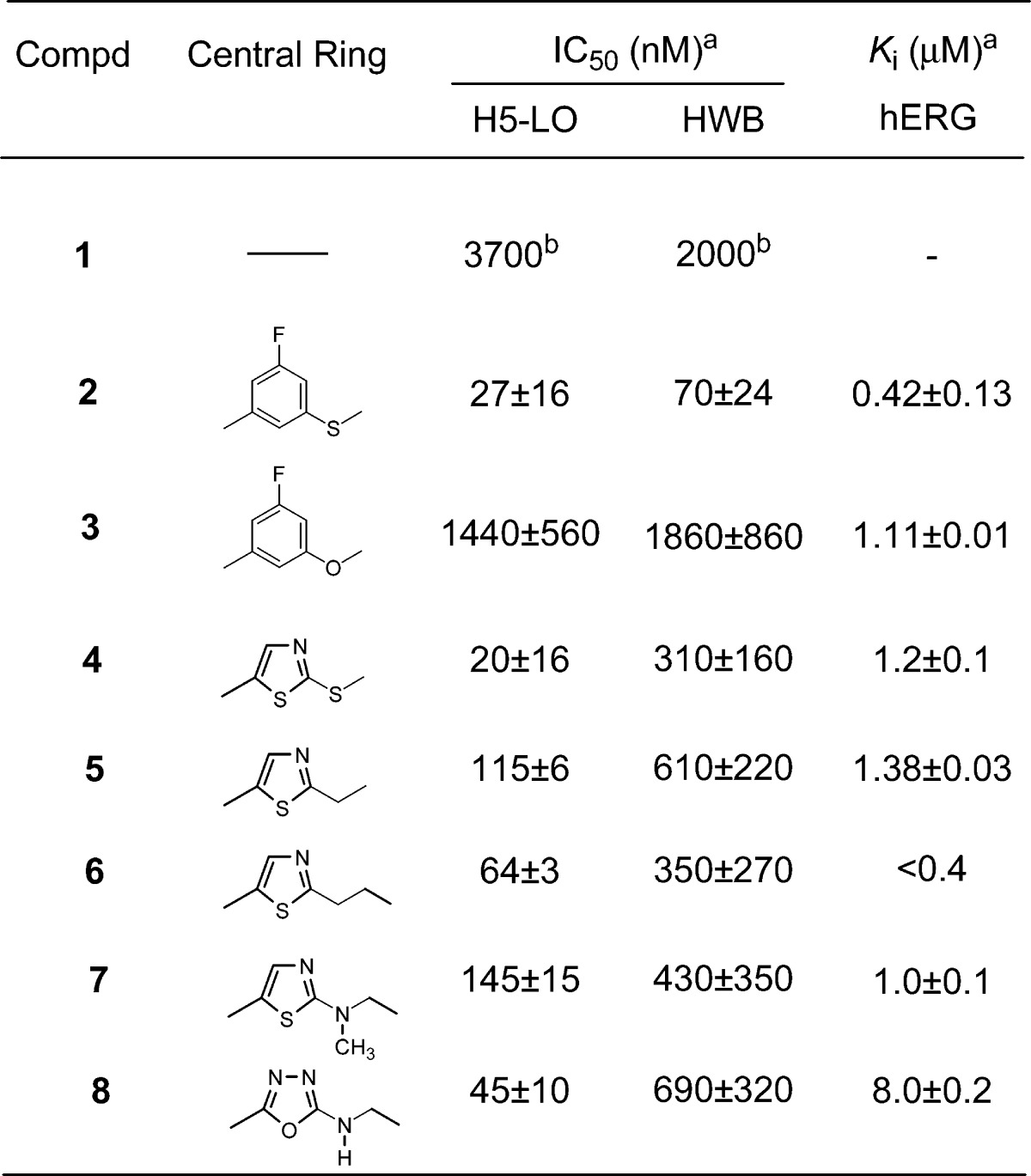
Table 1. SAR at the Central Ring Position.
 |
Each IC50 value corresponds to an average of at least two independent determinations (±SD).
Ref (19).
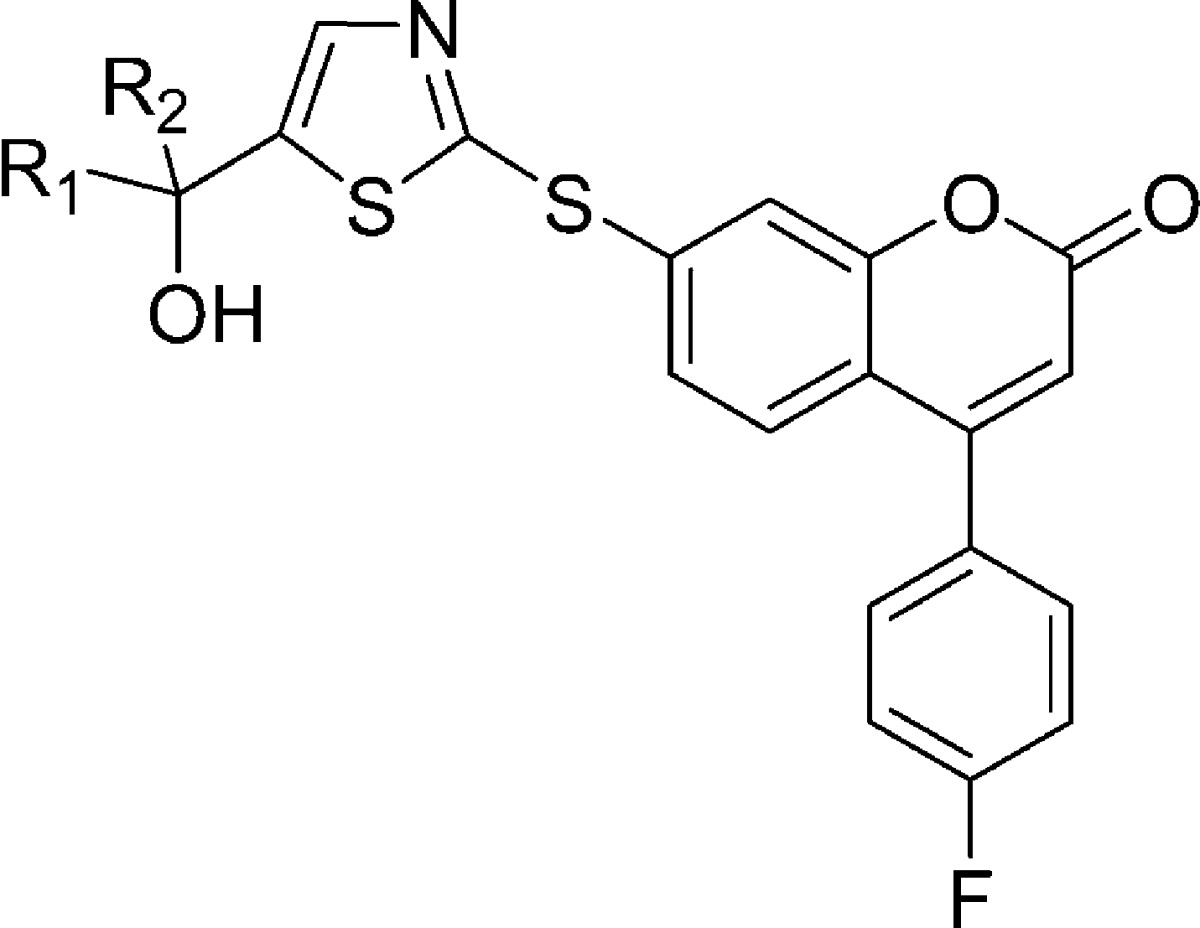
Our effort started with phenylthioether 2 (Table 1), a 5-LO inhibitor identified in a previous research campaign.13 This compound shows a high potency at inhibiting calcium ionophore-stimulated production of LTB4 in human whole blood (HWB) in vitro, with an IC50 of 70 nM. However, its development was hampered by the observation that two major long-lived and active metabolites, the corresponding sulfone and sulfoxide, were circulating in rats and monkeys. This observation triggered the search for structurally distinct 5-LO inhibitors. All compounds prepared were evaluated for their potency to inhibit the oxidation of arachidonic acid by recombinant human 5-LO (H5-LO),14 and the production of LTB4 in calcium ionophore-stimulated HWB.15 Each section of the molecule was investigated, but the most interesting modulation of structure−activity relationship (SAR) came from replacement of the central mercaptobenzene moiety of inhibitor 2. Most structural modifications of this moiety, even subtle ones like replacing the sulfur atom by an oxygen atom (3), were deleterious to potency and brought the HWB IC50 above 1 μM. The few analogues that retained submicromolar potency are depicted in Table 1. Among them, thiazole 4 rapidly caught our attention since it retained high potency in the H5-LO assay (IC50 = 20 nM) and showed only a 4-fold loss of activity in the HWB assay (IC50 = 310 nM) as compared to inhibitor 2. Many analogues of thiazole 4 were prepared, and it became clear that appropriate modification of the substitution pattern at the tertiary alcohol provided a major gain of potency in the HWB assay, as shown in Table 2. Replacing the two trifluoromethyl groups of compound 4 by two methyl groups brought a 2-fold loss of potency in the HWB assay (compound 9). Conversely, substitution by two ethyl groups was favorable, generating inhibitor 10 with an IC50 of 37 nM. Highly potent inhibitors were also obtained by an asymmetric substitution at the tertiary alcohol (compounds 11−14, all racemic). The optimal potency in this series was obtained by keeping one trifluoromethyl and one ethyl group to generate racemic inhibitor 12 (HWB IC50 = 50 nM). Both enantiomers of 12 were prepared, and it was observed that (R)-12, with an IC50 of 31 nM in the HWB assay, is more potent than isomer (S)-12. The stereochemical assignment was established by X-ray crystallography analysis. In contrast to what was observed for inhibitor 2,13 thiazole (R)-12 is orally bioavailable in rat, without having to rely on the administration of a hydroxy acid prodrug. Thus, upon dosing male rats with (R)-12 orally at 20 mg/kg in 0.5% methocel, good bioavailability (F = 37%) and a maximum concentration (Cmax) of 2.0 μM were observed, with an elimination half-life (t1/2) of 4.3 h. The potency of (R)-12 on the ex vivo generation of LTB4 in whole blood stimulated with calcium ionophore A23187 was measured in a dog model.16 In vitro, thiazole (R)-12 is 3-fold more potent in a dog whole blood assay (IC50 = 10 nM) than it is in the HWB assay (IC50 = 31 nM). Following oral dosing in dogs at a dose of 4 mg/kg, (R)-12 exhibited >94% inhibition of LTB4 in dog whole blood ex vivo up to 24 h. This level of inhibition is consistent with the potency of the compound in the dog whole blood assay and the measured plasma concentrations that are greater than 0.18 μM at all time points, up to 24 h. The remarkable profile of this 5-LO inhibitor triggered exploratory ancillary pharmacology studies. One of these studies evaluated the effect of (R)-12 on the cardiovascular system of anesthetized and vagotomized dogs, following the administration of 4 mg/kg/h of the drug by i.v. infusion, for 2 h. Unfortunately, upon examination of the electrocardiograms, a clear QTc interval prolongation was noted in treated dogs, beginning 45 min after initiation of the infusion. The maximum effect on mean QTc interval reached +9% (as compared to control) after 90 min of dosing, with an average plasma exposure of 10 μM. Given a targeted trough exposure of 100 nM for efficacy in the clinic, the likely therapeutic margin in humans was estimated to be too low to warrant the clinical development of (R)-12.
Table 2. SAR at the Tertiary Alcohol Position in the Thiazole Series.
| IC50 (nM)a |
Ki (μM)a | ||||
|---|---|---|---|---|---|
| compd | R1 | R2 | H5-LO | HWB | hERG |
| 4 | CF3 | CF3 | 20 ± 16 | 310 ± 160 | 1.2 ± 0.1 |
| 9 | Me | Me | 1230 ± 480 | 520 ± 310 | 2.9 ± 0.1 |
| 10 | Et | Et | 23 ± 6 | 37 ± 20 | 2.1 ± 0.2 |
| 11 | CF3 | Me | 30 ± 4 | 150 ± 40 | 1.5 ± 0.1 |
| 12 | CF3 | Et | 14 ± 4 | 50 ± 18 | 0.9 ± 0.1 |
| 13 | CF3 | i-Pr | 12 ± 4 | 88 ± 12 | 1.1 ± 0.1 |
| 14 | CF3 | t-Bu | 18 ± 1 | 210 ± 40 | |
| (S)-12 | CF3 | Et | 79 ± 15 | 83 ± 33 | 0.9 ± 0.1 |
| (R)-12 | Et | CF3 | 7 ± 2 | 31 ± 22 | 1.3 ± 0.2 |
Each IC50 value corresponds to an average of at least two independent determinations (±SD).
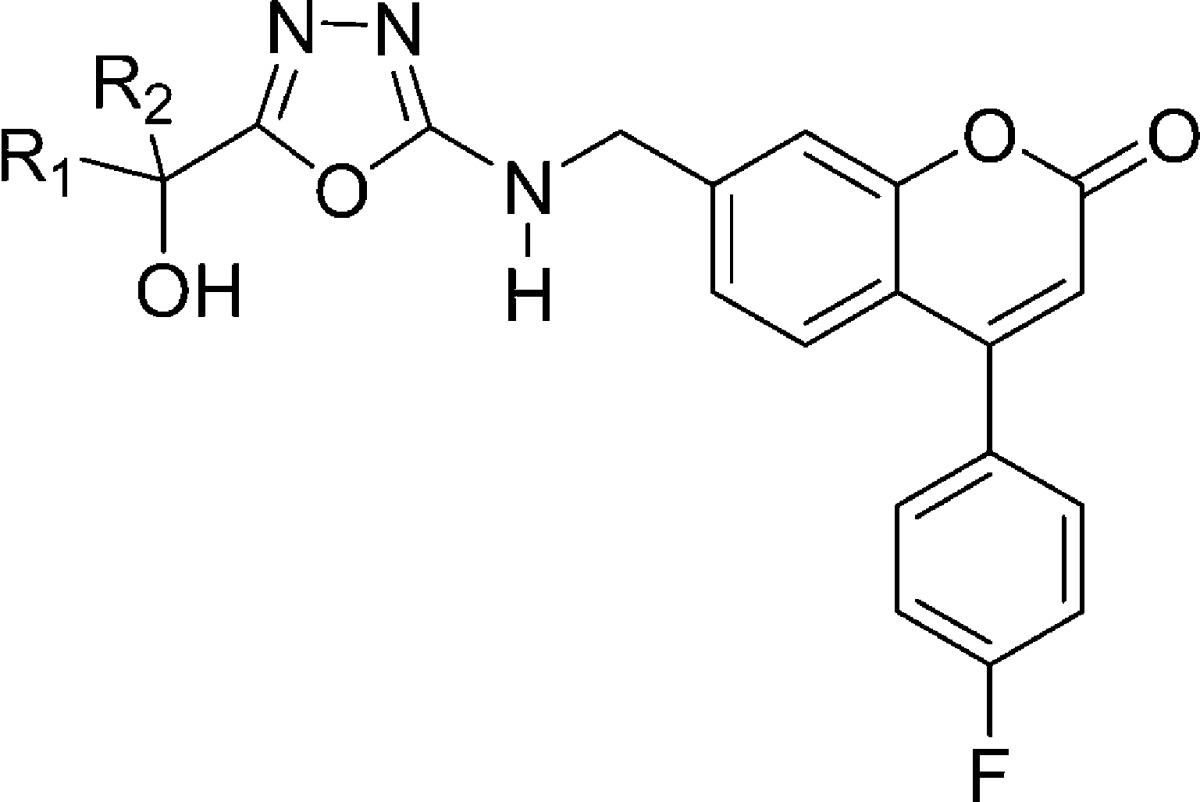
It is known that a variety of drugs that exhibit prolongation of the QTc interval tend to block the voltage-gated potassium channel encoded by the human ether-a-go-go gene (hERG).17 The affinity of molecules for this channel can be measured in vitro by using a MK-499 displacement binding assay.18 5-LO inhibitor (R)-12 exhibits a relatively high binding affinity in this assay with a Ki of 1.3 μM (Table 2). Because of the correlation between the prolongation of the QTc interval observed with (R)-12 in vivo in dogs and the potency of (R)-12 in the hERG binding assay, we have taken the binding affinity for the hERG potassium channel as measured in the MK-499 binding assay as an in vitro indicator for the potential for causing QTc interval prolongation. Therefore, our efforts focused on identifying those portions of the molecule that could be modified in such a way to decrease the affinity for the hERG channel while maintaining or improving the inhibitory potency against 5-LO. All of the thiazole derivatives described in Table 2 show a similar affinity for the hERG channel (Ki = 0.9−2.9 μM). On the other hand, modification of the central ring system seems to have a more profound effect on hERG binding affinity, as shown in Table 1. In this regard, oxadiazole derivative 8 is of particular interest with a hERG binding Ki of 8.0 μM, representing a 6-fold loss of affinity as compared to thiazole (R)-12. Unfortunately, oxadiazole 8 also shows a considerable loss of potency in the HWB assay (IC50 = 690 nM). By applying the lessons learned in the thiazole series to the oxadiazole case, we were hopeful that potency could be restored. The gem-diethyl derivative 15 was prepared and presented a potency equivalent to that shown by inhibitor (R)-12 in the HWB assay (IC50 = 36 nM), accompanied by a significantly lower affinity for the hERG channel (Ki = 14.5 μM, Table 3). The pharmacokinetic profile of this compound was not optimal though. Upon dosing male rats intraveneously with 15, a short elimination half-life of 1 h was observed. This problem was solved by preparing the asymmetric alcohol 16, bearing a trifluoromethyl and an ethyl group. Racemic inhibitor 16 presented good potency in the HWB assay with an IC50 of 52 nM. Both enantiomers of 16 were prepared, and the stereochemical assignment was again established by X-ray crystallography analysis. Enantiomer (S)-16, with an IC50 of 3.9 nM in the H5-LO assay and an IC50 of 52 nM in the HWB assay, is more potent than isomer (R)-16. This excellent 5-LO inhibitory profile is accompanied by a relatively low affinity for the hERG channel (Ki = 6.3 μM). Oxadiazole (S)-16 inhibits specifically 5-LO and is not active against 12-LO, 15-LO, and FLAP (>20 μM). The activity against FLAP was determined by measuring the displacement of a radiolabeled FLAP ligand from human polymorphonuclear cell membranes. Oxadiazole (S)-16 presents a good pharmacokinetic profile in many preclinical species (rat, 5 mg/kg iv, 20 mg/kg po, F = 66%, and t1/2 = 3.3 h; dog, 5 mg/kg iv, 4 mg/kg po, F = 64%, and t1/2 = 5.3 h; and rhesus monkey, 5 mg/kg iv, 4 mg/kg po, F = 54%, and t1/2 = 3.6 h). In vitro, 5-LO inhibitor (S)-16 is 2-fold more potent in a dog whole blood assay (IC50 = 21 nM) than it is in the HWB assay (IC50 = 52 nM). Following oral dosing in dogs at a dose of 2 mg/kg, (S)-16 exhibited >98% inhibition of LTB4 in dog whole blood ex vivo up to 6 h. This level of inhibition is consistent with its potency in dog whole blood and the measured concentration of drug in the plasma that is greater than 0.28 μM at all time points up to 6 h. Inhibition of LTB4 at 24 h was ∼60%, although the drug concentration in plasma was below the detection limit of 10 nM. Furthermore, and in contrast to what was observed with thiazole (R)-12, the oxadiazole inhibitor (S)-16 did not show adverse effects on the cardiovascular system of anesthetized and vagotomized dogs. Thus, following the administration of cumulative doses of 1, 3, and 10 mg/kg of the drug by i.v. infusion (each dose infused over 30 min), no treatment-related changes in the PR, QRS, and QTc intervals were observed, with an average plasma exposure of 9, 14, and 49 μM at those three doses, respectively.
Table 3. SAR at the Tertiary Alcohol Position in the Oxadiazole Series.
| IC50 (nM)a |
Ki (μM)a | ||||
|---|---|---|---|---|---|
| compd | R1 | R2 | H5-LO | HWB | hERG |
| 8 | CF3 | CF3 | 45 ± 10 | 690 ± 320 | 8.0 ± 0.2 |
| 15 | Et | Et | 130 ± 25 | 36 ± 17 | 14.5 ± 0.4 |
| 16 | CF3 | Et | 10 ± 3 | 52 ± 16 | 5.0 ± 0.8 |
| (R)-16 | CF3 | Et | 230 ± 70 | 100 ± 30 | 7.7 ± 0.8 |
| (S)-16 (MK-0633) | Et | CF3 | 3.9 ± 2.1 | 52 ± 21 | 6.3 ± 0.7 |
Each IC50 value corresponds to an average of at least two independent determinations (±SD).
The 5-LO inhibitors described in this study were prepared as shown in Schemes 1−5. The synthesis of diaryl ether 3 started by the addition of 3,4-dimethoxybenzyl alcoholate to 1-bromo-3,5-difluorobenzene (Scheme 1) to provide benzyl ether 17. Bromine−lithium exchange followed by treatment with hexafluoroacetone gave access to tertiary alcohol 18, which after deprotection afforded phenol 19. The desired diaryl ether 3 was finally obtained by engaging intermediate 19 with the known aryl bromide 20(13) under Ullmann ether synthesis conditions. The 2-thiazolyl thioethers 4 and 9−14 were prepared by two different pathways (Scheme 2). Deprotonation of 2-mercaptothiazole with lithium diisopropyl amide (LDA), followed by addition of hexafluoroacetone, acetone, 3-pentanone, or 1,1,1-trifluoro-2-butanone, gave tertiary alcohols 21a, b, c, and e, respectively. Alternatively, deprotonation of 2-mercaptothiazole with LDA followed by addition of ethyl trifluoroacetate gave ketone 22. Addition of methylmagnesium bromide, isopropylmagnesium chloride, or tert-butyllithium to 22 yielded alcohols 21d, f, and g, respectively. The desired thioethers were obtained by heating the appropriate 2-mercaptothiazoles 21 with bromocoumarin 20 in 1-methyl-2-pyrrolidinone (NMP). The two enantiomers of inhibitor 12 were obtained by submitting the racemic mixture of asymmetric alcohol 21e to a chiral separation on a Chiralpak AD column (Scheme 3). Each enantiomer was subsequently reacted with bromide 20 to afford inhibitors (S)-12 and (R)-12. The preparation of analogues 5, 6, and 7 is described in the Supporting Information. The preparation of 2-amino-oxadiazole derivatives 8, 15, and 16, described in Scheme 4, involved the synthesis of key intermediates 31a−c. Reaction of ester 30 [obtained by treatment of 2,2-bis(trifluoromethyl)-2-hydroxyacetic acid with diazomethane], first with hydrazine and then with cyanogen bromide, gave access to oxadiazole 31a. Alternatively, a similar sequence was applied to diethyloxalate and was followed by the addition of excess ethylmagnesium bromide to produce analogue 31b. As for asymmetric derivative 31c, its synthesis involved addition of ethylmagnesium bromide to ethyl trifluoropyruvate and subsequent reaction with hydrazine and with cyanogen bromide. The desired final products were then obtained by reductive amination reactions between the appropriate amines 31 and aldehyde 25 (see Supporting Information). The two enantiomers of inhibitor 16 were first obtained by submitting the racemic mixture to a chiral separation on a Chiralpak AD column. Afterward, larger quantities of the most active enantiomer (S)-16 were produced by following the synthetic route depicted in Scheme 5. Addition of ethylmagnesium bromide to ethyl trifluoropyruvate was followed by hydrolysis of the resulting hydroxy-ester to afford racemic acid 32. Coupling with (S)-2-phenylglycinol afforded a diastereomeric mixture that was purified by flash chromatography on silica gel to deliver diastereomerically pure amide (S,S)-33. Hydrolysis of the amide bond followed by esterification produced methyl ester (S)-34. The usual treatment with hydrazine and with cyanogen bromide gave oxadiazole (S)-35, and reductive amination with aldehyde 25 completed the preparation of multigram quantities of potent 5-LO inhibitor (S)-16.
Scheme 1.

Scheme 5.

Scheme 2.

Scheme 3.

Scheme 4.

In summary, we have discovered a series of novel 5-LO inhibitors that are potent, selective, and orally bioavailable. A major focus of the optimization effort was to preserve the 5-LO inhibitory potency while reducing the affinity for the hERG potassium channel. This work culminated in the identification of 4-(4-fluorophenyl)-7-[({5-[(2S)-1,1,1-trifluoro-2-hydroxybutan-2-yl]-1,3,4-oxadiazol-2-yl}amino)methyl]-2H-chromen-2-one [(S)-16, MK-0633, setileuton]. On the basis of its overall profile, setileuton was selected for clinical development, and its effect on the treatment of asthma and COPD will be reported in separate papers.
Acknowledgments
We acknowledge the contribution of Nancy N. Tsou (Merck Research Laboratory, Rahway) for the structure determination of compounds (R)-12 and (S,S)-33 by X-ray diffraction.
Supporting Information Available
Experimental synthetic procedures, spectroscopic characterization, and H5-LO assay protocol. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Peters-Golden M.; Henderson W. R. Jr. Leukotrienes. N. Engl. J. Med. 2007, 357, 1841–1854. [DOI] [PubMed] [Google Scholar]
- Rådmark O.; Werz O.; Steinhilber D.; Samuelsson B. 5-Lipoxygenase: Regulation of expression and enzyme activity. Trends Biochem. Sci. 2007, 32, 332–341. [DOI] [PubMed] [Google Scholar]
- Evans J. F.; Ferguson A. D.; Mosley R. T.; Hutchinson J. H. What's all the FLAP about?: 5-Lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol. Sci. 2008, 29, 72–78. [DOI] [PubMed] [Google Scholar]
- Werz O.; Steinhilber D. Therapeutic options for 5-lipoxygenase inhibitors. Pharmacol. Ther. 2006, 112, 701–718. [DOI] [PubMed] [Google Scholar]
- Fanta C. H. Asthma. N. Engl. J. Med. 2009, 360, 1002–1014. [DOI] [PubMed] [Google Scholar]
- Berger W.; De Chandt M. T.; Cairns C. B. Zileuton: Clinical implications of 5-lipoxygenase inhibition in severe airway disease. Int. J. Clin. Pract. 2007, 61, 663–676. [DOI] [PubMed] [Google Scholar]
- Diamant Z.; Timmers M. C.; van der Veen H.; Friedman B. S.; De Smet M.; Depré M.; Hilliard D.; Bel E. H.; Sterk P. J. The effect of MK-0591, a novel 5-lipoxygenase activating protein inhibitor, on leukotriene biosynthesis and allergen-induced airway responses in asthmatic subjects in vivo. J. Allergy Clin. Immunol. 1995, 95, 42–51. [DOI] [PubMed] [Google Scholar]
- Storms W. Update on montelukast and its role in the treatment of asthma, allergic rhinitis and exercise-induced bronchoconstriction. Expert Opin. Pharmacother. 2007, 8, 2173–2187. [DOI] [PubMed] [Google Scholar]
- Drakatos P.; Lykouras D.; Sampsonas F.; Karkoulias K.; Spiropoulos K. Targeting Leukotrienes for the treatment of COPD?. Inflammation Allergy: Drug Targets 2009, 8, 297–306. [DOI] [PubMed] [Google Scholar]
- Bäck M. Inhibitors of the 5-Lipoxygenase Pathway in Atherosclerosis. Curr. Pharm. Des. 2009, 15, 3116–3132. [DOI] [PubMed] [Google Scholar]
- Cortes-Burgos L. A.; Zweifel B. S.; Settle S. L.; Pufahl R. A.; Anderson G. D.; Hardy M. M.; Weir D. E.; Hu G.; Happa F. A.; Stewart Z.; Muthian S.; Graneto M. J.; Masferrer J. L. CJ-13610, an orally active inhibitor of 5-lipoxygenase is efficacious in preclinical models of pain. Eur. J. Pharmacol. 2009, 617, 59–67. [DOI] [PubMed] [Google Scholar]
- Poff C. D.; Balazy M. Drugs that target lipoxygenases and leukotrienes as emerging therapies for asthma and cancer. Curr. Drug Targets Inflammation Allergy 2004, 3, 19–33. [DOI] [PubMed] [Google Scholar]
- Grimm E. L.; Brideau C.; Chauret N.; Chan C. C.; Delorme D.; Ducharme Y.; Ethier D.; Falgueyret J.-P.; Friesen R. W.; Guay J.; Hamel P.; Riendeau D.; Soucy-Breau C.; Tagari P.; Girard Y. Substituted coumarins as potent 5-lipoxygenase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 2528–2531. [DOI] [PubMed] [Google Scholar]
- The protocol to perform the H5-LO assay is described in the Supporting Information.
- Brideau C.; Chan C.; Charlson S.; Denis D.; Evans J. F.; Ford-Hutchinson A. W.; Fortin R.; Gillard J. W.; Guay J.; Guevremont D.; Hutchinson J. H.; Jones T. R.; Léger S.; Mancini J. A.; McFarlane C. S.; Pickett C.; Piechuta H.; Prasit P.; Riendeau D.; Rouzer C. A.; Tagari P.; Vickers P. J.; Young R. N.; Abraham W. M. Pharmacology of MK-0591 (3-[1-(4-chlorobenzyl)-3-(t-butylthio)-5-(quinolin-2-yl-methoxy)-indol-2-yl]-2,2-dimethyl propanoic acid), a potent, orally active leukotriene biosynthesis inhibitor. Can. J. Physiol. Pharmacol. 1992, 70, 799–807. [DOI] [PubMed] [Google Scholar]
- Tagari P.; Becker A.; Brideau C.; Frenette R.; Sadl V.; Thomas E.; Vickers P.; Ford-Hutchinson A. Leukotriene generation and metabolism in dogs: Inhibition of biosynthesis by MK-0591. J. Pharmacol. Exp. Ther. 1993, 265, 416–25. [PubMed] [Google Scholar]
- Jamieson C.; Moir E. M.; Rankovic Z.; Wishart G. Medicinal Chemistry of hERG Optimizations: Highlights and Hang-ups. J. Med. Chem. 2006, 49, 5029–5046. [DOI] [PubMed] [Google Scholar]
- Friesen R. W.; Ducharme Y.; Ball R. G.; Blouin M.; Boulet L.; Côté B.; Frenette R.; Girard M.; Guay D.; Huang Z.; Jones T. R.; Laliberté F.; Lynch J. J.; Mancini J.; Martins E.; Masson P.; Muise E.; Pon D. J.; Siegl P. K.; Styhler A.; Tsou N. N.; Turner M. J.; Young R. N.; Girard Y. Optimization of a Tertiary Alcohol Series of Phosphodiesterase-4 (PDE4) Inhibitors: Structure−Activity Relationship Related to PDE4 Inhibition and Human Ether-a-Go-Go Related Gene Potassium Channel Binding Affinity. J. Med. Chem. 2003, 46, 2413–2426 and references therein. [DOI] [PubMed] [Google Scholar]
- Delorme D.; Ducharme Y.; Brideau C.; Chan C. C.; Chauret N.; Desmarais S.; Dubé D.; Falgueyret J.-P.; Fortin R.; Guay J.; Hamel P.; Jones T. R.; Lépine C.; Li C.; McAuliffe M.; McFarlane C. S.; Nicoll-Griffith D. A.; Riendeau D.; Yergey J. A.; Girard Y. Dioxabicyclooctanyl Naphthalenenitriles as Nonredox 5-Lipoxygenase Inhibitors: Structure−Activity Relationship Study Directed toward the Improvement of Metabolic Stability. J. Med. Chem. 1996, 39, 3951–3970. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.