

Abstract
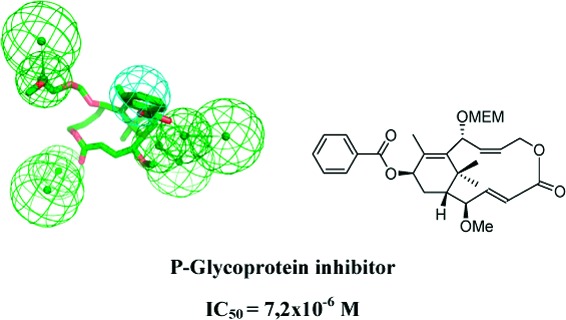
Three simplified “non-natural” natural taxanes, related to taxuspine X, were synthetized and assayed as P-glycoprotein (P-gp) inhibitors. One of them (6) proved to be a very efficient P-gp inhibitor with an IC50 = 7.2 × 10−6 M. In addition, to rationalize biological data, a pharmacophoric model was built through a ligand-based approach. This model represents the first example of a pharmacophore, which describes interactions of taxanes with P-gp.
Keywords: Taxanes, MDR reversing agents, P-glycoprotein, macrolactone, non-natural natural product
The diterpenoide paclitaxel (Taxol) 1 from the yew (Taxus) species and its derivative docetaxel 2 are two of the best anticancer agents in clinical use today for the treatment of ovarian, breast, and lung cancer, as well as of Kaposi's sarcoma. Paclitaxel and docetaxel belong to the antimitotic drugs that bind to β-tubulin.1−5 Clinical use of these anticancer drugs has disclosed that paclitaxel and docetaxel have a number of undesired side effects and, in particular, that their use can result in the development of cross resistance to a broad range of structurally and functionally unrelated compounds (multidrug resistance, MDR). MDR induced by taxoid treatment in tumor cells is a significant obstacle to the success of chemotherapy in different types of cancers.6 The most supported mechanism of MDR is the overexpression of transmembrane ABC transporters, which efflux a variety of chemotherapeutic drugs out of tumor cells. The ABC transporter mostly involved in MDR is ABCB1 (ABCB subfamily), well-known as P-glycoprotein (P-gp).7−9 Different strategies have been devised to overcome drug resistance mediated by multidrug transporters (P-gp): (1) the development of reversal agents that block the efflux pumps, preventing the extrusion of anticancer drugs from cross-resistant cells; (2) the development of cytotoxic agents that are not substrates for multidrug transporters and are not extruded from resistant cells; and (3) the development of cytoprotective agents that are pumped out from resistant cancer cells in place of the cytotoxic drug.10,11 Several natural and semisynthetic taxoids, devoid of cytotoxicity and tubulin affinity, are powerful inhibitors of P-gp activity, acting as efficient reversing agents and allowing accumulation of paclitaxel in MDR cancer cells.12−19 A number of taxol-related compounds were isolated from the Japanese yew Taxus cuspidata; among these, taxuspine X 3 has been shown to be a potent MDR reversing agent, increasing the accumulation of vincristine in MDR cell lines.20−23 These natural products were isolated in very poor yields from the natural source, thus requiring the intervention of synthetic chemistry to generate a sufficient amount of compound to fully explore the biological potential of taxuspine analogues. However, the total synthesis of biological active natural compounds such as taxuspine X shows intrinsic difficulties and high costs. As a result, we focused our efforts on the rational design and the synthesis of simplified taxuspine analogues with similar or improved biological properties. Even if natural compounds have several functional groups, not all of them are involved in the interaction with the biological target, and consequently, not all of them are fundamental for biological activity. It is known, for instance, that the phenylisoserine side chain is crucial for the cytotoxic activity of paclitaxel.24 On the contrary, replacement of the C13 phenylisoserine side chain with different acyl groups affords noncytotoxic taxanes, which possess MDR reversing activity.15,20−23 On the basis of this assumption, we have been involved in the past decade in the design and synthesis of simplified taxuspine X analogues. We have recently reported the synthesis of simplified diterpenoid 4 through a ring-closing metathetis approach.25−27 Taxane 4 still presents structural complexity and a synthetic challenge, due in part to the presence of five stereocenters. Hence, a further simplified taxuspine 3 analogue, compound 5, has been recently designed and synthesized with the help of computational methodologies, and it proved to be a good P-gp ATPase modulator.28 However, because it was not clear from the previous study if the ATPase modulator activity of 5 was due to its interaction only with P-gp or also with MRP1 and MRP2 transporters, herein, we decided to resynthesize 5 and assay it only as a P-gp inhibitor. In addition, on the basis of latter encouraging results,28 two novel taxane analogues, 6 and 7, strictly related to 5, were rationally designed and assayed as P-gp inhibitors (Figure 1). Compound 6 was designed on the basis of our previous studies on taxinine analogues. We demonstrated that taxanes bearing an acyloxy moiety at C13, in particular a benzoyloxy moiety, possess high MDR reversal activity.15 On the other hand, we rationalized that carbocycle 7 could be easily synthesized from the same substrates used in the synthesis of 6 and could be of interest for a structure−activity relationship (SAR) evaluation. These derivatives may be described as “non-natural” natural products,29 as they retain most of the (two-dimensional) structural features of the natural product lead 3. The presence of a few functional moieties as well as a bioisosteric macrolactone ring could allow an easy synthesis of these molecules, maintaining at the same time the biological activity of the natural compound.
Figure 1.

Natural taxanes and simplified analogues.
Compound 5 was reprepared according to our previous work.28 Accordingly, derivative 6 was prepared through a synthetic strategy similar to that used for 5 via Yamaguchi macrolactonization (Scheme 1).28−31 The structure of 6 consists of a 12-membered macrolactone condensed with ring A of paclitaxel in which the macrocyclic C6 of taxuspine X has been replaced by its bioisosteric oxygen atom.
Scheme 1. Synthesis of Lactone 6.

Reagents and conditions: (i) SeO2, pyridine, 80 °C, 1 h (30%). (ii) BzCl, pyridine, BuNI, DCM, room temperature, 12 h (86%). (iii) TBAF, THF, 40 °C, 1 h (90%). (iv) TPAP, NMO, DCM, room temperature, 2 h (quant.). (v) Propargylalcohol-OTBDPS, BuLi, THF, −78 °C, 1 h (90%). (vi) H2, Lindlar's catalyst, quinoline, AcOEt, 1 h (80%). (vii) MEMCl, DIPEA, DCM, 40 °C, 12 h (83%). (viii) DDQ, phosphate buffer, pH 7.2, 1 h (97%). (ix) NMO, TPAP, DCM, 1 h (quant.). (x) NaClO2, tBuOH, 2-methyl-2-butene, pH 7, 1 h (84%). (xi) TBAF, THF, 40 °C, 1 h (quant.). (xii) Trichlorobenzoyl chloride, Et3N, DMAP, THF/toluene, 75 °C, 3 h (50%).
Aldehyde 8 was converted into compound 9 in five steps.28 Compound 9 was chosen as the substrate to introduce a hydroxy moiety on the cyclohexene ring through allylic oxidation. Reaction of 9 with SeO2 at 80 °C led to the desired alcohol 10 in good yield. The relative configuration of the new stereocenter was established through nuclear Overhauser effect spectroscopy (NOESY) experiments. No nuclear Overhauser effect (NOE) was observed between H1 and H5 on compound 10. The chirality of the stereocenter was inverted by oxidation of 10 to ketone 11 followed by reduction with NaBH4, leading to diastereoisomer 12. NOESY experiment on 12 revealed a NOE effect between H1 and H5, confirming the assignment of the relative configuration.32
Allylic alcohol 10 was then reacted with benzoyl chloride affording derivative 13. This latter compound was desilylated and converted into aldehyde 14. Diastereoselective alkynylation of 14 gave compound 15, which was then partially hydrogenated into the alkene 16. The secondary alcohol 16 was protected with MEMCl affording derivative 17. Deprotection of the PMB protecting group led to alcohol 18, which was oxidized in a two step sequence to afford carboxylic acid 20. Desilylation of 20 led to hydroxyl acid 21, which was finally converted into the desired macrolactone 6 by Yamaguchi macrocyclization (50% yield). The synthesis of carbacyclic derivative 7 is reported in Scheme 2. Compound 9 was converted into aldehyde 22, which in turn was diastereoselectively alkynylated to give alkyne 23. Alkyne was selectively reduced with Lindlar's catalyst to give Z-alkene 24, which was in turn converted into MEM derivative 25. Desilylation of 25 with TBAF followed by PMB deprotection with DDQ led to diol 26. This latter compound was oxidized with TPAP/NMO affording dialdehyde 27, which represents the key synthon for macrocyclization via pinacol coupling. Reaction of 27 with TiCl4 and Zn dust in the presence of pyridine led to diol 28 in 49% yield.33 Compound 28 was obtained as a mixture of two diastereoisomers, in almost a 1.5:1 ratio, which were not separable by simple column chromoatography. Finally, diol 28 was converted into target compound diacetyl derivative 7.
Scheme 2. Synthesis of Carbacycle 7.

Reagents and conditions: (i) TBAF, THF, 40 °C, 1 h (93%). (ii) TPAP, NMO, DCM, 1 h (quant.). (iii) Propargyl alcohol OTBDPS, BuLi, −78 °C, 1 h (90%). (iv) H2, Lindlar's catalyst, quinoline, AcOEt, 1 h (80%). (v) MEMCl, DIPEA, DCM, 40 °C, 12 h (75%). (vi) DDQ, phosphate buffer, pH 7.2, 1 h (97%). (vii) TBAF, THF, 40 °C, 1 h (quant.). (viii) TPAP, NMO, DCM, 1 h (quant.). (ix) TiCl4, Zn dust, pyridine, THF, 1 h (49%). (x) (AcO)2O, Et3N, DMAP, DCM, 30 min (quant.).
Compounds 5, 6, and 7 were then investigated as P-gp inhibitors by measuring cyto-fluorimetrically the retention of the P-gp substrate rhodamine 123 (R123) in human MDR1 gene transfected mouse T-lymphoma L5178 cells (see the Supporting Information). In our previous work,28 compound 5 was assayed as a possible P-gp-ATPase modulator with the use of rat small intestine, brush border membrane vesicles by assaying both basal and verapamil stimulated ATPase activity. Compound 5 showed a bimodal effect on basal ATPase activity. In fact, it inhibited ATPase activity in a concentration-dependent fashion up to 1 μM concentration. At higher concentrations, however, inhibition vanished; the ATPase activity recovered, in fact, at 10 μM, a value even greater than that observed under control conditions (no inhibitor). Compound 5 at ≤1 μM concentration fully antagonized stimulation of ATPase activity operated by verapamil. This suggested that at ≤1 μM concentrations, compound 5 was behaving like an inhibitor, while at higher concentrations, it was behaving like a substrate of the enzyme. Immunoblot analysis, however, proved that ABC transporters of rat intestinal mucosa vesicles were mostly represented by MRP1.34 Hence, it was not clear if the activity of 5 could be ascribed to its interaction with P-gp or rather with MRP1 and/or MRP2. Consequently, we decided to focus on the capability of the novel compounds and compound 5 as well to inhibit P-gp activity. For this purpose, we employed murine T-lymphoma cells L5178 transfected with the human MDR1 gene, thus overexpressing P-gp. In the present study, we investigated with this cell line the extent of the inhibition of P-gp-mediated transport of R123 and the structure−activity relationships of 5−7. The fluorescent compound R123 was shown to be a substrate of P-gp,35 and its concentration in the cell, positively correlated to the degree of the inhibition of the outward transporter P-gp, may represent the response of the model system here employed to P-gp inhibitors. For each of the three compounds under scrutiny (5−7), we have provided experimental concentration−response curves. The concentration−inhibition curves of the compounds tested are shown in Figure 2, where IC50 and αmax values are also reported.
Figure 2.

Pharmacological evaluation. Concentration−response curve of the inhibition of P-gp-mediated R123 efflux from L5178 MDR1 cells by 5−7.
Compound 5 appeared to be the weakest inhibitor of this set of compounds characterized by an αmax value of 0.37 at the highest concentration used (1.0 × 10−4 M). Compounds 6 and 7 turned out to be effective inhibitors of P-gp-mediated R123 cell efflux, their αmax values approaching 1 (αmax expresses the efficacy of the inhibitor and varies between 0 (in the absence of the inhibitor) and 1 (when the amount of R123 found in L5178 MDR1 cells was comparable to that determined in presence of 5 × 10−3 M Vi). However, while 6 inhibited P-gp with an IC50 = 7.2 × 10−6 M, 7 showed a higher IC50 value, averaging 2.4 × 10−5 M [IC50 measures the potency of the inhibitor and represents the concentration that causes a half-maximal increase (α = 0.5) in the intracellular concentration of R123]. Under the same experimental conditions, cyclosporine A, a well-known P-gp inhibitor and MDR reversal agent, exhibited an IC50 = 6.7 × 10−7 M (data not shown). From the present findings, we can suggest that the introduction of the benzoyl group at C13 position of 5 leading to 6 turned out to be meaningful, since the latter compound was characterized by a much higher inhibitory activity on P-gp as compared to the former one. Moreover, the carbocycle structure is noteworthy in conferring to 7 a good capability to inhibit P-gp as the IC50 value of 7 was 2.4 × 10−5 M.
To rationalize these biological data, modeling studies were developed. P-gp inhibition constitutes a very difficult target for molecular modeling. Although the 3D structures of the protein are available in the RCSB Protein Databank from 2009,36 a structure-based approach for the design of novel P-gp inhibitors shows several difficulties mainly due to the high flexibility and adaptability of the protein itself. To overcome this problem, we generated a pharmacophoric model through a ligand-based approach.
The pharmacophoric model generated was then used to align the molecules proposed in this work as well as taxuspine X. Taxuspine X showed a full mapping of the pharmacophore with the cinnamoyl moiety in correspondence of the hydrophobic features and the hydrogen bond acceptor features mapped by the acetyl substituents. On the contrary, 5 showed a poor mapping of the pharmacophoric features especially for the hydrophobic one. Insertion of a benzoyl moiety in position 13 of 6 allowed the full mapping of the pharmacophore and induced a 180° rotation of the molecule along the longest axis. This rotation permitted also a best mapping of the polar features by the OMe and OMEM groups and restored the same relative stereochemistry observable in taxuspine X, suggesting also a solution for the better activity. In both 5 and 6, the six ring region of the molecule is in the opposite position of that of taxuspine. Finally, compound 7 having a smaller carbacyclic ring proved to still be able to map correctly the pharmacophore. Also, in this case, the six-membered fused ring is in the opposite position with regard to the position in taxuspine X; in this manner, it is able to map the hydrophobic features. The hydrogen bond acceptor features were mapped by 7 in a similar way to that of 6 (Figure 3). Although this pharmacophoric model is not predictive, it is able to give a rationale to the activity of the presented compounds and to support further SAR studies.
Figure 3.
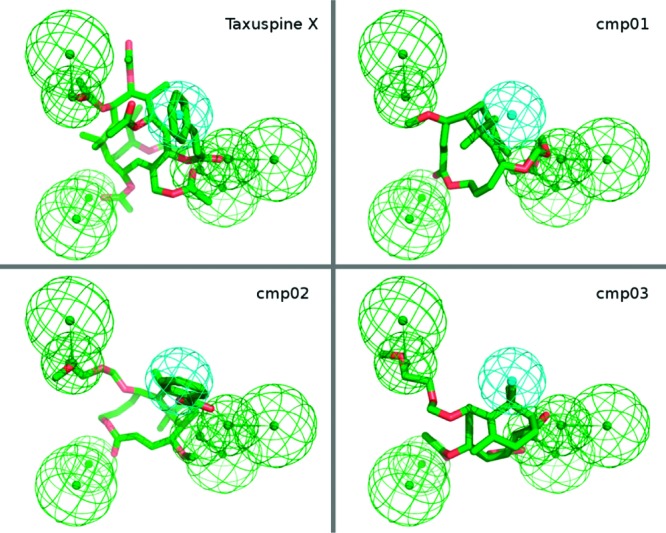
Pharmacophoric model. Cmp01 = 5, Cmp02 = 6, and Cmp03 = 7.
In conclusion, three novel “non-natural” natural taxanes 5−7 related to taxuspine X have been successfully synthesized and assayed as P-gp inhibitors. Compound 5 did not show any activity as a P-gp inhibitor, demonstrating that its P-gp-ATPase modulatory activity is ascribed to its interaction with MRP transporters. Derivative 6, bearing a bezoyloxy moiety on C13 of the taxane skeleton, proved to be a good inhibitor of P-gp. This latter result, in agreement with our previous studies,15 demonstrates the importance of the acyloxy side chain at C13. Carbocyclic taxane 7 also showed a promising P-gp inhibitory activity. Moreover, to rationalize biological data, a pharmacophoric model able to describe the interactions of taxanes with P-gp was computationally built. Compounds 6 and 7 could represent two interesting hit compounds, which can be further manipulated to lead to novel taxanes with improved activity. Studies on these compounds are currently in progress in our laboratory.
Acknowledgments
We thank Dr. Franco Zunino (Istituto Nazionale dei Tumori) and Dr. Sylvie Ducki (Centre for Molecular Drug Design, University of Salford).
Supporting Information Available
General methods, experimental procedures, 1H NMR and 13C NMR data, and computational and pharmacological methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The University of Siena is acknowledged for financial support.
Supplementary Material
References
- Crown J.; O'Leary M. The taxanes: An update. Lancet 2000, 335, 1176–1178. [DOI] [PubMed] [Google Scholar]
- Wani M. C.; Taylor H. L.; Wall M. E.; Coggon P.; McPhail A. T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [DOI] [PubMed] [Google Scholar]
- Rowinsky E. K.; Cazenave L. A.; Donehower R. C. Taxol: A novel investigational antimicrotubule agent. J. Natl. Cancer Inst. 1990, 82, 1247–1259. [DOI] [PubMed] [Google Scholar]
- Slichenmyer W. J.; Von Hoff D. D. Taxol: A new and effective anti-cancer drug. Anticancer Drugs 1991, 2, 519–530. [PubMed] [Google Scholar]
- Horwitz S. B. Mechanism of action of taxol. Trends Pharmacol. Sci. 1992, 13, 134–136. [DOI] [PubMed] [Google Scholar]
- Galletti E.; Magnani M.; Renzulli M. L.; Botta M. Paclitaxel and docetaxel resistance: Molecular mechanisms and development of new generation taxanes. ChemMedChem 2007, 2, 920–942. [DOI] [PubMed] [Google Scholar]
- Perez-Tomas R. Multidrug resistance: Retrospect and prospects in anti-cancer drug treatment. Curr. Med. Chem. 2006, 13, 1859. [DOI] [PubMed] [Google Scholar]
- Teodori E.; Dei S.; Martelli S.; Scapecchi F.; Gualtieri F. The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR). Curr. Drug Targets 2006, 7, 893. [DOI] [PubMed] [Google Scholar]
- Leslie E. M.; Deeley R. G.; Cole S. P. C. Multidrug Resistance Proteins: Role of P-glycoprotein, MRP1, MRP2 and BCRP (ABCG2) in Tissue Defense. Toxicol. Appl. Pharmacol. 2005, 204, 216. [DOI] [PubMed] [Google Scholar]
- Szakacs G.; Paterson J. K.; Ludwig J. A.; Booth-Genthe C.; Gottesman M. M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discovery 2006, 5, 219–234. [DOI] [PubMed] [Google Scholar]
- Nobili S.; Landini I.; Giglioni B.; Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr. Drug Targets 2006, 7, 861–879. [DOI] [PubMed] [Google Scholar]
- Kobayashi J.; Hosoyama H.; Wang X. X.; Shigemori H.; Koiso Y.; Iwasaki S.; Sasaki T.; Naito M.; Tsuruo T. Effects of taxoids from Taxus cuspidata on microtubule depolymerization and vincristine accumu- lation in MDR cells. Bioorg. Med. Chem. Lett. 1997, 7, 393. [Google Scholar]
- Ojima I.; Bounaud P. Y.; Takeuchi C.; Pera P.; Bernacki R. J. New taxanes as highly efficient reversal agents for multi-drug resistance in cancer cells. Bioorg. Med. Chem. Lett. 1998, 8, 189. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Gu J.; Yin D.; Chen X. Synthesis and biological evaluation of taxinine analogues as orally active multidrug resistance reversal agents in cancer. Bioorg. Med. Chem. Lett. 2004, 14, 4767. [DOI] [PubMed] [Google Scholar]
- Botta M.; Armaroli S.; Castagnolo D.; Fontana G.; Pera P.; Bombardelli E. Synthesis and biological evaluation of new taxoids derived from 2-deacetoxytaxinine. J. Bioorg. Med. Chem. Lett. 2007, 17, 1579–1583. [DOI] [PubMed] [Google Scholar]
- Ojima I.; Borella C. P.; Wu X.; Bounaud P.-Y.; Fumero Oderda C.; Sturm M.; Miller M. L.; Chakravarty S.; Chen J.; Huang Q.; Pera P.; Brooks T. A.; Baer M. R.; Bernacki R. J. Design, Synthesis and Structure-Activity Relationships of Novel Taxane-Based Multidrug Resistance Reversal Agents. J. Med. Chem. 2005, 48, 2218–2228. [DOI] [PubMed] [Google Scholar]
- Brooks T. A.; Kennedy D. R.; Gruol D. J.; Ojima I.; Baer M. R.; Bernacki R. J. Structure-Activity Analysis of Taxane-Based Broad-Spectrum Multidrug Resistance Modulators. Anticancer Res. 2004, 24, 409–415. [PubMed] [Google Scholar]
- Brooks T. A.; Minderman H.; O'Loughlin K. L.; Ojima I.; Baer M. R.; Bernacki R. J. Taxane-Based Reversal Agent Modulation of P-glycoprotein-, Multidrug Resistance Protein- and Breast Cancer Resistance Protein-Mediated Drug Transport. Mol. Cancer Ther. 2003, 2, 1195–1205. [PubMed] [Google Scholar]
- Minderman H.; Brooks T. A.; O'Loughlin K. L.; Ojima I.; Bernacki R. J.; Baer M. R. Modulation of ATP-Binding-Cassette Transport Proteins by the Taxane Derivatives BAY 59-8862 and tRA96023. Cancer Chemother. Pharmacol. 2004, 53, 363–369. [DOI] [PubMed] [Google Scholar]
- Zamir L. O.; Zhou Z. H.; Caron G.; Nedea M. E.; Sauriol F.; Mamer O. Isolation of a putative biogenetic taxane precursor from Taxus canadensis needles. J. Chem. Soc., Chem. Commun. 1995, 529–530. [Google Scholar]
- Begley M. J.; Jackson C. B.; Pattenden G. Total synthesis of verticillene, the putative biogenetic precursor of the taxane alkaloids. Tetrahedron Lett. 1985, 26, 3393–3396. [Google Scholar]
- Begley M. J.; Jackson C. B.; Pattenden G. Investigation of transannular cyclisations of verticillanes to the taxane ring system. Tetrahedron Lett. 1985, 26, 3397–3400. [Google Scholar]
- Hosoyama H.; Inubushi A.; Katsui T.; Shigemori H.; Kobayashi J. Taxuspines U, V, and W, new taxane and related diterpenoids from the Japanese yew Taxus cuspidate. Tetrahedron 1996, 52, 13145–13150. [Google Scholar]
- Gupta M. L.; Bode C. J.; Georgt G.; Himes R. H. Understanding Tubulin-Taxol Interactions: Mutations That Impart Taxol Binding to Yeast Tubulin. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 6394–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manetti F.; Maccari L.; Corelli F.; Botta M. 3-D QSAR models of interactions between beta-tubulin and microtubule stabilizing antimitotic agents (MSAA): A survey on taxanes and epothilones. Curr. Top. Med. Chem. 2004, 4, 203–217. [DOI] [PubMed] [Google Scholar]
- Renzulli M. L.; Rocheblave L.; Avramova S. I.; Galletti E.; Castagnolo D.; Tafi A.; Corelli F.; Botta M. Studies towards the synthesis of the bicyclic 3,8-secotaxane diterpenoid system using a ring closing metathesis strategy. Tetrahedron 2007, 63, 497–509. [Google Scholar]
- Galletti E.; Avramova S. I.; Renzulli M. L.; Corelli F.; Botta M. Stereoselective synthesis of advanced intermediates en route to Taxuspine U and X: a study of macrocyclization via ring closing metathesis to highly constrained twelve-membered rings. Tetrahedron Lett. 2007, 48, 751–754. [Google Scholar]
- Avramova S. I.; Galletti E.; Renzulli M. L.; Giorgi G.; Sgaragli G.; Alderighi D.; Ghiron C.; Corelli F.; Radi M.; Botta M. Synthesis of an original oxygenated taxuspine X analogue: A versatile “non-natural” natural product with remarkable P-gp modulating activity. ChemMedChem 2008, 3, 745–748. [DOI] [PubMed] [Google Scholar]
- Tietze L. F.; Bell H. P.; Chandrasekar S. Natural product hybrids as new leads for drug discovery. Angew. Chem., Int. Ed. 2003, 42, 3996–4028. [DOI] [PubMed] [Google Scholar]
- Inanaga J.; Hirata K.; Sacki H.; Katsuki T.; Yamaguchi M. Rapid esterification by means of mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar]
- Hitchcock S. A.; Houldsworth S. J.; Pattenden G.; Pryde D. C.; Thomson N. M.; Blake A. J. A tandem radical macrocyclisation-transannular cyclization approach towards the taxanes. J. Chem. Soc. Perkin Trans. 1 1998, 3181–3206. [Google Scholar]
- Kitagawa I.; Tsujii S.; Fujioka K.; Kajiwara A.; Yamamoto Y.; Shibuya H. Chemical transformation of terpenoids. VI: Syntheses of chiral segments, key building-blocks for the left half of taxane-type diterpenoids. Chem. Pharm. Bull. 1984, 3241294–1302. [Google Scholar]
- Swindell C. S.; Fan W. Taxane Synthesis through Intramolecular Pinacol Coupling at C-1-C-2. Highly Oxygenated C-Aromatic Taxanes. J. Org. Chem. 1996, 61, 1109–1118. [Google Scholar]
- Martelli C.; Alderighi D.; Coronnello M.; Dei S.; Frosini M.; Le Bozec B.; Manetti D.; Neri A.; Romanelli M. N.; Salerno M.; Scapecchi S.; Mini E.; Sgaragli G.; Teodori E. N,N-bis(cyclohexanol)amine aryl esters: A new class of highly potent transporter-dependent multidrug resistance inhibitors. J. Med. Chem. 2009, 52, 807–817. [DOI] [PubMed] [Google Scholar]
- Kessel D. Exploring multidrug resistance using rhodamine 123. Cancer Commun. 1989, 1, 145–149. [PubMed] [Google Scholar]
- Aller S. G.; Yu J.; Ward A.; Weng Y.; Chittaboina S.; Zhuo R.; Harrell P. M.; Trinh Y. T.; Zhang Q.; Urbatsch I. L.; Chang G. Structure of P-glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science 2009, 323, 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.