

Abstract
The early occurrence of β‐cell dysfunction has been broadly recognized as a critical determinant of the development and progression of type 2 diabetes. β‐cell dysfunction might be induced by insufficient β‐cell mass, by a dysfunction of the β‐cells, or both. Whether or not β‐cell dysfunction constitutes a cause of reduced β‐cells or vice‐versa currently remains unclear. The results of some studies have measured the loss of β‐cells in type 2 diabetic patients at between 22 and 63% by planimetric measurements. Because β‐cell hypertrophy has been noted in type 2 diabetic patients, the loss of β‐cell number should prove more profound than what has thus far been reported. Furthermore, β‐cell volumes are reduced even in patients with impaired fasting glucose. Such defects in β‐cell mass are associated with increased apoptosis rather than insufficient replication or neogenesis of β‐cells. With these results, although they still require clarification, the peak β‐cell mass might be determined at quite an early stage of life, and then might decline progressively over time as the result of exposure to harmful environmental influences over one’s lifetime. In this review, we have summarized the relevant studies regarding β‐cell mass in patients with type 2 diabetes, and then presented a review of the various causes of β‐cell loss in adults. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2010.00072.x, 2010)
Keywords: Diabetes, β‐cell mass, Hypertrophy
Introduction
Type 2 diabetes is the most common form of diabetes in humans. In the past few decades, the global incidence and prevalence of diabetes has increased dramatically. Additionally, the number of people with diabetes is expected to exceed 350 million individuals in 20251. Even in only one country, China2, the number of diabetic patients has recently approached 90 million individuals, and a comparable number of patients also exists in another Asian countries – namely, India3. The increase in type 2 diabetes in Asia differs from that reported in other regions of the world; it has evolved over a much shorter time, in a younger age group and in people with substantially lower body mass indices (BMI)4. Because the degree of obesity and aging is closely related to the degree of insulin resistance, insulin secretory defects might carry out a more important function in the development and progression of diabetes in our region.
β‐cell dysfunction might be induced by the loss of β‐cell mass, by functional defects in β‐cells, or both. Reductions in β‐cell mass and β‐cell dysfunction have both been shown in patients with type 2 diabetes5. Whether or not β‐cell dysfunction is the cause of reduced β‐cell mass or vice‐versa remains to be determined. We will focus on the β‐cell mass of people with type 2 diabetes.
A genetic element clearly underlies β‐cell dysfunction and insufficient β‐cell mass; however, a number of modifiable factors are also linked to β‐cell deterioration, most notably chronic hyperglycemia and elevated free fatty acid (FFA) levels. Evidence has also been found for a link between increased pro‐inflammatory cytokines and the impairment of insulin‐signaling pathways in the β‐cells, as well as the potential roles of islet amyloid deposition and fibrotic islet destruction.
In the present review, we provide an overview of the characteristic features and underlying pathogenesis of β‐cell mass defects in patients with type 2 diabetes mellitus, and then describe the morphological characteristics of the pancreatic islets. Finally, we address the pathogenic mechanisms and possible clinical implications of preventing β‐cell mass destruction in the prevention and delay of the progression of this disease.
β‐Cell Mass: Facts and Unresolved Questions
β‐Cell Mass in Diabetic Patients
β‐cell mass is determined as the sum of replication, neogenesis and hypertrophy minus the rate of apoptosis. Normally, obesity, pregnancy and increases in insulin resistance are the principal causes of β‐cell mass increases, through enhanced replication, neogenesis and hypertrophy. However, the progression from an insulin resistance condition to diabetes is inevitably associated with β‐cell dysfunction and reduced β‐cell mass5,6. Many previous studies have reported that β‐cell mass in type 2 diabetic subjects tends to be reduced relative to normal subjects. Saito et al.7 reported previously that the total islet number was approximately 30% lower in subjects with type 2 diabetes relative to the non‐diabetic subjects. In the same year, Westermark and Wilander8 also noted a 30% reduction in the total islet volumes of diabetic subjects. The islet volume of the diabetic patients was 1.01 ± 0.12 cm3 and that of the non‐diabetic patients was 1.60 ± 0.16 cm3. In 2002, Sakuraba et al.9 reported a 22–30% reduction in β‐cell volume, as well as a 22% reduction in the quantity of islets in Japanese type 2 diabetic patients. In 2003, Butler et al. reported that obese humans with type 2 diabetes evidenced a 63% deficit in relative β‐cell volume relative to non‐diabetic obese subjects, although the relative β‐cell volumes were increased in obese vs lean non‐diabetic cases. Lean subjects with type 2 diabetes also evidenced a 41% deficit in relative β‐cell volumes10. In 2003, we also showed that β‐cell mass was reduced in Korean type 2 diabetes patients. The mean relative volume of β‐cells was reduced by approximately 50% relative to normal subjects (Figure 1)11. Recently, Rahier et al.12 clearly showed that β‐cell mass decreased by approximately 39%, occurring similarly in the body and tail of the pancreatic islets in European subjects with type 2 diabetes. All together, we could agree that 30–60% of β‐cell mass was decreased in patients with type 2 diabetes mellitus and we also observed unexpected broad heterogeneity of β‐cell mass in each patient in many studies.
Figure 1.
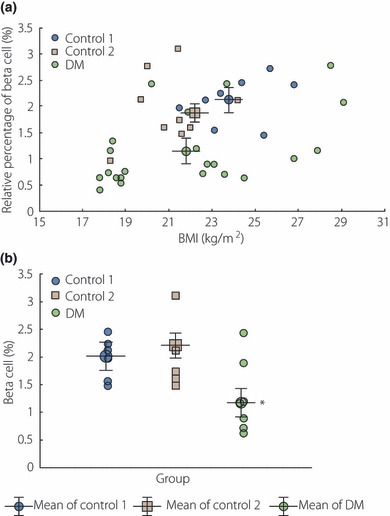
(a) The body mass index (BMI) and β‐cell mass were linearly correlated in control group 1 (r2 = 0.64, P = 0.003) and diabetic patients (r2 = 0.55, P < 0.05). Remarkably, the mean value of the relative volume of β‐cells in diabetic patients was lower than those of other control groups (adapted fromYoon et al.). (b) The pattern of distribution of β‐cells (%) among groups whose BMI ranges were between 21 and 25 kg/m2. The mean value of the relative volume of β‐cells in diabetic patients was lower than those of other control groups (adapted from Yoon et al., Copyright 2003, The Endocrine Society).
Interestingly, a good linear correlation between β‐cell mass and BMI has been reported in normal subjects and type 2 diabetic patients, as well by our group11, and such a trend was also reproduced by another group12. These results suggest appropriate β‐cell mass is critically important to maintain a certain amount of body mass in normal subjects and diabetic patients as well.
The evidence for β‐cell regeneration after various pancreatic injuries occurring in humans is minimal5, and the accelerated loss of β‐cell by apoptosis in diabetic patients had been shown10. A 40% deficit in β‐cell mass was reported even in obese humans with impaired fasting glucose13, which implies that the deficit in the β‐cell volume is an early occurrence in the development of type 2 diabetes and is likely to be of primary importance rather than simply occurring secondary to hyperglycemia. With these results, it has been unclear, yet we could imagine that the peak β‐cell mass could be determined at quite an early stage of life by genetic influences and nutritional status during the intrauterine and/or early postnatal period and progressively declined over time as a result of harmful environments during the lifetime.
β‐Cell Hypertrophy
Recently, we observed hypertrophy of the β‐cells in type 2 diabetic subjects (JH Cho, unpublished data). The average β‐cell size was approximately 30% larger in type 2 diabetic patients compared with normal subjects and the ratio of cytoplasm per nucleus area was also significantly higher in diabetic subjects. As we described earlier, β‐cell volume was significantly reduced in type 2 diabetic subjects. Along with this observation, we must consider the methods of β‐cell volume measurements used in previous studies. The β‐cell volume was assessed by the measurement of total β‐cell area in islets, where each β‐cell size was not considered. Therefore, the increase in each β‐cell size implies that β‐cell number is more reduced even in the same β‐cell volume. Thus, in these cases, not only is the β‐cell volume reduced, but the number of β‐cells is also considerably decreased. For example, if β‐cell size increased by 30% on average, in an individual with reduced β‐cell mass by approximately 50%, β‐cell number would actually be reduced by more than 60%. Reduced β‐cell numbers can lead to more severe compensatory loading onto each β‐cell, and can finally induce accelerated β‐cell loss in people with type 2 diabetes. Actually, Bagust and Beale14 reported a model of decline in β‐cell function combining two phases in which a long, slow, gradual loss of β‐cell function leads to a crisis in metabolic regulation, precipitating a much more rapid decay phase. Therefore, we propose a hypothesis about morphological alterations in pancreatic islets and how β‐cell loss could be more accelerated. First, factors that increase insulin resistance, such as obesity, increased calorie intake and decreased physical activity, could stimulate an increase in β‐cell mass through β‐cell replication, β‐cell neogenesis or β‐cell hypertrophy, maintaining a compensatory phase and normoglycemia. However, a sustained increase in insulin demand could cause β‐cell apoptosis through glucose toxicity, lipid toxicity, chronic inflammation and increased oxidative stress and so on, leading to an uncompensated phase with reduced β‐cell mass and hyperglycemia. An uncompensated phase could progress to an accelerating phase with severe β‐cell loss over time through more reduced β‐cell mass with increased fibrosis and amyloidosis. Furthermore, an increase in α‐cell mass could aggravate hyperglycemia (Figure 2).
Figure 2.
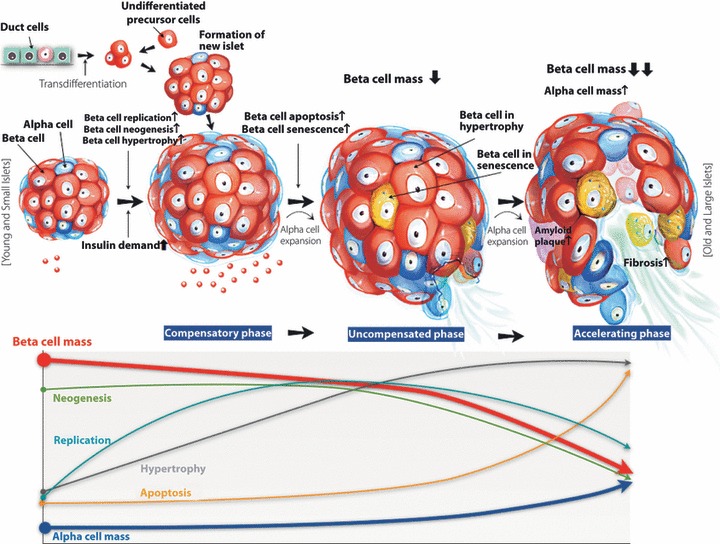
Hypothesis for morphological alterations of islets and β‐cell reductions in diabetic patients. Factors that increase insulin resistance, such as obesity, increased calorie intake and decreased physical activity, could stimulate an increase in β‐cell mass through β‐cell replication, β‐cell neogenesis or β‐cell hypertorphy, maintaining a compensatory phase and normoglycemia. However, a sustained increase in insulin demand could cause β‐cell apoptosis through glucose toxicity, lipid toxicity, and chronic inflammation and increased oxidative stress resulting in an uncompensated phase with reduced β‐cell mass and hyperglycemia. Genetic background or intrauterine nutrition could limit β‐cell expansion. Pharmacological agents, such as sulfonylurea, could play a role to increase β‐cell apoptosis. An uncompensated phase could progress to an accelerating phase with severe β‐cell loss over time through increased fibrosis and amyloidosis, as well as increased β‐cell toxicity as described earlier. Furthermore, an increase in α‐cell mass could aggravate hyperglycemia. Finally, the decline in β‐cell mass caused by apoptosis and increased α‐cell mass could aggravate hyperglycemia in diabetic subjects over time.
β‐Cell Proliferation vs Apoptosis
It is also necessary to identify the factors contributing to the relatively reduced β‐cell mass noted in type 2 diabetes patients. Butler et al. previously determined the frequency of new islet formation from exocrine ducts (neogenesis), as well as β‐cell replication in islets in order to evaluate the compensatory increases in β‐cell mass. There were no differences in the frequency of β‐cell replication and new islet formation between type 2 diabetic and non‐diabetic individuals10. In our preliminary unpublished data, we observed that the contribution of the β‐cell area of single β‐cell units, defined as islets composed of less than three cells and recognized as neogenetic loci15,16, to total β‐cell area tended to be greater in type 2 diabetic cases (10 ± 6%) compared with non‐diabetic subjects (7 ± 5%), although there was no significant difference. These results showed that new islet formation, the predominant input into the β‐cell mass in humans and β‐cell replication, which was relatively low in humans, appeared to be normal or slightly increased even in type 2 diabetic patients. Therefore, we summarize that the major deficit resulting in a reduction in β‐cell mass was related to increased apoptosis. Actually, the frequency of β‐cell apoptosis was increased by 10‐fold in the lean cases and threefold in the obese cases of type 2 diabetes, relative to their respective non‐diabetic control groups10.
Morphological Alterations of Islets in Patients with Type 2 Diabetes
Systemic morphological classification of islets is needed to understand the fate of islet over one’s lifetime. We could classify the observed islets into five different types (types 1, 2a, 2b, 3a and 3b) according to islet size and the β‐cell fraction in the islet (Figure 3). Type 1 consisted of single β‐cell units, defined as islets composed of less than three cells, and were recognized as neogenetic loci described earlier15,16. Type 2 consisted of small islets (smaller than 6415 μm2, which is the median size of islets in normal subjects11). Type 3 consisted of large islets (larger than 6415 μm2). An ‘a’ signified islets with normal β‐cell fractions in the islets (more than 0.64, which was the value for the 75th percentile of the total islets in the control group) and a ‘b’ signified β‐cell‐depleted islets (<0.64). The five types of islets are shown in Figure 3. We also measured the islet size and β‐cell areas of all the islets existing in the slide section randomly selected in five subjects with type 2 diabetes (DM group) and nine normal subjects (control group). From these results, we calculated the contribution rate of the β‐cell area within each islet type to the total β‐cell area. The results are shown in Figure 4. The contribution of the type 1 β‐cell area to the total β‐cell area tended to be higher in the DM group than in the control group (10.2 ± 6.0%vs 7.19 ± 4.98%, respectively), whereas the contribution of type 2a was lower in the DM group than in the control group (36.0 ± 3.51%vs 40.0 ± 11.8%, respectively). The contribution of type 3a was significantly lower in the DM group than in the control group (13.4 ± 6.7%vs 31.3 ± 14.6%, respectively; P = 0.025), whereas the contribution of type 3b was significantly higher in the DM group than in the control group (33.9 ± 4.87%vs 17.3 ± 13.2%, respectively; P = 0.020).
Figure 3.
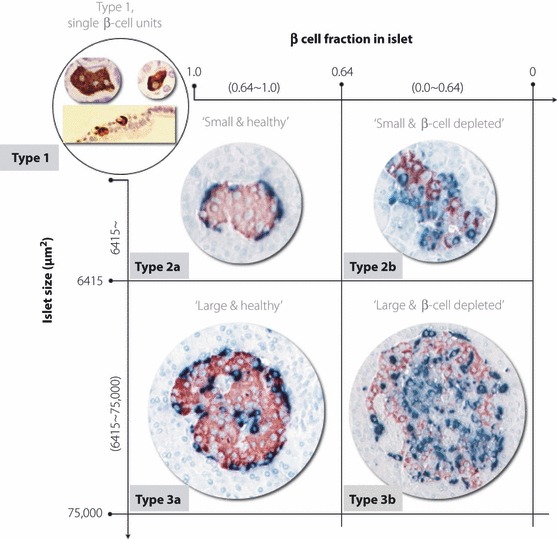
Islet classification according to islet size and β‐cell fraction in the islet: type 1, single β‐cell units or scattered β‐cells; type 2a, small healthy islets; type 2b, small β‐cell‐depleted islets; type 3a, large healthy islets; type 3b, large β‐cell‐depleted islets.
Figure 4.
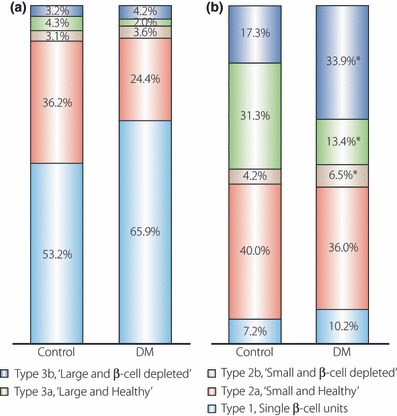
(a) Relative number of each islet type in the total islet number. (b) Relative contribution of the β‐cell area within each islet type to the total β‐cell area. *P < 0.05 between the control and diabetes mellitus (DM) groups.
Increased α‐Cell Ratio and Mass
Interestingly, in contrast to the changes in β‐cell mass, dysregulation of glucagon secretion or the disproportionately increased number of α‐cells relative to β‐cells in these individuals can contribute to hyperglycemia11,17–22. Müller et al.17 showed an unsuppressed glucagon response to a carbohydrate meal in type 2 diabetes. Unger et al. identified relative or absolute hyperglucagonemia in every form of endogeneous hyperglycemia and reported that such a glucagon excess could be a principal factor in the overproduction of glucose in diabetes8. We reported that the α‐cell mass was clearly increased in type 2 diabetic patients relative to the normal subjects. The ratio of α‐cell area to β‐cell area was far more profoundly increased in type 2 diabetic patients (Figure 5)11. As a mechanism of increase in α‐cell mass, O’Reilly et al.23 previously observed α‐cell neogenesis in an animal model. Ellingsgaard et al. reported that α‐cell expansion was regulated by IL‐6 in human islets, which is systemically elevated in obesity and is a predictive factor to developing type 2 diabetes. IL‐6 was associated with α‐cell proliferation and the prevention of α‐cell apoptosis24. We suggest α‐cell neogenesis or replication also could be developed together with β‐cell neogenesis or replication (Figure 2). However, it is not fully understood why α‐cell mass is increased. It is also not known why increased α‐cell mass is closely associated with hyperglucagonemia or unsuppressed glucagon response. So further studies on α‐cell mass and dysfunction are needed.
Figure 5.
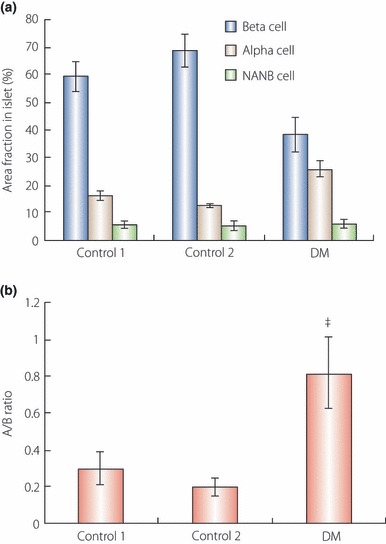
(a,b) Comparisons of the relative volumes of α‐and β‐cells and the A/B ratio (α‐cell area/β‐cell area) in islets among groups, respectively. (a) The β‐cell fraction in the islet area in diabetic patients was significantly decreased compared with other groups (*P < 0.05). but the α‐cell fraction was increased in diabetic patients (†P < 0.05) (adapted from Yoon et al.). Non‐α and non‐β endocrine cell area in islets did not differ between groups. (b) A/B ratio was increased in diabetic patients compared with other groups (‡P < 0.05) (adapted from Yoon et al., Copyright 2003, The Endocrine Society).
Determinant of β‐Cell Mass
Hypothesis: Thrifty Phenotype
The thrifty phenotype hypothesis proposes that there are epidemiological associations between poor intrauterine and early postnatal growth, and the subsequent development of type 2 diabetes25. Since the hypothesis was proposed, many studies have confirmed the initial epidemiological evidence26,27, although the strength of the relationships has varied from one study to another. In considering the downstream effects of poor intrauterine and early postnatal nutrition, poor development of pancreatic β‐cell mass and/or function were key elements linking poor early nutrition to later type 2 diabetes. The hypothesis also proposed that the emergence of pathological changes after undernutrition in early life was critically dependent on the superimposition of other factors, notably obesity, aging and physical inactivity. In other words, a β‐cell dysfunction induced by poor nutrition in early life period could not compensate increased insulin resistance in adult period. Whilst there is now little doubt that indices of poor early growth are linked to increased risk of type 2 diabetes, the extent to which genes or the early environment underlie the relationship remains controversial.
Genetics
Type 2 diabetes has strong genetic components28,29. A great deal of progress has been made in our understanding of the genetics of this disease. In early studies, genetic variants in the peroxisome proliferator‐activated receptor‐γ gene (PPARG)30 and the ATP‐sensitive potassium channel Kir6•2 (KCNJ11) were reproducibly associated with type 2 diabetes31. In Asian populations, the protective effects of the PPARG*A12Ala allele on insulin resistance and the risk of type 2 diabetes were not consistently seen32. Polymorphisms in the gene encoding for transcription factor‐7‐like protein 2 (TCF7L2) were reported in 2006 to be associated with type 2 diabetes33. This gene exerts the strongest effect on type 2 diabetes in Asian populations34. However, different genetic variations in TCF7L2 are associated with type 2 diabetes in Asian populations. Several other genetic variants have been identified through genome‐wide association studies, which involve the genotyping of hundreds of thousands of single‐nucleotide polymorphisms on a single array. Ramachandran et al.35 reported that these variants are associated with type 2 diabetes in different Asian groups, including Chinese, Japanese, Korean and Indian populations. Two recent Japanese genome‐wide association studies replicated several loci that had been identified previously in Europeans, and reported variants in the KCNQ1 gene that were associated with type 2 diabetes in Japanese and other east Asian populations36,37. The majority of genetic variants that were associated with type 2 diabetes appear to be related to insulin secretion rather than to insulin resistance, and several of the risk alleles are associated with reduced β‐cell function (Figure 6)38–41. Maturity Onset Diabetes of the Young (MODY) is a clinically heterogeneous group of disorders characterized by non‐ketotic diabetes mellitus and a group of monogenic diabetes disorders causing 2–5% of cases of type 2 diabetes42. One of these genes encodes the glycolytic enzyme glucokinase (associated with MODY 2)43, and the other five encode transcription factors: hepatocyte nuclear factor (HNF) 4 (associated with MODY 1)44, HNF‐1 (MODY 3)45, insulin promoter factor 1 (IPF‐1 [MODY 4])46, HNF‐1 (MODY 5)47 and neurogenic differentiation factor 1 (NeuroD1), also known as β‐cell E‐box transactivator 2 (BETA2 [MODY 6])48. All these genes are expressed in β‐cells, and mutation of any of them leads to β‐cell dysfunction and diabetes mellitus (Figure 6). The present catalogue of type 2 diabetes risk variants most likely accounts for only a small proportion of the genetic basis of type 2 diabetes. Nevertheless, the identification of these variants has provided us with valuable insights into the pathogenesis of type 2 diabetes.
Figure 6.
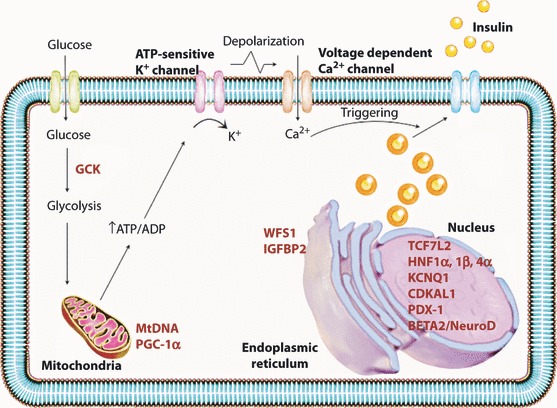
Schematic diagram of the pancreatic β‐cell showing the proposed subcellular localization of proteins encoded by diabetes‐associated genes. HNF4a, HNF1α, HNF1β and TCF7L2 encode transcription factors produced in the β‐cell and are implicated in pancreatic development. WFS1 encodes wolframin, a protein that regulates calcium transport in the endoplasmic reticulum. CDKAL1 and CDKN2A/B are involved in the cyclin‐dependent kinase pathway, and might thus influence β‐cell regeneration.
Causes of β‐Cell Loss in Adults
Glucose Toxicity
The results of several studies have shown that the chronic elevation of blood glucose concentration impairs β‐cell function and insulin sensitivity, a phenomenon referred to as glucotoxicity49. Several mechanisms have been previously reported to increase β‐cell programmed death. One common and powerful mechanism is the activation of oxidative stress as a result of increased mitochondrial generation of reactive oxygen species (ROS), a phenomenon that arises pursuant to excessive glucose metabolism50. The β‐cells are quite sensitive to oxidative stress, owing to very low expression and activity of anti‐oxidant enzymes51. Del Guerra et al.52 have reported that high glucose levels hamper glucose‐stimulated insulin secretion, activate apoptosis, induce alterations in mitochondrial morphology and density volume, and are associated with increased intracellular nitrotyrosine content. Even glucose fluctuations, as they might arise in response to meal ingestion under prediabetic conditions, might accelerate the loss of functional β‐cell mass. Chronic hyperglycemia results in chronic β‐cell stimulation and insulin biosynthesis can induce endoplasmic reticulum (ER) stress53. The ER is responsible for the synthesis, modification and delivery of proteins to their target sites. These processes might be impaired under a variety of physiological and pathological conditions, thereby resulting in ER stress54,55. Marchetti et al.56 recently reported that the β‐cells of type 2 diabetic subjects evidence only modest signs of ER when pancreatic islets are incubated in the presence of normal concentrations of glucose. However, exposure to higher glucose levels induced much more profound increases in ER stress markers relative to control islets. Thus, we could agree that glucose toxicity is one of the most important mechanisms of β‐cell loss in diabetic patients. However, it is difficult to explain the β‐cell loss observed in the prediabetic stage13.
Lipid Toxicity
Some FFA and lipoproteins might exert a pro‐apoptotic effect, whereas others appear to carry out a protective function. Thus, long‐term exposure to saturated fatty acids, such as palmitate, is associated with toxic effects, whereas monounsaturated fatty acids, such as oleate, protect against palmitate‐ and glucose‐induced β‐cell apoptosis57,58. FFA activates β‐cell apoptosis in a caspase‐dependent manner and through the downregulation of Bcl‐2 mRNA expression58. Additionally, FFA‐induced apoptosis is linked to the downregulation of Akt phosphorylation. By way of contrast, Akt activation inhibits the activation of apoptosis machinery, exerting a restraining effect on Bad, cytochrome c release and caspase‐959. Interestingly, a functional defect in Akt has been associated with a predisposition toward β‐cell apoptosis and the development of diabetes in the presence of defective insulin signaling60. FFA also induce NO synthase expression and have been shown to cause a fourfold increase in NO production. Again, the use of inhibitors of inducible NO synthase (iNOS) minimizes losses in insulin secretion61. Lipotoxicity is strictly linked to glucotoxicity and, according to the results reported by Poitout and Robertson62, the former is unlikely to occur in the absence of the latter.
Glucolipotoxicity
The idea that neither glucose nor FFA alone cause β‐cell toxicity is consistent with the ‘glucolipotoxicity’ hypothesis63. In our laboratory, the glucolipotoxicity was associated with the gradual decrease in β‐cell viability and increase in β‐cell death in a time‐dependent manner. When the islets were exposed to glucolipotoxicity, the fold increase of glucose‐stimulated insulin secretion was blunted and insulin gene expression was suppressed (Figure 7). Once chronic hyperglycemia is established, it can affect pancreatic β‐cell function and survival64. However, elevated levels of circulating and intracellular lipids also play an important role in inducing β‐cell dysfunction and decreased β‐cell mass63,65,66. Impaired insulin secretion in vivo coincides with major alterations in carbohydrate and lipid metabolism in β‐cells64. Therefore, stabilization of metabolic changes induced by glucolipotoxicity in β‐cells represents a potential new avenue for the treatment of patients with type 2 diabetes mellitus. Kim et al.67 have reported that PGC‐1α inhibits insulin and BETA2/NeuroD transcription levels and that attenuating PGC‐1α overexpression protects against glucolipotoxicity‐induced β‐cell dysfunction. We suggest that PGC‐1α plays an important key role in intracellular fuel regulation, which could herald a new era in the treatment of patients with type 2 diabetes mellitus by providing protection from glucolipotoxicity, which is an important cause of the development and progression of the disease.
Figure 7.
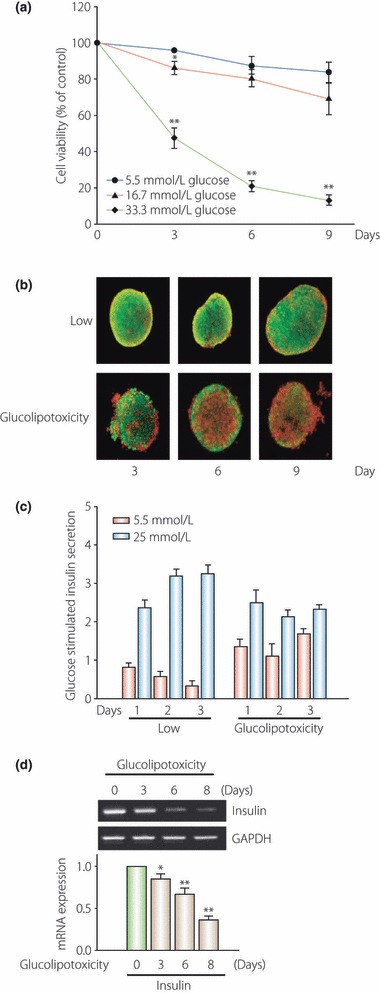
To investigate the effect of glucolipotoxicity on β‐cell dysfunction, isolated islets were incubated with or without 0.6 mmol/L free fatty acid (a mixture of palmitate and oleate) in the presence of a high concentration of glucose (33.3 mmol/L) for 3 days to induce glucolipotoxicity. The glucolipotoxicity was associated with (a) a gradual decrease in β‐cell viability and (b) an increase in β‐cell death in a time‐dependent manner. Under normal conditions, the fold‐increase of glucose‐stimulated insulin secretion is increased. (c) However, when the islets were exposed to glucolipotoxicity, the fold‐increase of glucose‐stimulated insulin secretion was blunted. (d) Insulin gene expression was suppressed in glucolipotoxicity. GAPDH, Glyceraldehyde 3‐phosphate dehydrogenase.
Insulin Resistance in β‐Cells
Insulin resistance is a common pathological state that is associated with many health disorders68. Environmental and physiological stress appears to cause insulin resistance through heterologous signaling cascades. Many studies have shown a variety of factors secreted from adipose tissue that inhibit insulin signaling, FFA, tumor necrosis factor‐alpha (TNF‐α) and resistin, or factors that promote insulin signaling, adipocyte complement‐related protein of 30 kDa (adiponectin) and leptin69. A common theme to explain insulin resistance could emerge when we understand how these diverse signals interface with the insulin signaling cascade. Signaling cascades activated during acute trauma, or chronic metabolic or inflammatory stress dysregulate insulin receptor substrate (IRS) proteins through various mechanisms, including proteasome‐mediated degradation, phosphotyrosine phosphatases and S/T‐phosphorylation69,70. Also, dysregulation of IRS protein signaling could be a common cause of peripheral insulin resistance71. Furthermore, dysregulation of IRS2 in β‐cells shows a mechanism of pancreatic β‐cell failure that contributes to diabetes69,72. Regardless of the complexity, S/T‐phosphorylation of IRS1 and IRS2 provides a plausible framework to understand the loss of compensatory β‐cell function during progressive peripheral insulin resistance.
Amyloid Deposition and Fibrotic Destruction in the Islets of Diabetic Patients
Islet‐amyloid polypeptide (IAPP), also referred to as amylin, is a normal secretory product of the pancreatic β‐cells. A potential role for IAPP in the development of IR, β‐cell dysfunction and type 2 diabetes has been proposed73, although these studies have yielded conflicting and inconclusive results74–76. By way of contrast with studies in which no association was detected between amyloid deposits and the duration of type 2 diabetes77, others have reported an association between deposits and apoptosis, replacement of β‐cell mass and declines in β‐cell function78–80. Some investigators have concluded that up to 90% of patients with type 2 diabetes harbor amyloid deposits in their islets81 and that the degree of amyloidosis is correlated with the duration and severity of the disease82. Human amyloid is toxic to β‐cells83 and contributes to losses of β‐cell mass84. In an animal model, islet amyloidosis evidences diffuse distribution throughout the pancreas, with a progressive diminution in endocrine mass occurring in tandem with increases in amyloid mass. In other words, as the amyloid deposits expand, the β‐cell mass shrinks, thus impairing β‐cell function and inducing glucose intolerance85. Although amylin deposition might be the attractive primary cause of β‐cell loss in diabetic patients, all the data we have so far is not yet clear.
Previously, we proposed that fibrotic islet destruction prominently observed in type 3b of Figure 3 might be one of the more important pathogenic mechanisms underlying diabetic patients’ limited β‐cell proliferation capacity (Figure 2)86. We have determined that pancreatic stellate cells (PSC) are involved in the progression of islet fibrosis in an animal model of type 2 diabetes and, possibly, in humans suffering from type 2 diabetes. Both high concentrations of glucose and insulin in the islets contribute to PSC activation and proliferation in diabetic patients, although the exact mechanisms underlying these effects remain to be confirmed. The results of both in vitro and in vivo studies have shown that angiotensin‐converting enzyme (ACE) inhibitor (ACEi) attenuates the islet destruction caused by fibrosis, and that this attenuation exerts some beneficial effects on glucose tolerance by suppressing the activation and proliferation of PSC87. We suggest that PSC might play an important role in the pathogenesis of fibrotic islet destruction observed in conjunction with type 2 diabetes.
Chronic Inflammation as a Cause of β‐Cell Loss
Schuster88 recently reported that the ability to store and limit fatty acid deposition to adipose tissue is a key component in remaining insulin sensitive, controlling the inflammatory cascade and reducing the risk of developing obesity‐related comorbidities, such as DM. The pancreatic islet could also be a target of inflammation. Inflammation in a tissue is classically defined by tissue damage, impaired function, the presence of increased numbers of immune cells and/or activation of local tissue immune cells, and local production of cytokines and chemokines. An accumulating body of evidence also indicates that this is the case in the human pancreatic islet in type 2 diabetes. The β‐cells of patients with type 2 diabetes express elevated levels of IL‐1β and a variety of chemokines89. Based on tissue histology, islets from type 2 diabetes patients express increased amounts of IL‐1β90, increased caspase‐1 (required for the cleavage of proIL‐1β to active IL‐1β)91 and reduced amounts of IL‐1Ra (the IL‐1 receptor antagonist)92, and are infiltrated with macrophages93,94. Thus, an islet inflammation might be involved in β‐cell compensation for insulin resistance, but if the inflammation were chronic, β‐cell demise would be the expected result.
Sulfonylurea
Glibenclamide and chlorpropamide treatment have been associated with improvements in glycemic control and lower incidence of microangiopathic complications95. Nonetheless, as mentioned previously, improvements in glycemic control were associated with an initial increase of the HOMA‐B, an index of β‐cell function, followed by a progressive linear decline96. Losses of β‐cell mass and function have raised concerns regarding the use of sulfonylureas for the treatment of type 2 diabetes. The results of some previous animal and cell studies have shown that these agents might induce β‐cell apoptosis. Studies carried out in isolated human islets suggest that glibenclamide, but not repaglinide, might activate apoptosis in β‐cells97. Nateglinide at low concentrations was not observed to induce β‐cell apoptosis. The incubation of pancreatic islets and β‐cell cells from ob/ob mice and Wistar rats with glucose and sulfonylureas has also been show to induce apoptotic β‐cell death98. Therefore, the loss of β‐cell function was determined to not be unique to sulfonylureas, as it occurred at an identical rate in patients treated with metformin or conventional treatments, thus suggesting that factors other than treatment might contribute to the process.
Discussion
The impact of a reduction in β‐cell mass in terms of the alterations of insulin secretion that characterize type 2 diabetes has yet to be clearly elucidated. Still, clinical observations and experimental data support a close interrelationship between the two parameters. Thus, a large proportion of living, related pancreatic donors who underwent a 50% pancreatectomy ultimately developed diabetes99. Pharmacological or surgical reduction of β‐cell mass in rodents has been shown to uniformly impair insulin secretion100. More recently, Matveyenko et al.101 have carefully analyzed the effects of a 50% pancreatectomy in normal dogs, and reported that partial pancreatectomy resulted in IFG and IGT. Partial pancreatic resection was associated with reductions of both basal and glucose‐stimulated insulin secretion. Collectively, these data support a mechanistic role of reduced β‐cell mass in the development of alterations in glucose homeostasis and progression toward type 2 diabetes. Actually, many authors have noted reduced β‐cell mass in human patients suffering from type 2 diabetes9–12. Additionally, such reductions in β‐cell mass are strongly associated with increased β‐cell apoptosis, whereas new islet formation rates and β‐cell replication remain relatively normal10. Furthermore, the results of our recent study regarding β‐cell size in humans with type 2 diabetes showed that the β‐cells in these patients were approximately 30% larger than the β‐cells of normal subjects. The ratio of cytoplasmic area to nuclear area was also far higher in the type 2 diabetic patients relative to normal controls. Most studies on β‐cell mass in humans with type 2 diabetes have been based on measurements of the total β‐cell area in the islets. Thus, considering that β‐cell size itself was also increased, we surmised that the numbers of β‐cells were even lower in the type 2 diabetic subjects than had been estimated. Reduced β‐cell mass, which includes decreased β‐cell numbers, might exacerbate disease burdens and aggravate β‐cell loss, causing continuous progression of the disease state. Several genetic factors and acquired factors, such as glucose toxicity, lipid toxicity, inflammation and exogenous stimuli, have been shown to cause β‐cell defects. Thus, in order to develop more fundamental treatments for type 2 diabetes, more advanced approaches focusing on the β‐cells themselves in order to directly prevent damage or restore functional defects must be developed, along with efforts to minimize or ameliorate acquired conditions, such as increases in insulin resistance, glucotoxicity, lipotoxicity and exogeneous factors relevant to β‐cell injury. Additionally, greater efforts must be made in the future to gain insight into alterations and disruptions of the microenvironment in the pancreatic islets of type 2 diabetic subjects, and to find ways of improving that environment in order to minimize such disruptions.
Acknowledgements
This study was supported by a grant of the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A092258).
References
- 1.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001; 414: 782–787 [DOI] [PubMed] [Google Scholar]
- 2.Gu D, Reynolds K, Duan X, et al. Prevalence of diabetes and impaired fasting glucose in the Chinese adult population: International Collaborative Study of Cardiovascular Disease in Asia (InterASIA). Diabetologia 2003; 46: 1190–1198 [DOI] [PubMed] [Google Scholar]
- 3.Ramachandran A. Epidemiology of diabetes in India – three decades of research. J Assoc Physicians India 2005; 53: 34–38 [PubMed] [Google Scholar]
- 4.Yoon KH, Lee JH, Cho JH, et al. Epidemic obesity and type 2 diabetes in Asia. Lancet 2006; 368: 681–688 [DOI] [PubMed] [Google Scholar]
- 5.Kahn SE. The relative contribution of insulin resistance and beta‐cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia 2003; 46: 3–19 [DOI] [PubMed] [Google Scholar]
- 6.Ferrannini E, Mari A. Beta‐cell function and its relation to insulin action in humans: a critical appraisal. Diabetologia 2004; 47: 943–956 [DOI] [PubMed] [Google Scholar]
- 7.Saito K, Yaginuma N, Takahashi T. Differential vometry of A, B and D cells in the pancreatic islets of diabetic and non‐diabetic subjects. Tohoku J Exp Med 1979; 129: 273–283 [DOI] [PubMed] [Google Scholar]
- 8.Westermark P, Wilander E. The influence of amyloid deposits on the islet volume in maturity onset diabetes mellitus. Diabetologia 1978; 15: 417–421 [DOI] [PubMed] [Google Scholar]
- 9.Sakuraba H, Mizukami H, Yagihashi N, et al. Reduced beta cell mass and expression of oxidative stress‐related DNA damage in the islets of Japanese type 2 diabetic patients. Diabetologia 2002; 45: 86–96 [DOI] [PubMed] [Google Scholar]
- 10.Butler AE, Janson J, Bonner‐Weir S, et al. Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52: 102–110 [DOI] [PubMed] [Google Scholar]
- 11.Yoon KH, Ko SH, Cho JH, et al. Selective beta cell loss and alpha cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 2003; 88: 2300–2308 [DOI] [PubMed] [Google Scholar]
- 12.Rahier J, Guiot Y, Goebbels RM, et al. Pancreatic beta‐cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008; 10(Suppl 4): 32–42 [DOI] [PubMed] [Google Scholar]
- 13.Ritzel RA, Butler AE, Rizza RA, et al. Relationship between beta cell mass and fasting blood glucose concentration in humans. Diabetes Care 2006; 29: 717–718 [DOI] [PubMed] [Google Scholar]
- 14.Bagust A, Beale S. Deteriorating beta‐cell function in type 2 diabetes: a long‐term model. Q J Med 2003; 96: 281–288 [DOI] [PubMed] [Google Scholar]
- 15.Bouwens L, Pipeleers DG. Extra‐insular beta‐cells associated with ductules are frequent in adult human pancreas. Diabetologia 1998; 41: 629–633 [DOI] [PubMed] [Google Scholar]
- 16.Bouwens L. Transdifferentiation versus stem cell hypothesis for the regeneration of islet beta‐cells in the pancreas. Microsc Res Tech 1998; 43: 332–336 [DOI] [PubMed] [Google Scholar]
- 17.Müller WA, Faloona GR, Aguilar‐Parada E, et al. Abnormal alpha‐cell function in diabetes. Response to carbohydrate and protein ingestion. N Engl J Med 1970; 283: 109–115 [DOI] [PubMed] [Google Scholar]
- 18.Unger RH, Orci L. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet 1975; 1: 14–16 [DOI] [PubMed] [Google Scholar]
- 19.Deng S, Vatamaniuk M, Huang X, et al. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes 2004; 53: 624–632 [DOI] [PubMed] [Google Scholar]
- 20.Donath MY, Ehses JA, Maedler K, et al. Mechanisms of beta‐cell death in type 2 diabetes. Diabetes 2005; 54(Suppl 2): S108–S113 Review. [DOI] [PubMed] [Google Scholar]
- 21.Rahier J, Goebbels RM, Henquin JC. Cellular composition of the human diabetic pancreas. Diabetologia 1983; 24: 366–371 [DOI] [PubMed] [Google Scholar]
- 22.Gromada J, Franklin I, Wollheim CB. Alpha‐cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev 2007; 28: 84–116 Epub 2007 Jan 16. [DOI] [PubMed] [Google Scholar]
- 23.O’Reilly LA, Gu D, Sarvetnick N, et al. Alpha cell neogenesis in animal model of IDDM. Diabetes 1997; 46: 599–606 [DOI] [PubMed] [Google Scholar]
- 24.Ellingsgaard H, Ehses JA, Hammar EB, et al. Interleukin‐6 regulates pancreatic alpha‐cell mass expansion. Proc Natl Acad Sci USA 2008; 105: 13163–13168 Epub 2008 Aug 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hales CN, Barker DJP. Type 2 (non‐insulin‐dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 1992; 35: 595–601 [DOI] [PubMed] [Google Scholar]
- 26.Hales CN, Barker DJP, Clark PMS, et al. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 1991; 303: 1019–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barker DJP, Hales CN, Fall CHD, et al. Type 2 (non‐insulin dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 1993; 36: 62–67 [DOI] [PubMed] [Google Scholar]
- 28.Viswanathan M, McCarthy MI, Snehalatha C, et al. Familial aggregation of type 2 (non‐insulin dependent) diabetes mellitus in south India; absence of excess maternal transmission. Diabetes Med 1996; 13: 232–237 [DOI] [PubMed] [Google Scholar]
- 29.Ng MC, Lee SC, Ko GTC, et al. Familial early onset type 2 diabetes in Chinese: the more significant roles of obesity and genetics than autoimmunity. Diabetes Care 2001; 24: 667–671 [DOI] [PubMed] [Google Scholar]
- 30.Altshuler D, Hirschhorn JN, Klannemark M, et al. The common PPAR gamma Pro12 Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 2000; 26: 76–80 [DOI] [PubMed] [Google Scholar]
- 31.Gloyn AL, Weedon MN, Owen KR, et al. Large‐scale association studies of variants in genes encoding the pancreatic beta‐cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003; 52: 568–572 [DOI] [PubMed] [Google Scholar]
- 32.Radha V, Vimaleswaran KS, Babu HN, et al. Role of genetic polymorphism peroxisome proliferators‐activated receptor‐gamma2 Pro12Ala on ethnic susceptibility to diabetes in South‐Asian and Caucasian subjects: evidence for heterogeneity. Diabetes Care 2006; 29: 1046–1051 [DOI] [PubMed] [Google Scholar]
- 33.Grant SF, Thorleifsson G, Reynisdottir I, et al. Variant of transcription factor 7‐like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 2006; 38: 320–323 [DOI] [PubMed] [Google Scholar]
- 34.Chang YC, Chang TJ, Jiang YD, et al. Association study of the genetic polymorphisms of the transcription factor 7‐like 2 (TCF7L2) gene and type 2 diabetes in the Chinese population. Diabetes 2007; 56: 2631–2637 [DOI] [PubMed] [Google Scholar]
- 35.Ramachandran A, Ma RC, Snehalatha C. Diabetes in Asia. Lancet 2010; 30: 375 [DOI] [PubMed] [Google Scholar]
- 36.Yasuda K, Miyake K, Horikawa Y, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 2008; 40: 1092–1097 [DOI] [PubMed] [Google Scholar]
- 37.Unoki H, Takahashi A, Kawaguchi T, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 2008; 40: 1098–1102 [DOI] [PubMed] [Google Scholar]
- 38.Lyssenko V, Lupi R, Marchetti P, et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest 2007; 117: 2155–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyssenko V, Jonsson A, Almgren P, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med 2008; 359: 2220–2232 [DOI] [PubMed] [Google Scholar]
- 40.Frayling TM. Genome‐wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet 2007; 8: 657–662 [DOI] [PubMed] [Google Scholar]
- 41.Florez JC. Clinical review: the genetics of type 2 diabetes. J Clin Endocrinol Metab 2008; 93: 4633–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity‐onset diabetes of the young. N Engl J Med 2001; 345: 971–980 [DOI] [PubMed] [Google Scholar]
- 43.Froguel P, Zouali H, Vionnet N, et al. Familial hyperglycemia due to mutations in glucokinase: definition of a subtype of diabetes mellitus. N Engl J Med 1993; 328: 697–702 [DOI] [PubMed] [Google Scholar]
- 44.Yagamata K, Furuta H, Oda N, et al. Mutations in the hepatocyte nuclear factor‐4 gene in maturity‐onset diabetes of the young (MODY1). Nature 1996; 384: 458–460 [DOI] [PubMed] [Google Scholar]
- 45.Yamagata K, Oda N, Kaisaki PJ, et al. Mutations in the hepatocyte nuclear factor‐1 gene in maturity‐onset diabetes of the young (MODY3). Nature 1996; 384: 455–458 [DOI] [PubMed] [Google Scholar]
- 46.Stoffers DA, Ferrer J, Clarke WL, et al. Early‐onset type‐II diabetes mellitus (MODY4) linked to IPF1. Nat Genet 1997; 17: 138–139 [DOI] [PubMed] [Google Scholar]
- 47.Horikawa Y, Iwasaki N, Hara M, et al. Mutation in hepatocyte nuclear factor‐1 gene (TCF2) associated with MODY. Nat Genet 1997; 17: 384–385 [DOI] [PubMed] [Google Scholar]
- 48.Malecki MT, Jhala US, Antonellis A, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet 1999; 23: 323–328 [DOI] [PubMed] [Google Scholar]
- 49.Kaiser N, Leibowitz G, Nesher R. Glucotoxicity and beta‐cell failure in type 2 diabetes mellitus. J Pediatr Endocrinol Metab 2003; 16: 5–22 [DOI] [PubMed] [Google Scholar]
- 50.Robertson RP, Harmon J, Tran PO, et al. Glucose toxicity in beta‐cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003; 52: 581–587 [DOI] [PubMed] [Google Scholar]
- 51.Zraika S, Aston‐Mourney K, Laybutt DR, et al. The influence of genetic background on the induction of oxidative stress and impaired insulin secretion in mouse islets. Diabetologia 2006; 49: 1254–1263 [DOI] [PubMed] [Google Scholar]
- 52.Del Guerra S, Grupillo M, Masini M, et al. Gliclazide protects human islet beta‐cells from apoptosis induced by intermittent high glucose. Diabetes Metab Res Rev 2007; 23: 234–238 [DOI] [PubMed] [Google Scholar]
- 53.Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 2002; 51: S455–S461 [DOI] [PubMed] [Google Scholar]
- 54.Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res 2005; 569: 29–63 [DOI] [PubMed] [Google Scholar]
- 55.Rabinovitch A, Suarez‐Pinzon WL. Roles of cytokines in the pathogenesis and therapy of type 1 diabetes. Cell Biochem Biophys 2007; 48: 159–163 [DOI] [PubMed] [Google Scholar]
- 56.Marchetti P, Bugliani M, Lupi R, et al. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007; 50: 2486–2494 [DOI] [PubMed] [Google Scholar]
- 57.Maedler K, Oberholzer J, Bucher P, et al. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta‐cell turnover and function. Diabetes 2003; 52: 726–733 [DOI] [PubMed] [Google Scholar]
- 58.Lupi R, Dotta F, Marselli L, et al. Prolonged exposure to free fatty acids has cytostatic and pro‐apoptotic effects on human pancreatic islets: evidence that beta‐cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl‐2 regulated. Diabetes 2002; 51: 1437–1442 [DOI] [PubMed] [Google Scholar]
- 59.Wrede CE, Dickson LM, Lingohr MK, et al. Protein kinase B/Akt prevents fatty acid‐induced apoptosis in pancreatic beta‐cells (INS‐1). J Biol Chem 2002; 277: 49676–49684 [DOI] [PubMed] [Google Scholar]
- 60.Shimabukuro M, Zhou YT, Levi M, et al. Fatty acid‐induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA 1998; 95: 2498–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shimabukuro M, Ohneda M, Lee Y, et al. Role of nitric oxide in obesity induced beta cell disease. J Clin Invest 1997; 100: 290–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and β‐cell dysfunction. Endocr Rev 2008; 29: 351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest 2006; 116: 1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laybutt DR, Kaneto H, Hasenkamp W, et al. Increased expression of antioxidant and antiapoptotic genes in islets that may contribute to β‐cell survival during chronic hyperglycemia. Diabetes 2002; 51: 413–423 [DOI] [PubMed] [Google Scholar]
- 65.Pick A, Clark C, Kubstrup M, et al. Role of apoptosis in failure of β‐cell mass compensation for insulin resistance and β‐cell defects in the male Zucker diabetic fatty rat. Diabetes 1998; 47: 358–364 [DOI] [PubMed] [Google Scholar]
- 66.Prentki M, Joly E, El‐Assaad W, et al. Malonyl‐CoA signaling, lipid partitioning, and glucolipotoxicity: role in beta‐cell adaptation and failure in the etiology of diabetes. Diabetes 2002; 51: S405–S413 [DOI] [PubMed] [Google Scholar]
- 67.Kim JW, You YH, Ham DS, et al. Suppression of peroxisome proliferator‐activated receptor gamma‐coactivator‐1alpha normalizes the glucolipotoxicity‐induced decreased BETA2/NeuroD gene transcription and improved glucose tolerance in diabetic rats. Endocrinology 2009; 150: 4074–4083 [DOI] [PubMed] [Google Scholar]
- 68.Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005; 9467: 1333–1346 [DOI] [PubMed] [Google Scholar]
- 69.White MF. Insulin signaling in health and disease. Science 2003; 5651: 1710–1711 [DOI] [PubMed] [Google Scholar]
- 70.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor‐alpha: direct role in obesity‐linked insulin resistance. Science 1993; 5091: 87–91 [DOI] [PubMed] [Google Scholar]
- 71.Schmitz‐Peiffer C, Whitehead JP. IRS‐1 regulation in health and disease. IUBMB Life 2003; 55: 367–374 [DOI] [PubMed] [Google Scholar]
- 72.Lin X, Taguchi A, Park S, et al. Dysregulation of insulin receptor substrate 2 in beta cells and brain causes obesity and diabetes. J Clin Invest 2004; 114: 908–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kahn SE, Verchere CB, Andrikopoulos S, et al. Reduced amylin release is a characteristic of impaired glucose tolerance and type 2 diabetes in Japanese Americans. Diabetes 1998; 47: 640–645 [DOI] [PubMed] [Google Scholar]
- 74.Leighton B, Cooper CJS. Pancreatic amylin and calcitonin gene‐related peptide cause resistance to insulin in skeletal muscle in vitro. Nature 1988; 335: 632–635 [DOI] [PubMed] [Google Scholar]
- 75.Silvestre RA, Peiró E, Dégano P, et al. Inhibitory effect of rat amylin on the insulin responses to glucose and arginine in the perfused rat pancreas. Regul Pept 1990; 31: 23–31 [DOI] [PubMed] [Google Scholar]
- 76.Cooper GJS, Day AJ, Willis AC, et al. Amylin and the amylin gene: structure, function and relationship to islet amyloid and to diabetes mellitus. Biochem Biophys Acta 1989; 1014: 247–258 [DOI] [PubMed] [Google Scholar]
- 77.Sempoux C, Guiot Y, Dubois D, et al. Human type 2 diabetes: morphological evidence for abnormal beta‐cell function. Diabetes 2001; 50: S172–S177 [DOI] [PubMed] [Google Scholar]
- 78.Bernard‐Kargar C, Ktorza A. Endocrine pancreas plasticity under physiological and pathological conditions. Diabetes 2001; 50: S30–S35 [DOI] [PubMed] [Google Scholar]
- 79.Porte D. Clinical importance of insulin secretion and its interaction with insulin resistance in the treatment of type 2 diabetes mellitus and its complications. Diabetes Metab Res Rev 2001; 17: 181–188 [DOI] [PubMed] [Google Scholar]
- 80.Kahn SE, Andrikopoulos S, Verchere CB. Islet amyloid: a long‐recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 1999; 48: 241–253 [DOI] [PubMed] [Google Scholar]
- 81.Finegood D, Topp B. β‐cell deterioration – prospects for reversal or prevention. Diabetes Obes Metab 2001; 3: S20–S27 [PubMed] [Google Scholar]
- 82.Hayden M, Tyagi S. Islet redox stress: the manifold toxicities of insulin resistance, metabolic syndrome and amylin derived islet amyloid in type 2 diabetes mellitus. Pancreas 2002; 3: 86–108 [PubMed] [Google Scholar]
- 83.Janson J, Ashley R, Harrison D, et al. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate‐sized toxic amyloid particles. Diabetes 1999; 48: 491–498 [DOI] [PubMed] [Google Scholar]
- 84.Lorenzo A, Razzaboni B, Weir GC, et al. Pancreatic islet cell toxicity of amylin associated with type‐2 diabetes mellitus. Nature 1994; 368: 756–760 [DOI] [PubMed] [Google Scholar]
- 85.Wang F, Hull R, Vidal J, et al. Islet amyloid develops diffusely throughout the pancreas before becoming severe and replacing endocrine cells. Diabetes 2001; 50: 2514–2520 [DOI] [PubMed] [Google Scholar]
- 86.Kim JW, Ko SH, Cho JH, et al. Loss of beta‐cells with fibrotic islet destruction in type 2 diabetes mellitus. Front Biosci 2008; 13: 6022–6033 [DOI] [PubMed] [Google Scholar]
- 87.Ko SH, Hong OK, Kim JW, et al. High glucose increases extracellular matrix production in pancreatic stellate cells by activating the renin‐angiotensin system. J Cell Biochem 2006; 98: 343–355 [DOI] [PubMed] [Google Scholar]
- 88.Schuster DP. Obesity and the development of type 2 diabetes: the effects of fatty tissue inflammation. Diabetes Metab Syndr Obes 2010; 3: 253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marselli L, Sgroi DC, Thorne J, et al. Evidence of inflammatory markers in beta cells of type 2 diabetic subjects. Diabetologia 2007; 50: S178 [Google Scholar]
- 90.Maedler K, Sergeev P, Ris F, et al. Glucoseinduced beta cell production of IL‐1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 2002; 110: 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maedler K, Schulthess FT, Bielman C, et al. Glucose and leptin induce apoptosis in human beta‐cells and impair glucose stimulated insulin secretion through activation of c‐Jun N‐terminal kinases. FASEB J 2008; 22: 1905–1913 [DOI] [PubMed] [Google Scholar]
- 92.Maedler K, Sergeev P, Ehses JA, et al. Leptin modulates beta‐cell expression of the IL‐1 receptor antagonist and release of IL‐1beta in human islets. Proc Nat Acad Sci U S A 2004; 101: 8138–8143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ehses JA, Perren A, Eppler E, et al. Increased number of islet‐associated macrophages in type 2 diabetes. Diabetes 2007; 56: 2356–2370 [DOI] [PubMed] [Google Scholar]
- 94.Richardson SJ, Willcox A, Bone AJ, et al. Islet‐associated macrophages in type 2 diabetes. Diabetologia 2009; 52: 1686–1688 [DOI] [PubMed] [Google Scholar]
- 95.UK Prospective Diabetes Study (UKPDS) Group . Intensive blood‐glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352: 837–853 [PubMed] [Google Scholar]
- 96.Heald AH, Anderson SG, Patel J, et al. Change in pancreatic B‐cell function (HOMA‐B) varies in different populations with similar genetic backgrounds but different environments. Diabet Med 2007; 24: 145–153 [DOI] [PubMed] [Google Scholar]
- 97.Maedler K, Carr RD, Bosco D, et al. Sulfonylurea induced beta‐cell apoptosis in cultured human islets. J Clin Endocrinol Metab 2005; 90: 501–506 [DOI] [PubMed] [Google Scholar]
- 98.Efanova IB, Zaitsev SV, Zhivotovsky B, et al. Glucose and tolbutamide induce apoptosis in pancreatic beta‐cells. A process dependent on intracellular Ca2+ concentration. J Biol Chem 1998; 273: 33501–33507 [DOI] [PubMed] [Google Scholar]
- 99.Kendall DM, Sutherland DE, Najarian JS, et al. Effects of hemipancreatectomy on insulin secretion and glucose tolerance in healthy humans. N Engl J Med 1990; 322: 898–903 [DOI] [PubMed] [Google Scholar]
- 100.Weir GC, Leahy JL, Bonner‐Weir S. Experimental reduction of B‐cell mass: implications for the pathogenesis of diabetes. Diabetes Metab Rev 1986; 2: 125–161 [DOI] [PubMed] [Google Scholar]
- 101.Matveyenko AV, Veldhuis JD, Butler PC. Mechanisms of impaired fasting glucose and glucose intolerance induced by an approximate 50% pancreatectomy. Diabetes 2006; 55: 2347–2356 [DOI] [PubMed] [Google Scholar]