

Abstract

Boceprevir (SCH 503034), 1, a novel HCV NS3 serine protease inhibitor discovered in our laboratories, is currently undergoing phase III clinical trials. Detailed investigations toward a second generation protease inhibitor culminated in the discovery of narlaprevir (SCH 900518), 37, with improved potency (∼10-fold over 1), pharmacokinetic profile and physicochemical characteristics, currently in phase II human trials. Exploration of synthetic sequence for preparation of 37 resulted in a route that required no silica gel purification for the entire synthesis.
Keywords: Hepatitis C virus NS3 serine protease inhibitor, α-ketoamide, narlaprevir, SCH 900518
Hepatitis C virus (HCV) infection is a global health crisis leading to liver cirrhosis, hepatocellular carcinoma and liver failure in humans.1 An estimated 3% of the human population worldwide is chronically infected with HCV.2 Currently the only available treatment regimens are subcutaneous α-interferon or long-acting pegylated-interferon, alone or in combination with oral antiviral agent ribavirin.3 The approved therapy is still far from ideal for the hard to treat genotype-1 patients4 and is frequently accompanied by adverse side effects. There is an unmet medical need to discover new, more effective and tolerable regimens for the treatment of HCV infection. Advances in the understanding of molecular pathways of HCV replication have resulted in several small molecule direct-acting antivirals entering clinical trials in the past few years.
Since identification of this virus, the NS3 serine protease contained within the N-terminal region of the NS3 protein has been studied extensively.5 This chymotrypsin-like serine protease plays a pivotal role in viral replication and, therefore, is an attractive target for HCV antiviral therapeutics.6,7 Intense efforts were focused in the past decade to discover novel small molecule agents that inhibit NS3 serine protease.8 Proof of concept studies in humans with BILN 2061, a noncovalent P1−P3 macrocyclic inhibitor, validated this hypothesis.9 Since then, several NS3 protease inhibitors have progressed to human clinical trials. Currently the most advanced among those are boceprevir (SCH 503034), 1,10,11 and telaprevir (VX950),12,13 from the slow-binding reversible α-ketoamide class, in phase III human evaluation. Inhibitors in phase II studies, from the structurally distinct noncovalent macrocyclic class, include ITMN-191,14 TMC-435350,15 and MK-7009 (P2−P4 macrocycle).16 Other NS3 protease inhibitors currently in clinical evaluation (structure not yet disclosed) include BI-201335, ABT-450, PHX-1766, ACH-1625 and VX-813.8
Inhibitor 1 exhibited Ki* = 14 nM in the enzyme binding assay,17 EC90 = 350 nM in the cell-based replicon assay,18 and acceptable pharmacokinetic profile in rats and dogs (Figure 1). In our efforts to discover a second generation HCV protease inhibitor, we focused mainly on improving the in vitro potency and preclinical pharmacokinetic profile of the inhibitor, specifically exposure in monkeys. Furthermore, to alleviate possible synthetic/purification issues that could arise during development, it was required to identify a molecule that existed as a single isomer, unlike 1, which was a mixture of diastereomers at P1.
Figure 1.

Profile of 1, and SAR toward new P4 moiety.
Based on the X-ray crystal structure of 1 bound to the NS3 protease, exploration of the P4 area seemed attractive, since additional interaction with the enzyme could potentially improve potency. Previously, we explored introduction of carbamate, keto, ester, imide or sulfonamide capped P4 moieties onto the core of 1.8,19−22 While we were able to improve the replicon potency and rat pharmacokinetic properties in most cases, monkey exposure turned out to be a challenge. However, a cyclohexyl moiety at P4 with an appended tert-butyl sulfone group provided encouraging results (Figure 1). Thus, inhibitor 3 exhibited improved replicon potency (EC90 = 100 nM) and displayed good monkey plasma exposure (AUC = 3.2 μM·h, 3 mpk, po). SAR studies toward the discovery of sulfone containing P4 cap is being communicated separately. Improvement in potency and monkey exposure in this series was accompanied with disproportionate loss in rat PK. Herein, we describe our efforts in the P4 sulfone capped series that culminated in the discovery of 37 (narlaprevir, SCH 900518), our second generation HCV NS3 serine protease inhibitor currently in phase II studies.
Synthesis of the sulfone capped cyclohexyl P4 moiety and further processing to target structures are shown in Scheme 1. Alkylation of ester 4 with benzyloxymethyl chloride afforded 5. Removal of the protecting group and subsequent activation of the alcohol provided mesylate 6. Displacement of the mesylate group with sodium tert-butylthiolate resulted in thioether 7. Hydrolysis of the ester moiety, followed by oxidation of the thioether, provided 8. Curtius rearrangement of acid 8 resulted in isocyanate 9, which was reacted with previously described intermediates of type 10,11 followed by Dess−Martin oxidation to provide target compounds of type 11 as a mixture of P1 diastereomers. Alternatively, isocyanate 9 was reacted with previously described P3−P2 ester 12.11 Hydrolysis of the resultant ester to acid 13, and subsequent coupling with P1−P′ intermediate of type 14, followed by Dess−Martin oxidation provided the target compounds. HPLC separation of the mixture afforded the required (S)-P1 diastereomer of type 15.
Scheme 1.

Synthesis of representative P1−P′ moiety is shown in Scheme 2. Thus, reduction of L-norleucine 16 followed by amino protection provided 17. Mild oxidation conditions employing bleach/TEMPO resulted in aldehyde 18. Passerini reaction of 18 with cyclopropylisonitrile 19 afforded the protected intermediate 20. Sequential deprotection unraveled the required P1−P′ intermediate 21 in approximately 60% overall yield.
Scheme 2.

Our efforts toward an improved synthesis of target compounds, specifically 37, are shown in Scheme 3. While the synthetic sequence (Scheme 1) described earlier was useful for SAR studies, we were challenged to develop a better route that was amenable for scale-up. Lewis acid mediated alkylation of 4 with thioether 30 provided 7 in one step. Hydrolysis of the methyl ester to acid 31, followed by Oxone oxidation, afforded sulfone 8 in 62% yield (3 steps). Treatment of isocyanate 9 described above, with L-tert-leucine 32, in a biphasic medium gave 33 which crystallized from ethyl acetate. Acid 33 was coupled with dimethylcyclopropylproline methyl ester 34, resulting in the P4−P2 methyl ester 35. Hydrolysis of 35 afforded the fully assembled P4−P2 acid 13, after crystallization, in 86% yield. The two advanced intermediates, P4−P2 acid 13 and P1−P′ amine hydrochloride 21, were coupled employing essentially conditions described above. After a brief study, we identified appropriate conditions for the final oxidation. Thus, intermediate 36 was oxidized using bleach/TEMPO under buffered conditions. The product obtained after workup was crystallized from acetone−water mixture to provide 37 in high yield. It is noteworthy to mention that the entire sequence for the synthesis of 37 was carried out without the need for silica gel purification, while maintaining the stereochemical integrity at the P1 center.
Scheme 3.

Target compounds synthesized in these studies were tested for HCV NS3 serine protease inhibition using the continuous spectrophotometric assay,17 and inhibition of HCV replication in vitro using the subgenomic replicon assay.18 All compounds were tested for inhibition of human neutrophil elastase (HNE) as a measure of selectivity. Selected compounds were then subjected to in vivo studies.
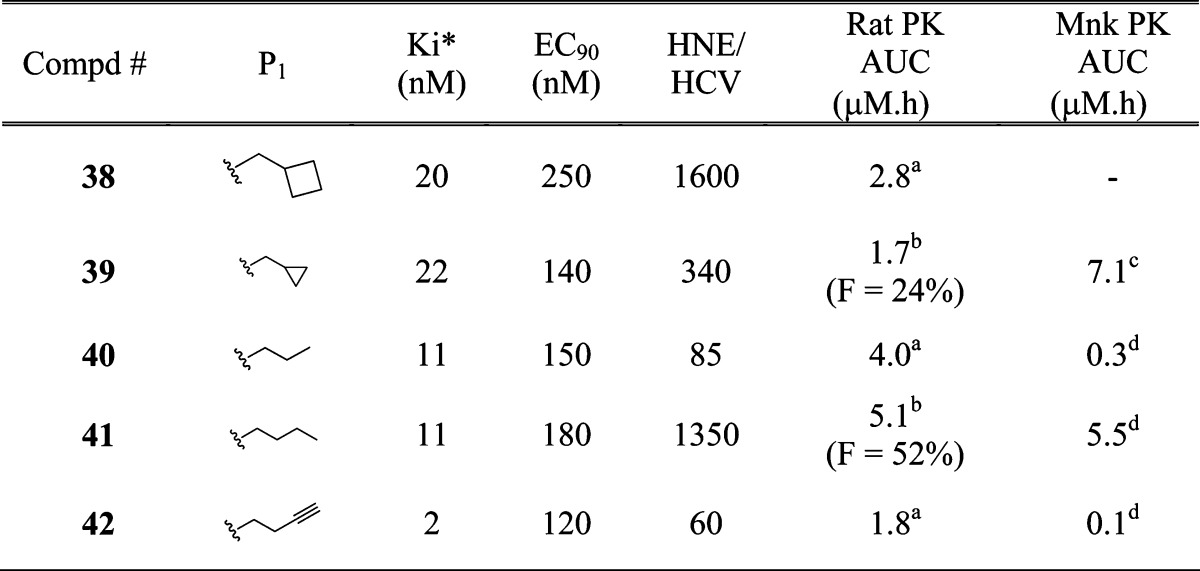
As described above, inhibitor 3, with tert-butyl sulfone capped cyclohexyl moiety at P4 exhibited improved replicon potency and monkey oral exposure compared to 1. However, this series of inhibitors containing P′ primary amide residue displayed poor rat PK profile. During our studies that led to the discovery of 1, it was observed that P′ secondary amides, specifically allyl amides, provided inhibitors with good rat PK profile. In order to improve rat plasma exposure of P4 sulfone capped inhibitors, we then set out to introduce allyl residue at the P′ position. We also knew from our previous studies that P1 cyclobutyl alanine moiety, in general, was not tolerated with allyl (or small alkyl) residue at P′ position. Hence, introduction of P′ allyl amide was carried out with various P1 surrogates to identify the appropriate P1−P′ combination (Table 1).
Table 1. P1 SAR of Inhibitors of Type 11.
 |
po (10 mpk).
iv/po (2/10 mpk).
iv/po (1/3 mpk).
po (3 mpk). Vehicle: po, 0.4% HPMC or 20% HPBCD; iv, 40% HPBCD.
Synthesis of P′ allyl amide containing inhibitor corresponding to 3 afforded 38. While 38 lost replicon potency compared to 3, there was a measurable improvement in rat oral exposure (AUC = 2.8 μM·h). Inhibitor 39 containing P1 cyclopropyl alanine moiety exhibited good replicon potency (EC90 = 140 nM) and good rat and monkey PK (AUC = 1.7 and 7.1 μM·h, respectively). Inhibitors 40 and 42 showed similar profile; good potency and rat PK, with suboptimal monkey AUC and selectivity. Inhibitor 41 containing norleucine P1 moiety demonstrated good overall profile, with EC90 = 180 nM, good selectivity (HNE/HCV = 1350), and oral exposure in rodent (AUC = 5.1 μM·h) and large animal (AUC = 5.5 μM·h). Based on the potency, selectivity and pharmacokinetic profile, including higher rat oral bioavailability, we decided to continue further studies with norleucine moiety at P1.
While exploring allyl residue as an appropriate P′ substituent to improve rat pharmacokinetic profile of the inhibitors, we were cognizant of the fact that a terminal double bond may potentially be a toxicological liability. In order to assess this potential liability, an analogue (structure not shown) containing an allyl residue at P′ was subjected to rat in vivo study. Results indicated that minor amounts of hydroxy-glutathione (GSH) adduct were formed. This observation raised the possibility of in vivo formation of a reactive epoxide from the terminal olefin, which would be highly undesirable and would have toxicological concerns. Thus, our next task was to find a suitable replacement for the allyl moiety. Furthermore, inhibitors described above were all diastereomeric mixtures at P1, similar to 1. A key requirement that we had set forth was identification and development of an appropriate inhibitor as a single isomer. Hence we conducted further SAR studies on the P′ region using norleucine P1 moiety as a single isomer (S-stereochemistry). Results of the P′ SAR studies are shown in Table 2.
Table 2. P′ SAR of Inhibitors of Type 15.
 |
po (10 mpk).
iv/po (4/10 mpk).
po (3 mpk).
iv/po (1/3 mpk). Vehicle: po, 0.4% HPMC or 20% HPBCD; iv, 40% HPBCD.
In our efforts to replace the allyl moiety, we introduced a methyl group at P′ resulting in compound 43, with desirable enzyme (Ki* = 4 nM) and replicon potency (EC90 = 55 nM), and very good selectivity (HNE/HCV = 1500). While the rat AUC was reasonable (2.2 μM·h) for 43, monkey AUC was rather low (0.4 μM·h). Extending the chain to P′ ethyl moiety provided 44 with slight loss in potency and selectivity, albeit moderate improvement in rat AUC. n-Propyl, 45, and cyclopropylmethyl analogue, 46, exhibited similar potency (EC90 = 100 nM). Introduction of cyclopropyl moiety at P′ afforded inhibitor 37, with significantly improved overall profile. Thus, 37 showed excellent potency (EC90 = 40 nM), ∼10-fold improvement over compound 1. Moreover, rat and monkey exposure were also superior to 1, with AUC = 6.5 and 1.1 μM·h, respectively. Increasing the P′ ring size to cyclobutyl (47) and cyclopentyl moiety (48) resulted in gradual loss in enzyme potency. Similarly, substitution of the cyclopropyl ring proved to be detrimental (49, Ki* = 300 nM). While ether containing P′, 50, exhibited good replicon potency (EC90 = 80 nM), the sulfone containing inhibitor, 51, showed no improvement in potency compared to 1. Aromatic and heteroaromatic substitutions at P′ position (inhibitors 52−61) displayed good replicon potency, except 53 and 57. Thiazole containing inhibitor 56 exhibited excellent potency (Ki* = 2.7 nM, EC90 = 40 nM, ∼10-fold improvement over 1) and selectivity (HNE/HCV = 1200). While monkey exposure for 56 was significantly improved (AUC = 1.9 μM·h), rat PK (AUC = 1.2 μM·h) was similar to that of 1. Amino acid derivatives as P′ surrogates (inhibitors 62−66) resulted in loss in potency, with the exception of 66 (EC90 = 50 nM). Unfortunately, 66 displayed no plasma exposure on oral dosing in rats.
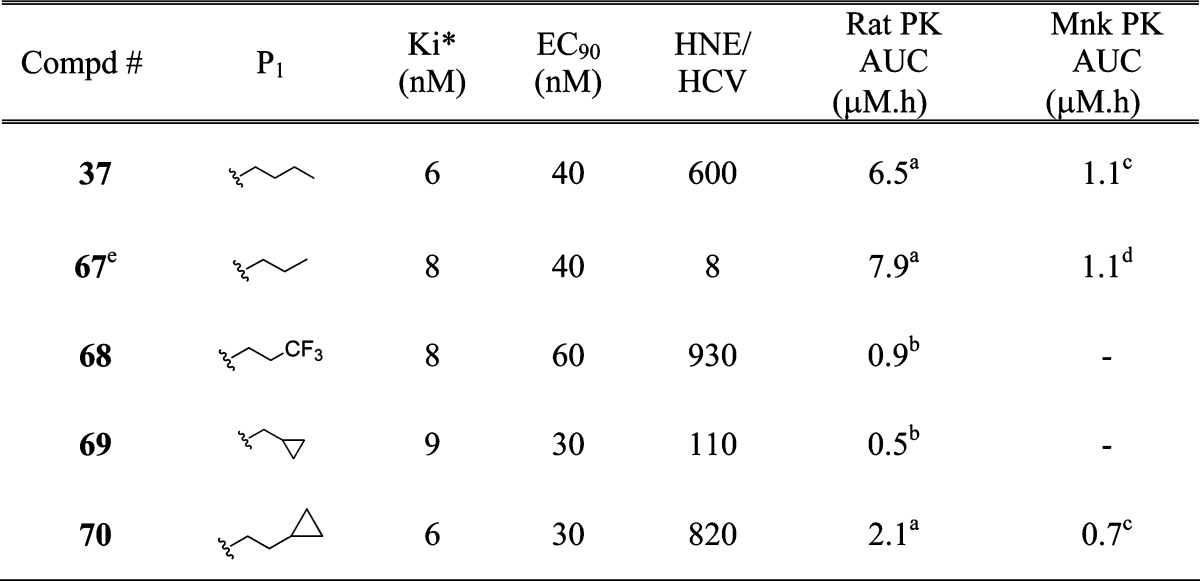
From these explorations, it was clear that the cyclopropyl moiety was the best P′ residue (37), providing a superior overall profile compared to 1. Results from Table 1 demonstrated that P1 variations could have a beneficial influence on the potency profile of the inhibitor. Having identified the appropriate P′ moiety, we then investigated the P1 area in order to find the most suitable combination (Table 3). Inhibitor 67, with norvaline P1, exhibited similar potency and PK profile as 37. Unfortunately, the selectivity (HNE/HCV) was poor. Introduction of trifluoronorvaline P1 residue, expected to improve the selectivity based on our previous studies, resulted in inhibitor 68. A measurable enhancement in selectivity was observed for 68, albeit with less than acceptable rat exposure. While P1 cyclopropylalanine containing inhibitor 69 was equipotent to 37, rat exposure and selectivity were not optimal. Incorporation of homocyclopropylalanine P1 residue, expected to enhance selectivity, resulted in inhibitor 70 with EC90 = 30 nM, good selectivity (HNE/HCV = 820) and monkey exposure similar to 37.
Table 3.
 |
iv/po (2 or 4/10 mpk).
po (10 mpk).
iv/po (1/3 mpk).
po (3 mpk).
P3 = β-MeChg. Vehicle: po, 0.4% HPMC or 20% HPBCD; iv, 40% HPBCD.
Thus, our efforts led to the identification of two promising compounds, 70 and 37, with desirable characteristics. While both the inhibitors were essentially equipotent, 37 demonstrated a better overall pharmacokinetic profile. When dosed orally in rats, 37 displayed very good AUC of 6.5 μM·h, and 46% bioavailability. 37 was well distributed in the target organ; rat liver concentration at 8 h post dosing (C8h) was 750 ng/g. In a similar rat in vivo study, 70 exhibited lower AUC and oral bioavailability (2.1 μM·h, and 21%), and liver C8h was only 90 ng/g. Dog oral exposure for 37 was moderate (AUC = 0.9 μM·h, F = 29%). The corresponding data for 70 were slightly lower, AUC = 0.5 μM·h and F = 16%. More importantly, in monkeys inhibitor 37 displayed an AUC of 1.1 μM·h, with 46% oral bioavailability, a significant improvement over 1. This improvement, while not apparent based on the structural features of 37, could have possibly occurred through some form of transport mechanism that was specific to monkeys. Based on the aforementioned pharmacokinetic profile in three species, inhibitor 37 was considered for further evaluation.
Preclinical resistance studies indicated that 37 was cross-resistant to mutations raised against 1, in both enzymatic and replicon assays. However, 37 retained more activity against many of the mutants due to its higher intrinsic potency compared to 1.23 Inhibitor 37 displayed no significant issues in the in vitro CYP or hERG assay. Unlike an early compound containing P′ allyl moiety, no glutathione or acyl glucuronide conjugates were observed with 37 in rats, dogs and monkeys, thus indicating no reactive metabolite issue. Plasma protein binding data (in vitro, hu) were essentially similar for both 1 and 37. Thus, based on all the preclinical investigations, and having achieved improvements in potency (∼10-fold over 1), pharmacokinetic profile (improved monkey and rat exposure), and physicochemical characteristics (crystalline, single isomer), inhibitor 37 was advanced as development candidate.
Binding of 37 to the active site was confirmed by X-ray crystal structure of the protease bound complex (Figure 2). As disclosed previously, the core (P3−P′) region of the inhibitor 37 was bound to the protease via a series of hydrogen bonding and hydrophobic interactions.11 The newly introduced P4 cyclohexyl moiety was positioned appropriately for additional hydrophobic contact with the enzyme S4 pocket. Moreover, one of the P4 sulfone oxygen atoms was in close proximity for hydrogen bonding interaction with Cys-159. These two additional favorable interactions with the enzyme resulted in improved potency.
Figure 2.
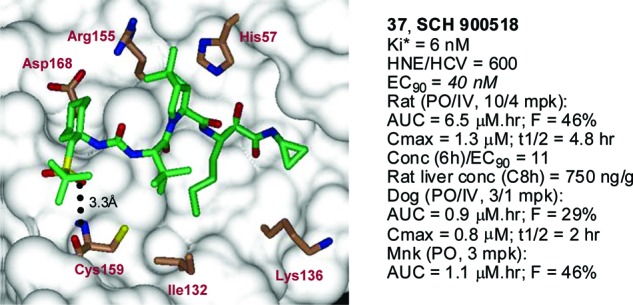
NS3 protease bound X-ray structure and profile of 37.
In conclusion, our efforts toward identification of a suitable second generation HCV NS3 protease inhibitor were initiated based on the P4 sulfone capped inhibitor, 3, exhibiting good replicon potency, good monkey exposure but low rat PK. Introduction of P′ allyl amide moiety, with concomitant P1 modifications, resulted in inhibitor 41, with enhanced rat exposure, while retaining monkey PK. Since replacement of allyl moiety was necessary to alleviate any potential toxicological issues, concerted efforts were directed at identification of a suitable nonolefin containing P′ residue. These investigations were carried out on P1 norleucine containing inhibitor, as a single isomer. P′ cyclopropyl amide turned out to be the optimal group giving rise to compound 37, with ∼10-fold improvement in replicon potency over 1. Furthermore, 37 displayed very good rat exposure, and significantly improved monkey exposure in comparison with 1. Inhibitor 37 was well distributed in the target organ (rat liver C8h/EC90 ∼25). X-ray crystal structure of the inhibitor (37)−protease complex revealed the importance of P4 sulfone capped cyclohexyl moiety in improving the potency. Optimization of the synthetic sequence resulted in a route that did not require any silica gel purification for the entire synthesis, with 37 being obtained in a crystalline form, thus solving a key developmental requirement. Having achieved all the criteria set forth for a suitable second generation HCV NS3/4A serine protease inhibitor, 37 was advanced for further toxicological studies and was found to be safe.24 Inhibitor 37 is currently undergoing phase II clinical studies in humans.
Acknowledgments
We thank Structural Chemistry Department at Schering-Plough Research Institute for NMR and MS analysis for all new compounds.
Supporting Information Available
Experimental details of synthetic procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
†Coordinates for 37 have been deposited with the RCSB Protein Data Bank under the code 3LON.
Supplementary Material
References
- Cohen J. The scientific challenge of hepatitis C. Science 1999, 285, 26–30. [DOI] [PubMed] [Google Scholar]
- Lavanchy D. The global burden of hepatitis C. Liver Int. 2009, 29s174–81. [DOI] [PubMed] [Google Scholar]
- Dymock B. W. Emerging therapies for hepatitis C infection. Emerging Drugs 2001, 6113–42. [DOI] [PubMed] [Google Scholar]
- Feld J. J.; Hoofnagle J. H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005, 436, 967–972. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A.; Kadow J. F.; Scola P. M. Progress toward the discovery and development of specifically targeted inhibitors of hepatitis C virus. Annu. Rep. Med. Chem. 2009, 44397–440. [Google Scholar]
- Kolykhalov A. A.; Mihalik K.; Feinstone S. M.; Rice C. M. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 2000, 74, 2046–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R.; Lohmann V. Replication of hepatitis C virus. J. Gen. Virol. 2000, 81, 1631–1648. [DOI] [PubMed] [Google Scholar]
- Chen K. X.; Njoroge F. G. A review of HCV protease inhibitors. Curr. Opin. Invest. Drugs 2009, 108821–837. [PubMed] [Google Scholar]
- Lamarre D.; Anderson P. C.; Bailey M.; Beaulieu P.; Bolger G.; Bonneau P.; Bös M.; Cameron D. R.; Cartier M.; Cordingley M. G.; Faucher A.-M.; Goudreau N.; Kawai S. H.; Kukolj G.; Lagacé L.; LaPlante S. R.; Narjes H.; Poupart M.-A.; Rancourt J.; Sentjens R. E.; George T, S.; Simoneau B.; Steinmann G.; Thibeault D.; Tsantrizos Y. S.; Weldon S. M.; Yong C.-L.; Llinàs-Brunet M. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003, 186–189. [DOI] [PubMed] [Google Scholar]
- Zeuzem S.; Sarrazin C.; Rouzier R.; Tarral A.; Brion N.; Forestier N.; Gupta S.; Deckman D.; Fellows K.; Hussain M.; Cutler D.; Zhang J. In 56th Annual Meeting of AASLD, San Francisco, CA, 2005. [Google Scholar]
- Venkatraman S.; Bogen S. L.; Arasappan A.; Bennett F.; Chen K.; Jao E.; Liu Y.-T.; Lovey R.; Hendrata S.; Huang Y.; Pan W.; Parekh T.; Pinto P.; Popov V.; Pike R.; Ruan S.; Santhanam B.; Vibulbhan B.; Wu W.; Yang W.; Kong J.; Liang X.; Wong J.; Liu R.; Butkiewicz N.; Chase R.; Hart A.; Agrawal S.; Ingravallo P.; Pichardo J.; Kong R.; Baroudy B.; Malcolm B.; Guo Z.; Prongay A.; Madison V.; Broske L.; Cui X.; Cheng K.-C.; Hsieh T. Y.; Brisson J.-M.; Prelusky D.; Korfmacher W.; White R.; Bogdanowich-Knipp S.; Pavlovsky A.; Bradley P.; Saksena A. K.; Ganguly A.; Piwinski J.; Girijavallabhan V.; Njoroge F. G. Discovery of (1R,5S)-N-[3-Amino-1-(cyclobutylmethyl)-2−3-dioxopropyl]-3-[2(S)-[[[(1,1-dimethylethyl)-amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2(S)-carboxamide (SCH 503034), a selective, potent, orally bioavailable, hepatitis C virus NS3 protease inhibitor: A potential therapeutic agent for the treatment of hepatitis C infection. J. Med. Chem. 2006, 49, 6074–6086. [DOI] [PubMed] [Google Scholar]
- Reesink H. W.; Zeuzem S.; Weegink C. J.; Forestier N.; van Vilet A.; Van de Wetering de Rooij J.; McNair L.; Purdy S.; Kauffmann R.; Alam J.; Jansen P. L. Rapid decline of viral RNA in hepatitis C patients treated with VX-950: A phase 1b placebo-controlled randomized study. Gastroenterology 2006, 131, 997–1002. [DOI] [PubMed] [Google Scholar]
- Perni R. B.; Almquist S. J.; Byrn R. A.; Chandorkar G.; Chaturvedi P. R.; Courtney L. F.; Decker C. J.; Dinehart K.; Gates C. A.; Harbeson S. L.; Heiser A.; Kalkeri G.; Kolaczkowski E.; Lin K.; Luong Y.-P.; Rao B. G.; Taylor W. P.; Thomson J. A.; Tung R. D.; Wei Y.; Kwong A. D.; Lin C. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob. Agents Chemother. 2006, 50, 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiwert S. D.; Andrews S. W.; Jiang Y.; Serebryany V.; Tan H.; Kossen K.; Rajagopalan P. T. R.; Misialek S.; Stevens S. K.; Stoycheva A.; Hong J.; Lim S. R.; Qin X.; Rieger R.; Condroski K. R.; Zhang H.; Do M. G.; Lemieux C.; Hingorani G. P.; Hartley D. P.; Josey J. A.; Pan L.; Beigelman L.; Blatt L. M. Preclinical characteristics of the hepatitis C virus NS3/4A protease inhibitor ITMN-191 (R7227). Antimicrob. Agents Chemother. 2008, 52124432–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raboisson P.; de Kock H.; Rosenquist A.; Nilsson M.; Salvador-Oden L.; Lin T.-I.; Roue N.; Ivanov V.; Wahling H.; Wickstrom K.; Hamelink E.; Edlund M.; Vrang L.; Vendeville S.; Van de Vreken W.; McGowan D.; Tahri A.; Hu L.; Boutton C.; Lenz O.; Delouvroy F.; Pille G.; Surleraux D.; Wigerinck P.; Samuelsson B.; Simmen K. Structure-activity relationship study on a novel series of cyclopentane-containing macrocyclic inhibitors of the hepatitis C virus NS3/4A protease leading to the discovery of TMC435350. Bioorg. Med. Chem. Lett. 2008, 18, 4853–4858. [DOI] [PubMed] [Google Scholar]
- Liverton N. J.; Carroll S. S.; DiMuzio J.; Fandozzi C.; Graham D. J.; Hazuda D.; Holloway M. K.; Ludmerer S. W.; McCauley J. A.; McIntyre C. J.; Olsen D. B.; Rudd M. T.; Stahlhut M.; Vacca J. P. MK-7009: A potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob. Agents Chemother. 2010, 541305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Beyer B. M.; Durkin J.; Ingram R.; Njoroge F. G.; Windsor W. T.; Malcolm B. A. A continuous spectrophotometric assay for the hepatitis C virus serine protease. Anal. Biochem. 1999, 270, 268–275. [DOI] [PubMed] [Google Scholar]
- Lohmann V.; Körner F.; Koch J.-O.; Herian U.; Theilmann L.; Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [DOI] [PubMed] [Google Scholar]
- Arasappan A.; Padilla A. I.; Jao E.; Bennett F.; Bogen S. L.; Chen K. X.; Pike R. E.; Sannigrahi M.; Soares J.; Venkatraman S.; Vibulbhan B.; Saksena A. K.; Girijavallabhan V.; Tong X.; Cheng K.-C.; Njoroge F. G. Toward second generation hepatitis C virus NS3 serine protease inhibitors: Discovery of novel P4 modified analogues with improved potency and pharmacokinetic profile. J. Med. Chem. 2009, 52, 2806–2817. [DOI] [PubMed] [Google Scholar]
- Bogen S. L.; Pan W.; Ruan S.; Nair L. G.; Arasappan A.; Bennett F.; Chen K. X.; Jao E.; Venkatraman S.; Vibulbhan B.; Liu R.; Cheng K.-C.; Guo Z.; Tong X.; Saksena A. K.; Girijavallabhan V.; Njoroge F. G. Toward the back-up of boceprevir (SCH 503034): Discovery of new extended P4-capped ketoamide inhibitors of hepatitis C virus NS3 serine protease with improved potency and pharmacokinetic profiles. J. Med. Chem. 2009, 52, 3679–3688. [DOI] [PubMed] [Google Scholar]
- Venkatraman S.; Blackman M.; Wu W.; Nair L.; Arasappan A.; Padilla A.; Bogen S.; Bennett F.; Chen K.; Pichardo J.; Tong X.; Prongay A.; Cheng K.-C.; Girijavallabhan V.; Njoroge F. G. Discovery of novel P3 sulfonamide-capped inhibitors of HCV NS3 protease. Inhibitors with improved cellular potencies. Bioorg. Med. Chem. 2009, 17, 4486–4495. [DOI] [PubMed] [Google Scholar]
- Tong X.; Arasappan A.; Bennett F.; Chase R.; Feld B.; Guo Z.; Hart A.; Madison V.; Malcolm B.; Pichardo J.; Prongay A.; Ralston R.; Skelton R.; Xia E.; Njoroge F. G. Preclinical characterization of SCH 900518, a novel mechanism-based inhibitor of HCV NS3 protease. J. Hepatol. 2009, 50Suppl 1S351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reesink H. W.; Bergmann J. F.; de Bruijne J.; Weegink C. J.; van Lier J.; van Vliet A.; Keung A.; Li J.; O’Mara E.; Treitel M. A.; Hughes E. A.; Janssen H. L. A.; de Knegt R. J. Safety and antiviral activity of SCH 900518 administered as monotherapy and in combination with peginterferon alfa-2b to naive and treatment-experienced HCV-1 infected patients. J. Hepatol. 2009, 50Suppl 1S35–S36. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.