

Abstract
Diabetic complications are the major causes of morbidity and mortality in patients with diabetes. Microvascular complications include retinopathy, nephropathy and neuropathy, which are leading causes of blindness, end‐stage renal disease and various painful neuropathies; whereas macrovascular complications involve atherosclerosis related diseases, such as coronary artery disease, peripheral vascular disease and stroke. Diabetic complications are the result of interactions among systemic metabolic changes, such as hyperglycemia, local tissue responses to toxic metabolites from glucose metabolism, and genetic and epigenetic modulators. Chronic hyperglycemia is recognized as a major initiator of diabetic complications. Multiple molecular mechanisms have been proposed to mediate hyperglycemia’s adverse effects on vascular tissues. These include increased polyol pathway, activation of the diacylglycerol/protein kinase C pathway, increased oxidative stress, overproduction and action of advanced glycation end products, and increased hexosamine pathway. In addition, the alterations of signal transduction pathways induced by hyperglycemia or toxic metabolites can also lead to cellular dysfunctions and damage vascular tissues by altering gene expression and protein function. Less studied than the toxic mechanisms, hyperglycemia might also inhibit the endogenous vascular protective factors such as insulin, vascular endothelial growth factor, platelet‐derived growth factor and activated protein C, which play important roles in maintaining vascular homeostasis. Thus, effective therapies for diabetic complications need to inhibit mechanisms induced by hyperglycemia’s toxic effects and also enhance the endogenous protective factors. The present review summarizes these multiple biochemical pathways activated by hyperglycemia and the potential therapeutic interventions that might prevent diabetic complications. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2010.00018.x, 2010)
Keywords: Diabetic complications, Diabetes mellitus, Endogenous protective factors
Introduction
According to the recent edition of International Diabetes Federation Atlas in 2009, the estimated diabetes prevalence for 2010 had risen to 285 million, representing 6.6% of the world’s adult population, with a prediction that by 2030 the number of people with diabetes in the world will have risen to 438 million1, with the majority of the new diabetic population coming from Asia. Diabetes‐induced vascular dysfunction and pathologies are the major causes of morbidity and mortality in diabetic patients. Microvascular complications include retinopathy, nephropathy and neuropathy, which are the leading causes of blindness, renal failure, and nerve injuries that are associated with non‐healing ulcers and non‐traumatic amputation. Macrovascular complications involve atherosclerosis‐related diseases, such as coronary artery disease, peripheral vascular disease, stroke and possibly cognitive dysfunction.
Two large studies, the Diabetic Control and Complications Trial (DCCT) and the United Kingdom Prospective Diabetes Study (UKPDS) clearly showed that intensive treatment for hyperglycemia could reduce the progression of diabetic microvascular complications2,3. Furthermore, the long‐term follow‐up studies of DCCT showed that patients who received intensive blood glucose control decreased the incidence of cardiovascular diseases involving atherosclerosis4,5. These clinical observations indicate that hyperglycemia is a major responsible factor for the pathogenesis of diabetic complications. In contrast, it is known that multiple factors, such as fatty acid, lipid, insulin resistance, inflammatory cytokines and others also can increase the risk for atherosclerosis in diabetes.
Multiple potential molecular mechanisms have been proposed to explain hyperglycemia‐induced diabetic complications. Some of the most studied mechanisms include increased polyol pathway, activation of the diacylglycerol (DAG)/protein kinase C (PKC) pathway, increased oxidative stress, increased advanced glycation end products (AGE) formation and action, and increased hexosamine pathway. In addition, alterations of signal transduction pathways induced by hyperglycemia or toxic metabolites have been reported to cause multiple vascular and neurological dysfunctions, such as abnormal blood flow, increased rate of apoptosis, hyperpermeability and accumulation of extracellular matrix (ECM) in vasculature by alteration of gene expression or protein function. Recently, we have proposed that hyperglycemia can also inhibit endogenous protective factors in the vascular tissues, such as insulin, vascular endothelial growth factor (VEGF), platelet‐derived growth factor (PDGF), and activated protein C (APC), which play important roles in maintaining vascular homeostasis and neutralizing hyperglycemia‐induced toxic factors including oxidative stress, AGE or activation of nuclear factor‐κB (NF‐κB), resulting in the prevention and delaying of the progression of diabetic complications (Figure 1)6.
Figure 1.
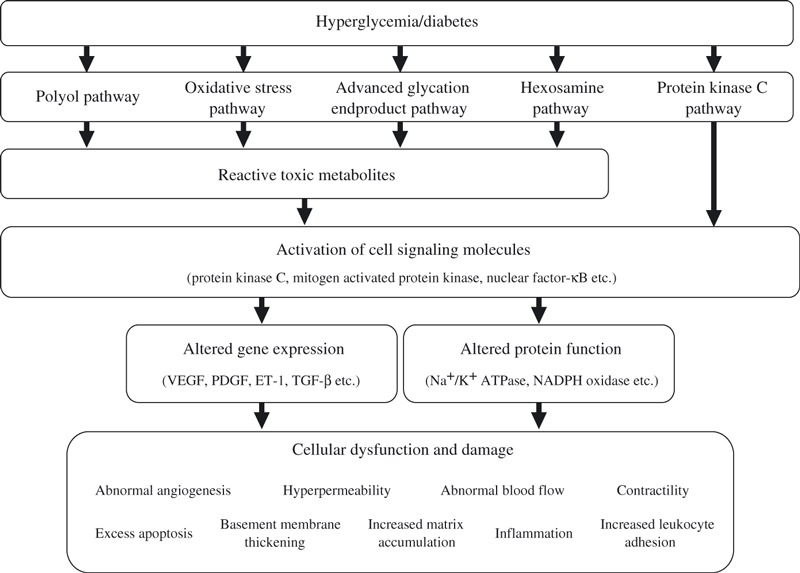
Mechanisms by which hyperglycemia induced diabetic vascular complications. ET‐1, endothelin‐1; NADPH, nicotinamide adenine dinucleotide phosphate; PDGF, platelet‐derived growth factor; TGF‐β, transforming growth factor‐β; VEGF, vascular endothelial growth factor.
Genetic factors also have been suggested as important risk markers for developing diabetic complications. It has been well established that merely 30–40% of type 1 diabetic patients develop chronic renal failure7,8. The risk of developing renal failure in diabetic patients decreases after 25–30 years of disease duration. Recently, we have reported that the results of the 50‐Year Medalist Study of the Joslin Diabetes Center (JDC), which was initiated to recognize JDC or non‐JDC patients who survived at least 50 years with insulin‐dependent diabetes or type 1 diabetes. The patients were questioned about the presence or absence of eye, kidney and peripheral neuropathies. The Medalist Study showed that significant numbers (40%) of diabetic patients could live with no or mild levels of microvascular complications, regardless of their HbA1c levels and other classical markers thought to be important and predictive markers for diabetic complications. These data suggest that they might possess endogenous protective factors that can neutralize the adverse effects of hyperglycemia9. Epigenetic factors are also important. The DCCT and Epidemiology of Diabetes Interventions and Complications (EDIC) studies reported that patients from the original DCCT study continue to have discordance in the development of microvascular complications, even 10 years after maintaining the same levels of glycemic control as shown by HbA1c. These findings showed that hyperglycemia might induce epigenetic changes that are not reversed easily10–12. Thus, diabetic complications are a result of interactions among systemic metabolic changes, such as hyperglycemia, differential local tissue responses to toxic metabolites of glucose metabolism, and genetic and epigenetic modulators.
Molecular mechanisms of diabetic vascular complications
Hyperglycemia is recognized as a major responsible factor for the development of diabetic complications, especially for microvascular diseases. For example, pathologies in the retina and renal glomeruli are specific to diabetes and not usually observed in elderly or insulin resistant people without diabetes. The most studied mechanisms include: (i) increased polyol pathway; (ii) increased DAG/activation of PKC pathway; (iii) increased oxidative stress; (iv) increased AGE formation and action; and (v) increased hexosamine pathway (Figure 1).
Increased Flux Through the Polyol Pathway
In the polyol pathway, intracellular glucose is converted to sorbitol by aldose reductase (AR), which is the rate‐limiting enzyme, in a nicotinamide adenine dinucleotide phosphate (NADPH) dependent reaction. Sorbitol is then oxidized to fructose by sorbitol dehydrogenase (SDH). In a normal glucose condition, only a small fraction of glucose is metabolized through this pathway, because Michaelis–Menten kinetics of AR for glucose is above normoglycemic levels. In diabetic states, elevation of intracellular glucose levels can cause an increased flux through AR13,14. The activation of the polyol pathway has been suggested to cause vascular pathologies by osmotic damage and reduced Na+‐K+‐ATPase activity15. AR and SDH use NADPH and NAD+ as a cofactor, respectively. Therefore, the decline in cellular NADPH and the increased NADH/NAD+ ratio changes the intracellular redox balance resulting in the reduced production of nitric oxide and increased oxidative stress16. Lenses specific AR overexpressed transgenic mice with diabetes showed a significant decrease in glutathione (GSH) level, leading to enhanced oxidative stress. In the AR null mutant mice, diabetes did not lead to any decrease in the nerve GSH level17.
Studies inhibiting the polyol pathway using aldose reductase inhibitors (ARI) in vivo have yielded inconsistent results. In animal studies, ARI have been shown to prevent some abnormalities in cataracts, retinopathy18,19, nephropathy20, neuropathy21,22 and cardiomyopathy. However, in a 5 year study in dogs, AR inhibition could prevent only neuropathy, but failed to prevent retinopathy and nephropathy23. In clinical studies, ARI have not been shown to be clearly effective in patients with diabetic retinopathy (DR) and nephropathy24. For diabetic neuropathy, some studies have suggested positive effects. In a double‐blind placebo controlled study, fidarestat showed improved nerve conduction velocity and a variety of subjective symptoms, such as numbness and spontaneous pain25. In addition, it has been reported that long‐term treatment with epalrestat also can effectively delay the progression of diabetic neuropathy and ameliorate the associated symptoms of the disease25,26. More large full studies in phase three trials are needed to show that ARI can clearly be effective for neuropathy.
Increased DAG/Activation of PKC Pathway
DAG and PKC are important intracellular signaling molecules that can regulate many vascular functions. Receptor‐mediated physiological PKC activation is mediated mostly by the activation of phospholipase C, which leads to an increase in Ca2+ and DAG levels27.
Intracellular hyperglycemia increases glycolytic pathway flux and leads to an elevation of glycolytic intermediate dihydroxyacetone phosphate. Increased levels of this intermediate can stimulate increases in the de novo synthesis of DAG through the reduction of the latter to glyceraldehydes‐3‐phospate and stepwise acylation28. In diabetes, many studies have showed that DAG levels in various tissues, such as retina29, glomeruli30,31, aorta and heart32 are increased. Furthermore, various cell culture studies also show that DAG levels are increased by the elevation of glucose levels from low to high concentration in retinal and aortic endothelial cells29,32, smooth muscle cells32, mesangial cells33,34 and other vascular cells. These chronically elevated levels of DAG can activate PKC. In addition, several PKC isoforms are also activated through other mechanisms, such as reactive oxygen species35,36 and free fatty acids (FFA)37,38.
Increased PKC activation has been associated with alterations in blood flow, basement membrane thickening, ECM expansion, increases in vascular permeability, abnormal angiogenesis, excessive apoptosis, increased leukocyte adhesion, and changes in enzymatic activity alterations, such as Na+‐K+‐ATPase, cPLA2, PI3K and mitogen activated protein kinase (MAPK)39. These effects are probably mediated through the altered gene expression for vasoactive and growth factors, such as VEGF40–42, endothelin‐1 (ET‐1)43,44, transforming growth factor (TGF)‐β45,46 and connective tissue growth factor (CTGF)41,46,47. Furthermore, PKC activation contributes to the overexpression of plasminogen activator‐1 (PAI‐1)48,49, the activation of NF‐κB and the activation of NADPH oxidase50,51 in many vascular cells including endothelial cells, smooth muscle cells, pericytes, mesangial cells, and others52,53.
PKC is a family of enzymes composed of at least 12 members54. Of the various PKC isoforms in vascular cells, PKC‐α, ‐β and ‐δ isoforms appear to be preferentially activated by immunoblotting studies in the aorta and heart of diabetic rodents, cultured aortic smooth muscle cells, and endothelial cells exposed to high levels of glucose32,55. However, increases in other isoforms, such as PKC‐α, ‐β2, ‐δ, and ‐ε in the retinal cells29,43 and PKC‐α, ‐β1/2, ‐δ, ‐ε, and ‐ζ in the glomerular cells45,56–59 exposed to high glucose or diabetes have also been shown to be activated. In animals with diabetes, ruboxistaurin mesylate (RBX), a PKC‐β isoform selective inhibitor, has been shown to prevent many vascular abnormalities associated with retinopathy, nephropathy and neuropathy31,45,57–59. Furthermore, we showed that PKC‐β null mice with streptozotocin (STZ)‐induced diabetes showed improvement in renal abnormalities including albuminuria, renal hypertrophy and mesangial expansion46. Recent studies by Harja et al. have also suggested that PKC‐β activation might also play a role in accelerating atherosclerosis60. Clinical studies showed that RBX improved endothelial dysfunction61, renal glomerular filtration rate62 and prevented loss of visual acuity63 in diabetic patients. However, RBX was not effective in patients with painful diabetic neuropathy. Thus, PKC activation involving several isoforms is likely to be responsible for some of the pathologies in DR (Figure 2), nephropathy and cardiovascular disease.
Figure 2.
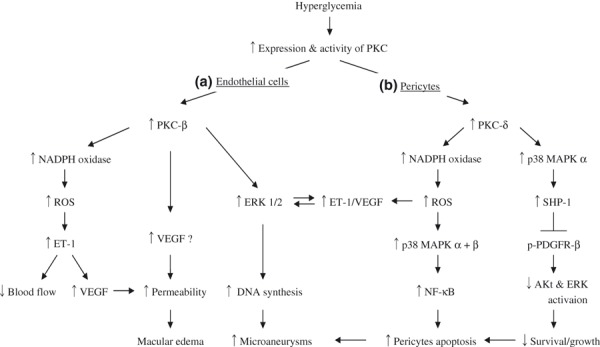
Outline for mechanisms of diabetic retinopathy. (A) Activation of protein kinase C (PKC)‐β in retinal endothelial cells contributes to increased vascular permeability and formation of microaneurisms. (B) Activation of PKC‐δ induce retinal pericytes apoptosis through activation of nuclear factor‐κB (NF‐κB) by oxidative stress and activating Src homology‐2 domain containing phosphatase‐1 (SHP‐1) to inhibit platelet‐derived growth factor’s (PDGF) survival actions. Akt, protein kinase B; ERK, extracellular signal regulated kinase; ET‐1, endothelin‐1; MAPK, mitogen activated protein kinase; NADPH, nicotinamide adenine dinucleotide phosphate; PDGF, platelet‐derived growth factor; ROS, reactive oxygen species; TGF‐β, transforming growth factor‐β; VEGF, vascular endothelial growth factor.
Increased Oxidative Stress
Recent studies have suggested increases in oxidative stress as being a main metabolic abnormality involved in the development of diabetic complications14,64–66. Oxidative stress occurs when the production of reactive oxygen species (ROS) exceeds the capability of antioxidant systems. There is substantial evidence showing that ROS production is increased in endothelial cells, kidney, retina either exposed to hyperglycemia or from diabetic animals51,67–69. Likewise, diabetic patients have elevated levels of isoprostanes, 8‐hydroxy‐deoxyguanosine and lipid peroxides in the plasma or urine70,71. The increased oxidative stress markers reflect increased production of ROS, decreased antioxidants, or both. Increased ROS production is a result of abnormal metabolism of glucose, FFA and other reactive metabolites in diabetes64. Several processes are sources for increased ROS, including gluco‐oxidants and AGE, which are created by non‐enzymatic glycolysis and mitochondrial oxidative phosphorylation14,36. Furthermore, byproducts of these processes can cause activation of certain signaling cascades, such as PKC, which can activate NADPH oxidase to increase ROS72. Elevated FFA levels can also increase ROS production by β‐oxidative phosphorylation through mitochondrial metabolism73. Therefore, increased ROS production in diabetes can originate from the metabolism of both glucose and FFA through multiple pathways. This provides an explanation for the findings of increased oxidative stress in insulin resistant non‐diabetic patients74. In contrast, decreased antioxidants have been shown in diabetic animals and patients. For example, GSH level was decreased in kidneys and red blood cells from STZ‐induced diabetic rats68,75. Some studies showed that plasma vitamin C and E levels were decreased, whereas others showed no changes76,77. However, it is possible that the plasma levels of antioxidants might not reflect those at the tissue levels.
Antioxidant therapies have been applied in animal experiments, such as vitamin C, vitamin E and α‐lipoic acids. All of them have showed improved biological and pathological changes, and prevented or slowed the progression of diabetic complications64,65. Overexpression of catalase or superoxide dismutase (SOD) protected the kidneys against hyperglycemia‐induced damage in mice78,79. However, large studies such as the Heart Outcomes Prevention Study using vitamin E and d‐α‐tocopherol, did not show the improvement of microvascular or cardiovascular damage80,81. Therefore, the efficacy of antioxidants in humans is still inconclusive.
Increased AGE Formation and Action
Non‐enzymatic reactions between glucose and proteins, known as the Maillard reaction, result in the formation of Schiff base. Over time, a series of chemical rearrangements lead to AGE82,83. In the diabetic condition, elevated levels of AGE can be found in serum84, glomerular tissue85 and retinal tissues86,87. Some AGE are stable, irreversible products that can be formed intracellularly and extracellularly. AGE can cause vascular damage through several mechanisms. Intracellular proteins, such as basic fibroblast growth factor88 and mitochondrial electron related proteins89, can be modified by AGE, which then alters their function. Glycation of ECM proteins, such as collagen I, IV and laminin90–92, can change their function and alter cell/ECM interactions. Lipoproteins can also be glycated and altered in their metabolism93,94.
AGE might also interact with cellular receptors, one of which is called receptor for AGE (RAGE), a transmembrane receptor that is a member of the immunogloblin superfamily of proteins95. The AGE/RAGE interactions have been reported in the development of diabetic complications. It is reported that the expression of RAGE is increased in glomeruli in diabetic patients compared with healthy control subjects96. Furthermore, detailed studies by Yamamoto et al. clearly showed AGE/RAGE interactions, leading to diabetic nephropathy97 in animal studies. They overexpressed RAGE in vascular endothelial cells in mice and induced diabetes by crossbreeding them with mice overexpressing inducible nitric oxide synthase (iNOS) under the control of the insulin promotor. These mice consistently developed hypoinsulinemic diabetes as a result of NO‐mediated selective destruction of insulin producing pancreatic β cells. The double transgenic mice showed the characteristics of diabetic nephropathy and progressive renal insufficiency, such as exacerbation of nephromegaly, mesangial expansion, albuminuria, glomerular hypertropthy and sclerosis.
Binding of AGE‐modified proteins to RAGE induces activation of cellular signaling cascades including NF‐κB98–100. ET‐1, vascular cell adhesion molecule‐1, intercellular adhesion molecule‐1, E‐selectin, VEGF, and proinflammatory cytokines including IL‐1α, IL‐6 and TNF‐α are induced by NF‐κB101,102. AGE/RAGE interactions have been also shown to induce vascular oxidative stress through the activation of NADPH oxidase103. In addition, other receptors, such as the macrophage scavenger receptor, p60, p90 and galectin‐3, have also been reported to bind AGE104,105.
An inhibitor of AGE formation, aminoguanidine, can prevent the development of diabetic complications, such as retinopathy and nephropathy in animal models106,107. However clinical trials using aminoguanidine have been inconclusive as a result of the presence of limiting toxicity. The cross‐link breakers, including ALT‐711 and N‐phenyl‐thiazolium bromide, improved arterial compliance and cardiac function108,109, atherosclerosis110 and diabetic nephropathy111,112. Blockade of the AGE/RAGE interaction by soluble RAGE has been shown to suppress atherosclerosis and neointimal formation113–115 and nephropathy in diabetic animals116.
Increased Flux Through the Hexosamine Pathway
In a normal glucose condition, only a small fraction (approximately 1–3%) of glucose is metabolized through the hexosamine pathway. Elevation of intracellular glucose levels can cause an increased flux through the hexosamine pathway. Fructose‐6‐phosphate, a glycolysis intermediate, is converted to glucosamine‐6‐phosphate by the rate‐limiting enzyme, glutamine: fructose‐6‐phosphate aminotransferase (GFAT)117. The major end‐product is uridine diphosphate N‐acetylglucosamine (UDP‐GlcNAc), which is a substrate for the subsequent O‐linked GlcNAc modification of target proteins at serine and threonine residues. Functional importance of O‐GlcNAc modification has been reported for several transcription factors, such as Sp‐1118–120. Some reports have showed that glucosamine or overexpression of GFAT increased the promoter activity and expression of PAI‐1 through the increased O‐GlcNAc modification of Sp‐1 in vascular endothelial cells121, smooth muscle cells122 and mesangial cells123. Furthermore, it is reported that glucosamine or GFAT overexpression stimulates the overexpression of TGF‐β1 through increased expression upstream stimulatory factors 1 and 2 (USF1 and 2), but not increased O‐GlcNAc modification of those transcription factors in mesangial cells124. In addition, hyperglycemia might inhibit endothelial nitric oxide synthase (eNOS) activity by the O‐GlcNAc modification at serine 1177 in endothelial cells125.
Altered expression and actions of endogenous protective factors
The discussion of the present review has focused on the mechanisms by which hyperglycemia could be mediating its toxic effects. However, very few studies have focused on the endogenous protective factors that might exist to neutralize hyperglycemia’s toxic actions. One clear example of endogenous protective factors is the antioxidative enzymes, which are generally activated by an elevated state of increased oxidant production126. Clinically, the pulmonary system appears to be protected from the toxic actions of hyperglycemia because patients with type 1 diabetes are relatively free from vascular pathologies of the pulmonary system. Furthermore, the 50‐Year Medalist Study shows that some protective factors might exist and can neutralize high glucose‐induced adverse effects in many diabetic patients9. In the following, we will propose that many of the changes in the elevation of cytokines are as a result of the body’s responses to protect itself from injury. Hyperglycemia, through several mechanisms, might be deactivating these cytokines and causing resistance to them.
Systemic metabolic changes in patients with diabetes lead to altered expression or action of several factors, such as insulin, PDGF, VEGF or APC, which are physiologically important factors for keeping the homeostasis of vasculatures. In this part, we will focus on these endogenous protective factors against diabetes‐induced vascular injuries and potential mechanisms induced by hyperglycemia, which are deactivating their actions.
Insulin
Insulin resistance is observed in patients with not only type 2 diabetes and obesity, but also type 1 diabetes. In addition to its important role for maintaining glycemic control, insulin has many vasotropic actions. Insulin resistance in vascular tissues is associated with endothelial dysfunction, leading to cardiovascular diseases including atherosclerosis127. Furthermore, microalbuminuria, which is known as not only the predictive marker for nephropathy but also as the independent risk factor for cardiovascular diseases, is also associated with endothelial dysfunction128–130. Physiologically, insulin has an important role in the maintenance of blood vessels through the activation of endothelium‐derived NO. Insulin increases endothelial NO production by rapid post‐translational mechanisms, which are mediated by the PI3K/protein kinase B (Akt) signaling pathway129,130 or slowly by increases in its transcription process. In insulin resistance states, the PI3K/Akt pathway is selectively inhibited, but another major pathway of insulin signaling, MAPK, is not inhibited131. This selective insulin resistance has been shown in skeletal muscle from obese people and patients with type 2 diabetes132, and in the vasculature and myocardium of obese Zucker rats 133, which are the animal models of insulin resistance. These are likely multiple mechanisms for inducing selective insulin resistance on the PI3K/Akt pathway. We have reported that PI3K activity is inhibited by PKC activation in endothelial cells134 and vascular tissues of obese Zucker rats130. Furthermore, we found that RBX can improve insulin signaling on NO production in the vasculature and myocardium of Zucker rats134. Insulin stimulates not only NO production from endothelial cells but also the expression of eNOS. The vascular endothelial cells specific insulin receptor knockout (VENIRKO) mice showed that eNOS expression in the aorta was decreased by 62%135. Thus, insulin regulation of NO might be an important factor for vascular homeostasis, which is reduced in diabetes or insulin resistance.
Because intensive glycemic treatment in clinical trials using insulin can delay the progression of retinopathy and other microvascular pathologies in type 1 diabetic patients, the loss of direct vasotropic actions of insulin might increase the risks of developing retinal disease in type 1 diabetic patients2. Recently, we found that insulin can inhibit oxidative stress‐induced retinal pericyte apoptosis through the induction of hemeoxygenase‐1 (HO‐1), which is a representative mediator of antioxidants and cytoprotectants against various stress stimuli, including oxidants in vascular tissues136. Furthermore, we showed that insulin induced the expression of HO‐1 through the PI3K/Akt pathway, but not through the MAPK pathway. Thus, insulin might exert vascular protective effects through the production of NO or the induction of HO‐1. Therefore, impairment of insulin action in vascular tissue might contribute to diabetic vascular complications.
PDGF‐B
PDGF‐B is essential for the recruitment of mural cells, such as pericytes, to the blood vessels137. PDGF‐B or PDGFR‐β deficient mice show a loss of retinal pericytes that resemble the early changes of DR138,139. Pericyte loss is known as a hallmark of human DR and might be causally involved in its pathogenesis140–142.These animal studies suggest that PDGF‐B deficiency might trigger the development for DR. However, paradoxically, it has been shown that the expression of PDGF‐B is increased in retinal tissues of diabetic rats44. Recently we found that hyperglycemia persistently activates PKC‐δ and p38 MAPK to increase Src homology‐2 domain containing phosphatase‐1 (SHP‐1), and leads to PDGFR‐β dephosphorylation and reduction downstream, resulting in pericytes apoptosis and acellular capillaries in diabetic retina. Interestingly, we observed that increased PKC‐δ and acellular capillaries were not reversible with insulin treatment that achieved normoglycemia143. These data showed that hyperglycemia can cause pericytes apoptosis through two pathways. One is the activation of NF‐κB by oxidative stress. The second is by activating SHP‐1 to inhibit PDGF’s important survival actions in the pericytes (Figure 2). These data show that PDGF‐B plays an important role for pericytes survival as a retinal vascular protecting factor. PDGF‐B resistance exists in retina in diabetes and could be an important contributor to DR.
Vascular Endothelial Growth Factor A
VEGF includes a family of growth factors that act on endothelial cells regulated by hypoxia and promote angiogenesis, increase permeability in vasculature, and is also known as a major regulator of endothelial proliferation, migration, and survival144.
Increased concentration of VEGF‐A has been reported in the ocular fluids145 and retinal tissues146,147 of diabetic patients and is associated with the severity of proliferative DR (PDR). Anti‐VEGF treatment, including intravitreal injection, can inhibit the progression of PDR148. However, it is likely that retinal VEGF levels are initially elevated as a result of a reaction against retinal hypoxia or ischemia in diabetes to maintain endothelial function and circulation, as a result of pericytes loss and acellular capillaries. This increase in VEGF is probably a tissue response to increase survival. Thus, the use of chronic anti‐VEGF therapies might have beneficial effects on the vasculature in the short‐term. Clinically, the loss of VEGF without good glycemic control or a decrease of metabolic demands, as by photocoagulation, might cause complications in the neural retina.
In early stage diabetic nephropathy, many reports have shown that the expression of VEGF‐A is increased in glomeruli of diabetic animals46,149,150 and proposed that inhibition of VEGF‐A might have beneficial effects against diabetic renal injuries. Treatment with VEGF‐A antibodies in STZ‐induced diabetic rats ameliorated renal changes, such as albuminuria, hyperfiltration, and glomerular hypertrophy151. Furthermore, in db/db mice also, administration of antibodies to VEGF improved renal abnormalities including kidney weight, glomerular volume, basement membrane thickness and albuminuria152. However, another study showed that treatment with VEGF‐A antibodies did not improve diabetic renal abnormalities in G‐K rats 153. At the early stage of human diabetic nephropathy, increased expression of VEGF accompanied glomerular endothelial cell proliferation and extra small vessel formations in the vascular pole154,155. At the late stage of nephropathy, the expression of VEGF‐A is decreased. Baelde et al. showed that the glomerular VEGF‐A expression was decreased by 2.5‐fold and coincided with endothelial cells and the reduction of podocyte makers in the moderate‐severe stage of type 2 diabetic nephropathy156. Other studies have shown also that the expression of VEGF‐A was decreased in sclerotic lesions of nephropathy157–159.
What is the physiological role of VEGF‐A in the kidney, especially glomeruli? It is reported that treatment with anti‐VEGF antibodies to patients with cancers160 or within patients with preeclampasia161 causes proteinuria and endothelial damage, suggesting that VEGF‐A plays an important role in maintaining endothelial cell function and the glomerular filtration barrier. Supporting this, detailed reports by Quaggin et al. clearly show that VEGF‐A is necessary for forming and maintaining the glomerular filtration barrier162,163. In their reports, using a conditional Cre‐loxP targeting system, podocyte‐specific VEGF null mice failed to form a glomerular filtration barrier as a result of defects in endothelial cell migration, survival and differentiation; resulting in perinatal lethality. Loss of a single VEGF‐A allele in podocytes leads to endotheliosis, a nephrotic syndrome accompanied by glomerulosclerosis, renal failure and death at 9–12 weeks‐of‐age162,163. Furthermore, they reported that adult mice with inducible podocyte‐specific knockout of VEGF developed proteinuria, hypertension with swollen endothelial cells and intracapillary thrombus in glomeruli after 4–5 weeks after induction of conditional knockout. The results from these mice were similar to renal abnormalities of patients with thrombotic microangiopathy as a result of treatment with anti‐VEGF, bevacizumab164. Furthermore, in several other glomerular diseases, a beneficial role of VEGF has been shown through the prevention of progressive capillary rarefaction, promotion of capillary repair/regeneration, improvement of glomerulosclerosis, and renal scarring165–168. Quaggin et al. have also reported that overexpression of VEGF‐A to mRNA levels 15–20‐fold higher than in wild‐type mice, leads to collapse of the glomerular tuft, proteinuria and death from renal failure within the first week of life162,163. Therefore, it is thought that tight regulation of VEGF‐A signaling is required for development and maintenance of the glomerular filtration barrier.
Is VEGF‐A a bad or good player for the progression of diabetic nephropathy? Hohenstein et al. determined VEGF expression and its bioactivity in glomeruli of type 2 diabetic patients using specific antibodies for VEGF‐A and VEGF‐VEGFR complex154. Although VEGF expression of glomeruli is upregulated during all stages (mild, moderate and severe) of nephropathy, VEGF bioactivity in endothelial cells is only increased in mildly injured glomeruli and decreased in moderate or severe lesions. Furthermore, they showed that glomerular capillary rarefaction was linked to the degree of glomerulosclerosis and endothelial cell proliferation, showing capillary repair was markedly increased only in mildly/moderately injured glomeruli, even if apoptosis was detected in all stages. They suggest that diabetic nephropathy is associated with glomerular capillary rarefaction by an imbalance of endothelial cell proliferation, repair and apoptosis, and injury; and reduced VEGF activity might be an indicator of an insufficient capillary repair reaction154. Therefore, if increased VEGF expression occurs as a reaction of compensation for the damage of glomerular endothelial cells, inhibition of VEGF should not be given as a treatment for diabetic nephropathy. However, further studies are needed to conclude whether VEGF‐A is or is not an endogenous protective factor for diabetic nephropathy.
Activated Protein C
APC is also an endogenous protective factor for endothelial cells. The production of APC is dependent on binding between thrombomodulin and thrombin, which occurs on the surface of the endothelial cells. The thrombin/thrombomodulin complex catalyses the conversion of protein C to its activation form, APC. APC acts directly on cells to exert multiple cytoprotective effects including anti‐inflammation, anti‐apoptotic activities and protection of endothelial barrier function through the endothelial protein C receptor, protease‐activated receptor‐1 or sphingosine‐1 receptor169.
It has been reported that plasma thrombomodulin levels, which are thought to reflect loss of thrombomodulin from the endothelium and reduced levels of APC, are elevated in patients with diabetes, and the impairment of thrombomodulin/protein C system is associated with diabetic complications, such as nephropathy and neuropathy170,171. Recently, Isermann et al. showed that impaired APC formation as a result of reduced thrombomodulin expression is associated with diabetic nephropathy; and the increased levels of APC can prevent diabetic nephropathy through anti‐apoptotic effects against diabetes‐induced endothelial cells and podocytes172.
Recent studies described in the present review identify various mechanisms by which hyperglycemia can induce adverse effects to cause diabetic complications. Inhibition of AR, PKC, AGE/RAGE interaction or oxidative stress should provide useful targets for treatment. Treatments for these targets have been successful in animal models with diabetes; however, many clinical trials using agents directly against these targets have not shown a robust effort to prevent or stop the various diabetic complications. The lack of efficacy of these agents suggests that other mechanisms are involved in the development of diabetic complications.
We proposed that the function of endogenous protective factors, including insulin, VEGF, PDGF and APC, are important for vascular homeostasis and might also be impaired by diabetes (Figure 3). Therefore, further studies are needed to understand the loss of protective factors in the development of diabetic complications. New therapies need to inhibit hyperglycemia’s toxic effect and enhance endogenous protective factors in order to be effective.
Figure 3.
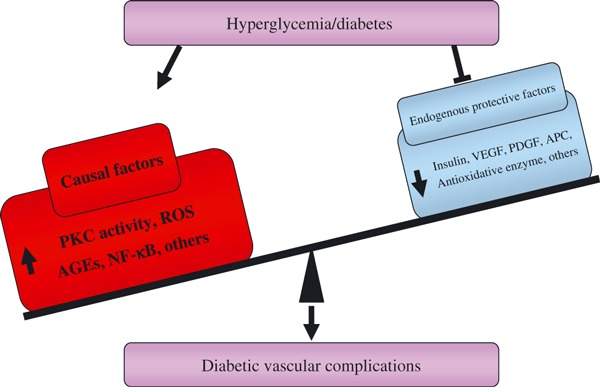
Dual action of hyperglycemia to induce diabetic vascular complications. Induction of toxic pathways and inhibition of endogenous protective factors. AGE, advanced glycation end‐products; APC, activated protein C; NF‐κB, nuclear factor‐κB; PDFG, platelet‐derived growth factor; PKC, protein kinase C; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor.
Acknowledgements
Dr King is supported by National Institutes of Health grants 5 R01 DK053105, R01 EY016150, American Diabetes Association 1‐08‐RA‐93 and by Joslin Diabetes Center DERC P30 DK036836. None of the authors have any financial support or relationships that might pose a conflict of interest.
References
- 1.International Diabetes Foundation , 2009. (Accessed 2009, at http://www.diabetesatlas.org/content/foreward)
- 2.The Diabetes Control and Complications Trial Research Group . The effect of intensive treatment of diabetes on the development and progression of long‐term complications in insulin‐dependent diabetes mellitus. N Engl J Med 1993; 329: 977–986 [DOI] [PubMed] [Google Scholar]
- 3.UK Prospective Diabetes Study (UKPDS) Group . Effect of intensive blood‐glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998; 352: 854–865 [PubMed] [Google Scholar]
- 4.Nathan DM, Cleary PA, Backlund JY, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353: 2643–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nathan DM, Lachin J, Cleary P, et al. Intensive diabetes therapy and carotid intima‐media thickness in type 1 diabetes mellitus. N Engl J Med 2003; 348: 2294–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rask‐Madsen C, King GL. Kidney complications: factors that protect the diabetic vasculature. Nat Med 2010; 16: 40–41 [DOI] [PubMed] [Google Scholar]
- 7.Krolewski AS, Warram JH, Christlieb AR, Busick EJ, Kahn CR. The changing natural history of nephropathy in type I diabetes. Am J Med 1985; 78: 785–794 [DOI] [PubMed] [Google Scholar]
- 8.Andersen AR, Christiansen JS, Andersen JK, Kreiner S, Deckert T. Diabetic nephropathy in Type 1 (insulin‐dependent) diabetes: an epidemiological study. Diabetologia 1983; 25: 496–501 [DOI] [PubMed] [Google Scholar]
- 9.Keenan HA, Costacou T, Sun JK, et al. Clinical factors associated with resistance to microvascular complications in diabetic patients of extreme disease duration: the 50‐year medalist study. Diabetes Care 2007; 30: 1995–1997 [DOI] [PubMed] [Google Scholar]
- 10.White NH, Sun W, Cleary PA, et al. Prolonged effect of intensive therapy on the risk of retinopathy complications in patients with type 1 diabetes mellitus: 10 years after the Diabetes Control and Complications Trial. Arch Ophthalmol 2008; 126: 1707–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group . Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA 2003; 290: 2159–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin CL, Albers J, Herman WH, et al. Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes Care 2006; 29: 340–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheetz MJ, King GL. Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. JAMA 2002; 288: 2579–2588 [DOI] [PubMed] [Google Scholar]
- 14.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001; 414: 813–820 [DOI] [PubMed] [Google Scholar]
- 15.Williamson JR, Chang K, Frangos M, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 1993; 42: 801–813 [DOI] [PubMed] [Google Scholar]
- 16.Tesfamariam B. Free radicals in diabetic endothelial cell dysfunction. Free Radic Biol Med 1994; 16: 383–391 [DOI] [PubMed] [Google Scholar]
- 17.Chung SS, Ho EC, Lam KS, Chung SK. Contribution of polyol pathway to diabetes‐induced oxidative stress. J Am Soc Nephrol 2003; 14(8 Suppl 3): S233–S236 [DOI] [PubMed] [Google Scholar]
- 18.Robison WG Jr, Nagata M, Tillis TN, Laver N, Kinoshita JH. Aldose reductase and pericyte‐endothelial cell contacts in retina and optic nerve. Invest Ophthalmol Vis Sci 1989; 30: 2293–2299 [PubMed] [Google Scholar]
- 19.Kador PF, Akagi Y, Takahashi Y, Ikebe H, Wyman M, Kinoshita JH. Prevention of retinal vessel changes associated with diabetic retinopathy in galactose‐fed dogs by aldose reductase inhibitors. Arch Ophthalmol 1990; 108: 1301–1309 [DOI] [PubMed] [Google Scholar]
- 20.Mauer SM, Steffes MW, Azar S, Brown DM. Effects of sorbinil on glomerular structure and function in long‐term‐diabetic rats. Diabetes 1989; 38: 839–846 [DOI] [PubMed] [Google Scholar]
- 21.Yue DK, Hanwell MA, Satchell PM, Turtle JR. The effect of aldose reductase inhibition on motor nerve conduction velocity in diabetic rats. Diabetes 1982; 31: 789–794 [DOI] [PubMed] [Google Scholar]
- 22.Cameron NE, Leonard MB, Ross IS, Whiting PH. The effects of sorbinil on peripheral nerve conduction velocity, polyol concentrations and morphology in the streptozotocin‐diabetic rat. Diabetologia 1986; 29: 168–174 [DOI] [PubMed] [Google Scholar]
- 23.Engerman RL, Kern TS, Larson ME. Nerve conduction and aldose reductase inhibition during 5 years of diabetes or galactosaemia in dogs. Diabetologia 1994; 37: 141–144 [DOI] [PubMed] [Google Scholar]
- 24.Sorbinil Retinopathy Trial Research Group . A randomized trial of sorbinil, an aldose reductase inhibitor, in diabetic retinopathy. Arch Ophthalmol 1990; 108: 1234–1244 [DOI] [PubMed] [Google Scholar]
- 25.Hotta N, Toyota T, Matsuoka K, et al. Clinical efficacy of fidarestat, a novel aldose reductase inhibitor, for diabetic peripheral neuropathy: a 52‐week multicenter placebo‐controlled double‐blind parallel group study. Diabetes Care 2001; 24: 1776–1782 [DOI] [PubMed] [Google Scholar]
- 26.Matsuoka K, Sakamoto N, Akanuma Y, et al. A long‐term effect of epalrestat on motor conduction velocity of diabetic patients: ARI‐Diabetes Complications Trial (ADCT). Diabetes Res Clin Pract 2007; 77(Suppl 1): S263–S268 [DOI] [PubMed] [Google Scholar]
- 27.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 1992; 258: 607–614 [DOI] [PubMed] [Google Scholar]
- 28.Das Evcimen N, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res 2007; 55: 498–510 [DOI] [PubMed] [Google Scholar]
- 29.Shiba T, Inoguchi T, Sportsman JR, Heath WF, Bursell S, King GL. Correlation of diacylglycerol level and protein kinase C activity in rat retina to retinal circulation. Am J Physiol 1993; 265(5 Pt 1): E783–E793 [DOI] [PubMed] [Google Scholar]
- 30.Craven PA, Davidson CM, DeRubertis FR. Increase in diacylglycerol mass in isolated glomeruli by glucose from de novo synthesis of glycerolipids. Diabetes 1990; 39: 667–674 [DOI] [PubMed] [Google Scholar]
- 31.Ishii H, Jirousek MR, Koya D, et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science 1996; 272: 728–731 [DOI] [PubMed] [Google Scholar]
- 32.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A 1992; 89: 11059–11063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayo SH, Radnik R, Garoni JA, Troyer DA, Kreisberg JI. High glucose increases diacylglycerol mass and activates protein kinase C in mesangial cell cultures. Am J Physiol 1991; 261(4 Pt 2): F571–F577 [DOI] [PubMed] [Google Scholar]
- 34.Studer RK, Craven PA, DeRubertis FR. Role for protein kinase C in the mediation of increased fibronectin accumulation by mesangial cells grown in high‐glucose medium. Diabetes 1993; 42: 118–126 [DOI] [PubMed] [Google Scholar]
- 35.Konishi H, Tanaka M, Takemura Y, et al. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci U S A 1997; 94: 11233–11237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404: 787–790 [DOI] [PubMed] [Google Scholar]
- 37.Go M, Sekiguchi K, Nomura H, Kikkawa U, Nishizuka Y. Further studies on the specificity of diacylglycerol for protein kinase C activation. Biochem Biophys Res Commun 1987; 144: 598–605 [DOI] [PubMed] [Google Scholar]
- 38.Kuroki T, Inoguchi T, Umeda F, Nawata H. Effect of eicosapentaenoic acid on glucose‐induced diacylglycerol synthesis in cultured bovine aortic endothelial cells. Biochem Biophys Res Commun 1998; 247: 473–477 [DOI] [PubMed] [Google Scholar]
- 39.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes 1998; 47: 859–866 [DOI] [PubMed] [Google Scholar]
- 40.Aiello LP, Bursell SE, Clermont A, et al. Vascular endothelial growth factor‐induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta‐isoform‐selective inhibitor. Diabetes 1997; 46: 1473–1480 [DOI] [PubMed] [Google Scholar]
- 41.Suzuma K, Naruse K, Suzuma I, et al. Vascular endothelial growth factor induces expression of connective tissue growth factor via KDR, Flt1, and phosphatidylinositol 3‐kinase‐akt‐dependent pathways in retinal vascular cells. J Biol Chem 2000; 275: 40725–40731 [DOI] [PubMed] [Google Scholar]
- 42.Suzuma K, Takahara N, Suzuma I, et al. Characterization of protein kinase C beta isoform’s action on retinoblastoma protein phosphorylation, vascular endothelial growth factor‐induced endothelial cell proliferation, and retinal neovascularization. Proc Natl Acad Sci U S A 2002; 99: 721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park JY, Takahara N, Gabriele A, et al. Induction of endothelin‐1 expression by glucose: an effect of protein kinase C activation. Diabetes 2000; 49: 1239–1248 [DOI] [PubMed] [Google Scholar]
- 44.Yokota T, Ma RC, Park JY, et al. Role of protein kinase C on the expression of platelet‐derived growth factor and endothelin‐1 in the retina of diabetic rats and cultured retinal capillary pericytes. Diabetes 2003; 52: 838–845 [DOI] [PubMed] [Google Scholar]
- 45.Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K, King GL. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor‐beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest 1997; 100: 115–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohshiro Y, Ma RC, Yasuda Y, et al. Reduction of diabetes‐induced oxidative stress, fibrotic cytokine expression, and renal dysfunction in protein kinase Cbeta‐null mice. Diabetes 2006; 55: 3112–3120 [DOI] [PubMed] [Google Scholar]
- 47.Way KJ, Isshiki K, Suzuma K, et al. Expression of connective tissue growth factor is increased in injured myocardium associated with protein kinase C beta2 activation and diabetes. Diabetes 2002; 51: 2709–2718 [DOI] [PubMed] [Google Scholar]
- 48.Feener EP, Xia P, Inoguchi T, Shiba T, Kunisaki M, King GL. Role of protein kinase C in glucose‐ and angiotensin II‐induced plasminogen activator inhibitor expression. Contrib Nephrol 1996; 118: 180–187 [DOI] [PubMed] [Google Scholar]
- 49.Ahn JD, Morishita R, Kaneda Y, et al. Transcription factor decoy for activator protein‐1 (AP‐1) inhibits high glucose‐ and angiotensin II‐induced type 1 plasminogen activator inhibitor (PAI‐1) gene expression in cultured human vascular smooth muscle cells. Diabetologia 2001; 44: 713–720 [DOI] [PubMed] [Google Scholar]
- 50.Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C‐‐dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000; 49: 1939–1945 [DOI] [PubMed] [Google Scholar]
- 51.Kitada M, Koya D, Sugimoto T, et al. Translocation of glomerular p47phox and p67phox by protein kinase C‐beta activation is required for oxidative stress in diabetic nephropathy. Diabetes 2003; 52: 2603–2614 [DOI] [PubMed] [Google Scholar]
- 52.Pieper GM, Riaz ul H. Activation of nuclear factor‐kappaB in cultured endothelial cells by increased glucose concentration: prevention by calphostin C. J Cardiovasc Pharmacol 1997; 30: 528–532 [DOI] [PubMed] [Google Scholar]
- 53.Yerneni KK, Bai W, Khan BV, Medford RM, Natarajan R. Hyperglycemia‐induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells. Diabetes 1999; 48: 855–864 [DOI] [PubMed] [Google Scholar]
- 54.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev 2008; 88: 1341–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kunisaki M, Bursell SE, Umeda F, Nawata H, King GL. Normalization of diacylglycerol‐protein kinase C activation by vitamin E in aorta of diabetic rats and cultured rat smooth muscle cells exposed to elevated glucose levels. Diabetes 1994; 43: 1372–1377 [DOI] [PubMed] [Google Scholar]
- 56.Dlugosz JA, Munk S, Ispanovic E, Goldberg HJ, Whiteside CI. Mesangial cell filamentous actin disassembly and hypocontractility in high glucose are mediated by PKC‐zeta. Am J Physiol Renal Physiol 2002; 282: F151–F163 [DOI] [PubMed] [Google Scholar]
- 57.Koya D, Haneda M, Nakagawa H, et al. Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC beta inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. FASEB J 2000; 14: 439–447 [DOI] [PubMed] [Google Scholar]
- 58.Kim H, Sasaki T, Maeda K, Koya D, Kashiwagi A, Yasuda H. Protein kinase Cbeta selective inhibitor LY333531 attenuates diabetic hyperalgesia through ameliorating cGMP level of dorsal root ganglion neurons. Diabetes 2003; 52: 2102–2109 [DOI] [PubMed] [Google Scholar]
- 59.Nakamura J, Kato K, Hamada Y, et al. A protein kinase C‐beta‐selective inhibitor ameliorates neural dysfunction in streptozotocin‐induced diabetic rats. Diabetes 1999; 48: 2090–2095 [DOI] [PubMed] [Google Scholar]
- 60.Harja E, Chang JS, Lu Y, et al. Mice deficient in PKCbeta and apolipoprotein E display decreased atherosclerosis. FASEB J 2009; 23: 1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beckman JA, Goldfine AB, Gordon MB, Garrett LA, Creager MA. Inhibition of protein kinase Cbeta prevents impaired endothelium‐dependent vasodilation caused by hyperglycemia in humans. Circ Res 2002; 90: 107–111 [DOI] [PubMed] [Google Scholar]
- 62.Tuttle KR. Protein kinase C‐beta inhibition for diabetic kidney disease. Diabetes Res Clin Pract 2008; 82(Suppl 1): S70–S74 [DOI] [PubMed] [Google Scholar]
- 63.PKC‐DRS Study Group . The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: initial results of the Protein Kinase C beta Inhibitor Diabetic Retinopathy Study (PKC‐DRS) multicenter randomized clinical trial. Diabetes 2005; 54: 2188–2197 [DOI] [PubMed] [Google Scholar]
- 64.Scott JA, King GL. Oxidative stress and antioxidant treatment in diabetes. Ann N Y Acad Sci 2004; 1031: 204–213 [DOI] [PubMed] [Google Scholar]
- 65.Xu J, Zou MH. Molecular insights and therapeutic targets for diabetic endothelial dysfunction. Circulation 2009; 120: 1266–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Newsholme P, Haber EP, Hirabara SM, et al. Diabetes associated cell stress and dysfunction: role of mitochondrial and non‐mitochondrial ROS production and activity. J Physiol 2007;583(Pt 1): 9–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Z, Apse K, Pang J, Stanton RC. High glucose inhibits glucose‐6‐phosphate dehydrogenase via cAMP in aortic endothelial cells. J Biol Chem 2000; 275: 40042–40047 [DOI] [PubMed] [Google Scholar]
- 68.Xu Y, Osborne BW, Stanton RC. Diabetes causes inhibition of glucose‐6‐phosphate dehydrogenase via activation of PKA, which contributes to oxidative stress in rat kidney cortex. Am J Physiol 2005; 289: F1040–F1047 [DOI] [PubMed] [Google Scholar]
- 69.Lee EA, Seo JY, Jiang Z, et al. Reactive oxygen species mediate high glucose‐induced plasminogen activator inhibitor‐1 up‐regulation in mesangial cells and in diabetic kidney. Kidney Int 2005; 67: 1762–1771 [DOI] [PubMed] [Google Scholar]
- 70.Mezzetti A, Cipollone F, Cuccurullo F. Oxidative stress and cardiovascular complications in diabetes: isoprostanes as new markers on an old paradigm. Cardiovasc Res 2000; 47: 475–488 [DOI] [PubMed] [Google Scholar]
- 71.Leinonen J, Lehtimaki T, Toyokuni S, et al. New biomarker evidence of oxidative DNA damage in patients with non‐insulin‐dependent diabetes mellitus. FEBS Lett 1997; 417: 150–152 [DOI] [PubMed] [Google Scholar]
- 72.Bedard K, Krause KH. The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007; 87: 245–313 [DOI] [PubMed] [Google Scholar]
- 73.Chinen I, Shimabukuro M, Yamakawa K, et al. Vascular lipotoxicity: endothelial dysfunction via fatty‐acid‐induced reactive oxygen species overproduction in obese Zucker diabetic fatty rats. Endocrinology 2007; 148: 160–165 [DOI] [PubMed] [Google Scholar]
- 74.Urakawa H, Katsuki A, Sumida Y, et al. Oxidative stress is associated with adiposity and insulin resistance in men. J Clin Endocrinol Metab 2003; 88: 4673–4676 [DOI] [PubMed] [Google Scholar]
- 75.Diaz‐Flores M, Ibanez‐Hernandez MA, Galvan RE, et al. Glucose‐6‐phosphate dehydrogenase activity and NADPH/NADP+ ratio in liver and pancreas are dependent on the severity of hyperglycemia in rat. Life Sci 2006; 78: 2601–2607 [DOI] [PubMed] [Google Scholar]
- 76.Ruiz C, Alegria A, Barbera R, Farre R, Lagarda MJ. Lipid peroxidation and antioxidant enzyme activities in patients with type 1 diabetes mellitus. Scand J Clin Lab Invest 1999; 59: 99–105 [DOI] [PubMed] [Google Scholar]
- 77.Will JC, Ford ES, Bowman BA. Serum vitamin C concentrations and diabetes: findings from the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Clin Nutr 1999; 70: 49–52 [DOI] [PubMed] [Google Scholar]
- 78.Brezniceanu ML, Liu F, Wei CC, et al. Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney Int 2007; 71: 912–923 [DOI] [PubMed] [Google Scholar]
- 79.El‐Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 2008; 205: 2409–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lonn E, Yusuf S, Hoogwerf B, et al. Effects of vitamin E on cardiovascular and microvascular outcomes in high‐risk patients with diabetes: results of the HOPE study and MICRO‐HOPE substudy. Diabetes Care 2002; 25: 1919–1927 [DOI] [PubMed] [Google Scholar]
- 81.Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin E supplementation and cardiovascular events in high‐risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 2000; 342: 154–160 [DOI] [PubMed] [Google Scholar]
- 82.Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 2006; 114: 597–605 [DOI] [PubMed] [Google Scholar]
- 83.Ahmad FK, He Z, King GL. Molecular targets of diabetic cardiovascular complications. Curr Drug Targets 2005; 6: 487–494 [DOI] [PubMed] [Google Scholar]
- 84.Berg TJ, Bangstad HJ, Torjesen PA, Osterby R, Bucala R, Hanssen KF. Advanced glycation end products in serum predict changes in the kidney morphology of patients with insulin‐dependent diabetes mellitus. Metabolism 1997; 46: 661–665 [DOI] [PubMed] [Google Scholar]
- 85.Suzuki D, Miyata T, Saotome N, et al. Immunohistochemical evidence for an increased oxidative stress and carbonyl modification of proteins in diabetic glomerular lesions. J Am Soc Nephrol 1999; 10: 822–832 [DOI] [PubMed] [Google Scholar]
- 86.Stitt AW, Li YM, Gardiner TA, Bucala R, Archer DB, Vlassara H. Advanced glycation end products (AGEs) co‐localize with AGE receptors in the retinal vasculature of diabetic and of AGE‐infused rats. Am J Pathol 1997; 150: 523–531 [PMC free article] [PubMed] [Google Scholar]
- 87.Hammes HP, Wellensiek B, Kloting I, Sickel E, Bretzel RG, Brownlee M. The relationship of glycaemic level to advanced glycation end‐product (AGE) accumulation and retinal pathology in the spontaneous diabetic hamster. Diabetologia 1998; 41: 165–170 [DOI] [PubMed] [Google Scholar]
- 88.Giardino I, Edelstein D, Brownlee M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity. A model for intracellular glycosylation in diabetes. J Clin Invest 1994; 94: 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rosca MG, Mustata TG, Kinter MT, et al. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am J Physiol Renal Physiol 2005; 289: F420–F430 [DOI] [PubMed] [Google Scholar]
- 90.Tanaka S, Avigad G, Brodsky B, Eikenberry EF. Glycation induces expansion of the molecular packing of collagen. J Mol Biol 1988; 203: 495–505 [DOI] [PubMed] [Google Scholar]
- 91.Haitoglou CS, Tsilibary EC, Brownlee M, Charonis AS. Altered cellular interactions between endothelial cells and nonenzymatically glucosylated laminin/type IV collagen. J Biol Chem 1992; 267: 12404–12407 [PubMed] [Google Scholar]
- 92.He Z, King GL. Microvascular complications of diabetes. Endocrinol Metab Clin North Am 2004; 33: 215–238, xi–xii. [DOI] [PubMed] [Google Scholar]
- 93.Bucala R, Makita Z, Koschinsky T, Cerami A, Vlassara H. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc Natl Acad Sci U S A 1993; 90: 6434–6438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bucala R, Mitchell R, Arnold K, Innerarity T, Vlassara H, Cerami A. Identification of the major site of apolipoprotein B modification by advanced glycosylation end products blocking uptake by the low density lipoprotein receptor. J Biol Chem 1995; 270: 10828–10832 [DOI] [PubMed] [Google Scholar]
- 95.Schmidt AM, Stern DM. RAGE: a new target for the prevention and treatment of the vascular and inflammatory complications of diabetes. Trends Endocrinol Metab 2000; 11: 368–375 [DOI] [PubMed] [Google Scholar]
- 96.Tanji N, Markowitz GS, Fu C, et al. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol 2000; 11: 1656–1666 [DOI] [PubMed] [Google Scholar]
- 97.Yamamoto Y, Kato I, Doi T, et al. Development and prevention of advanced diabetic nephropathy in RAGE‐overexpressing mice. J Clin Invest 2001; 108: 261–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)‐mediated neurite outgrowth and activation of NF‐kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem 1999; 274: 19919–19924 [DOI] [PubMed] [Google Scholar]
- 99.Bierhaus A, Schiekofer S, Schwaninger M, et al. Diabetes‐associated sustained activation of the transcription factor nuclear factor‐kappaB. Diabetes 2001; 50: 2792–2808 [DOI] [PubMed] [Google Scholar]
- 100.Yan SD, Schmidt AM, Anderson GM, et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem 1994; 269: 9889–9897 [PubMed] [Google Scholar]
- 101.Basta G, Lazzerini G, Massaro M, et al. Advanced glycation end products activate endothelium through signal‐transduction receptor RAGE: a mechanism for amplification of inflammatory responses. Circulation 2002; 105: 816–822 [DOI] [PubMed] [Google Scholar]
- 102.Schmidt AM, Hori O, Chen JX, et al. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule‐1 (VCAM‐1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest 1995; 96: 1395–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 2001; 280: E685–E694 [DOI] [PubMed] [Google Scholar]
- 104.Vlassara H, Li YM, Imani F, et al. Identification of galectin‐3 as a high‐affinity binding protein for advanced glycation end products (AGE): a new member of the AGE‐receptor complex. Mol Med 1995; 1: 634–646 [PMC free article] [PubMed] [Google Scholar]
- 105.el Khoury J, Thomas CA, Loike JD, Hickman SE, Cao L, Silverstein SC. Macrophages adhere to glucose‐modified basement membrane collagen IV via their scavenger receptors. J Biol Chem 1994; 269: 10197–10200 [PubMed] [Google Scholar]
- 106.Soulis‐Liparota T, Cooper M, Papazoglou D, Clarke B, Jerums G. Retardation by aminoguanidine of development of albuminuria, mesangial expansion, and tissue fluorescence in streptozocin‐induced diabetic rat. Diabetes 1991; 40: 1328–1334 [DOI] [PubMed] [Google Scholar]
- 107.Hammes HP, Martin S, Federlin K, Geisen K, Brownlee M. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci U S A 1991; 88: 11555–11558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bakris GL, Bank AJ, Kass DA, Neutel JM, Preston RA, Oparil S. Advanced glycation end‐product cross‐link breakers. A novel approach to cardiovascular pathologies related to the aging process. Am J Hypertens 2004; 17(12 Pt 2): 23S–30S [DOI] [PubMed] [Google Scholar]
- 109.Candido R, Forbes JM, Thomas MC, et al. A breaker of advanced glycation end products attenuates diabetes‐induced myocardial structural changes. Circ Res 2003; 92: 785–792 [DOI] [PubMed] [Google Scholar]
- 110.Forbes JM, Yee LT, Thallas V, et al. Advanced glycation end product interventions reduce diabetes‐accelerated atherosclerosis. Diabetes 2004; 53: 1813–1823 [DOI] [PubMed] [Google Scholar]
- 111.Forbes JM, Thallas V, Thomas MC, et al. The breakdown of preexisting advanced glycation end products is associated with reduced renal fibrosis in experimental diabetes. FASEB J 2003; 17: 1762–1764 [DOI] [PubMed] [Google Scholar]
- 112.Lassila M, Seah KK, Allen TJ, et al. Accelerated nephropathy in diabetic apolipoprotein e‐knockout mouse: role of advanced glycation end products. J Am Soc Nephrol 2004; 15: 2125–2138 [DOI] [PubMed] [Google Scholar]
- 113.Bucciarelli LG, Wendt T, Qu W, et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E‐null mice. Circulation 2002; 106: 2827–2835 [DOI] [PubMed] [Google Scholar]
- 114.Sakaguchi T, Yan SF, Yan SD, et al. Central role of RAGE‐dependent neointimal expansion in arterial restenosis. J Clin Invest 2003; 111: 959–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou Z, Wang K, Penn MS, et al. Receptor for AGE (RAGE) mediates neointimal formation in response to arterial injury. Circulation 2003; 107: 2238–2243 [DOI] [PubMed] [Google Scholar]
- 116.Wendt TM, Tanji N, Guo J, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol 2003; 162: 1123–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Buse MG. Hexosamines, insulin resistance, and the complications of diabetes: current status. Am J Physiol Endocrinol Metab 2006; 290: E1–E8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Han I, Kudlow JE. Reduced O glycosylation of Sp1 is associated with increased proteasome susceptibility. Mol Cell Biol 1997; 17: 2550–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Roos MD, Su K, Baker JR, Kudlow JE. O glycosylation of an Sp1‐derived peptide blocks known Sp1 protein interactions. Mol Cell Biol 1997; 17: 6472–6480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang X, Su K, Roos MD, Chang Q, Paterson AJ, Kudlow JE. O‐linkage of N‐acetylglucosamine to Sp1 activation domain inhibits its transcriptional capability. Proc Natl Acad Sci U S A 2001; 98: 6611–6616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen YQ, Su M, Walia RR, Hao Q, Covington JW, Vaughan DE. Sp1 sites mediate activation of the plasminogen activator inhibitor‐1 promoter by glucose in vascular smooth muscle cells. J Biol Chem 1998; 273: 8225–8231 [DOI] [PubMed] [Google Scholar]
- 122.Du XL, Edelstein D, Rossetti L, et al. Hyperglycemia‐induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor‐1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A 2000; 97: 12222–12226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.James LR, Fantus IG, Goldberg H, Ly H, Scholey JW. Overexpression of GFAT activates PAI‐1 promoter in mesangial cells. Am J Physiol Renal Physiol 2000; 279: F718–F727 [DOI] [PubMed] [Google Scholar]
- 124.Weigert C, Brodbeck K, Sawadogo M, Haring HU, Schleicher ED. Upstream stimulatory factor (USF) proteins induce human TGF‐beta1 gene activation via the glucose‐response element‐1013/‐1002 in mesangial cells: up‐regulation of USF activity by the hexosamine biosynthetic pathway. J Biol Chem 2004; 279: 15908–15915 [DOI] [PubMed] [Google Scholar]
- 125.Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest 2001; 108: 1341–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med 2009; 47: 1304–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rask‐Madsen C, King GL. Mechanisms of Disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab 2007; 3: 46–56 [DOI] [PubMed] [Google Scholar]
- 128.Mogensen CE, Keane WF, Bennett PH, et al. Prevention of diabetic renal disease with special reference to microalbuminuria. Lancet 1995; 346: 1080–1084 [DOI] [PubMed] [Google Scholar]
- 129.Montagnani M, Chen H, Barr VA, Quon MJ. Insulin‐stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). J Biol Chem 2001; 276: 30392–30398 [DOI] [PubMed] [Google Scholar]
- 130.Kuboki K, Jiang ZY, Takahara N, et al. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo: a specific vascular action of insulin. Circulation 2000; 101: 676–681 [DOI] [PubMed] [Google Scholar]
- 131.Jiang ZY, Lin YW, Clemont A, et al. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest 1999; 104: 447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cusi K, Maezono K, Osman A, et al. Insulin resistance differentially affects the PI 3‐kinase‐ and MAP kinase‐mediated signaling in human muscle. J Clin Invest 2000; 105: 311–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.He Z, Opland DM, Way KJ, et al. Regulation of vascular endothelial growth factor expression and vascularization in the myocardium by insulin receptor and PI3K/Akt pathways in insulin resistance and ischemia. Arterioscler Thromb Vasc Biol 2006; 26: 787–793 [DOI] [PubMed] [Google Scholar]
- 134.Naruse K, Rask‐Madsen C, Takahara N, et al. Activation of vascular protein kinase C‐beta inhibits Akt‐dependent endothelial nitric oxide synthase function in obesity‐associated insulin resistance. Diabetes 2006; 55: 691–698 [DOI] [PubMed] [Google Scholar]
- 135.Vicent D, Ilany J, Kondo T, et al. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J Clin Invest 2003; 111: 1373–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Geraldes P, Yagi K, Ohshiro Y, et al. Selective regulation of heme oxygenase‐1 expression and function by insulin through IRS1/phosphoinositide 3‐kinase/Akt‐2 pathway. J Biol Chem 2008; 283: 34327–34336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Andrae J, Gallini R, Betsholtz C. Role of platelet‐derived growth factors in physiology and medicine. Genes Dev 2008; 22: 1276–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF‐B‐deficient mice. Science 1997; 277: 242–245 [DOI] [PubMed] [Google Scholar]
- 139.Enge M, Bjarnegard M, Gerhardt H, et al. Endothelium‐specific platelet‐derived growth factor‐B ablation mimics diabetic retinopathy. EMBO J 2002; 21: 4307–4316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cogan DG, Toussaint D, Kuwabara T. Retinal vascular patterns. IV. Diabetic retinopathy. Arch Ophthalmol 1961; 66: 366–378 [DOI] [PubMed] [Google Scholar]
- 141.Kuwabara T, Cogan DG. Retinal vascular patterns. VII. Acellular change. Invest Ophthalmol 1965; 4: 1049–1064 [PubMed] [Google Scholar]
- 142.Bresnick GH, Davis MD, Myers FL, de Venecia G. Clinicopathologic correlations in diabetic retinopathy. II. Clinical and histologic appearances of retinal capillary microaneurysms. Arch Ophthalmol 1977; 95: 1215–1220 [DOI] [PubMed] [Google Scholar]
- 143.Geraldes P, Hiraoka‐Yamamoto J, Matsumoto M, et al. Activation of PKC‐delta and SHP‐1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med 2009; 15: 1298–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev 2004; 25: 581–611 [DOI] [PubMed] [Google Scholar]
- 145.Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 1994; 331: 1480–1487 [DOI] [PubMed] [Google Scholar]
- 146.Hammes HP, Lin J, Bretzel RG, Brownlee M, Breier G. Upregulation of the vascular endothelial growth factor/vascular endothelial growth factor receptor system in experimental background diabetic retinopathy of the rat. Diabetes 1998; 47: 401–406 [DOI] [PubMed] [Google Scholar]
- 147.Boulton M, Foreman D, Williams G, McLeod D. VEGF localisation in diabetic retinopathy. Br J Ophthalmol 1998; 82: 561–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Simo R, Hernandez C. Intravitreous anti‐VEGF for diabetic retinopathy: hopes and fears for a new therapeutic strategy. Diabetologia 2008; 51: 1574–1580 [DOI] [PubMed] [Google Scholar]
- 149.Cooper ME, Vranes D, Youssef S, et al. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR‐2 in experimental diabetes. Diabetes 1999; 48: 2229–2239 [DOI] [PubMed] [Google Scholar]
- 150.Braun L, Kardon T, Reisz‐Porszasz ZS, Banhegyi G, Mandl J. The regulation of the induction of vascular endothelial growth factor at the onset of diabetes in spontaneously diabetic rats. Life Sci 2001; 69: 2533–2542 [DOI] [PubMed] [Google Scholar]
- 151.de Vriese AS, Tilton RG, Elger M, Stephan CC, Kriz W, Lameire NH. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol 2001; 12: 993–1000 [DOI] [PubMed] [Google Scholar]
- 152.Flyvbjerg A, Dagnaes‐Hansen F, De Vriese AS, Schrijvers BF, Tilton RG, Rasch R. Amelioration of long‐term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes 2002; 51: 3090–3094 [DOI] [PubMed] [Google Scholar]
- 153.Schrijvers BF, De Vriese AS, Tilton RG, et al. Inhibition of vascular endothelial growth factor (VEGF) does not affect early renal changes in a rat model of lean type 2 diabetes. Horm Metab Res 2005; 37: 21–25 [DOI] [PubMed] [Google Scholar]
- 154.Hohenstein B, Hausknecht B, Boehmer K, Riess R, Brekken RA, Hugo CP. Local VEGF activity but not VEGF expression is tightly regulated during diabetic nephropathy in man. Kidney Int 2006; 69: 1654–1661 [DOI] [PubMed] [Google Scholar]
- 155.Kanesaki Y, Suzuki D, Uehara G, et al. Vascular endothelial growth factor gene expression is correlated with glomerular neovascularization in human diabetic nephropathy. Am J Kidney Dis 2005; 45: 288–294 [DOI] [PubMed] [Google Scholar]
- 156.Baelde HJ, Eikmans M, Lappin DW, et al. Reduction of VEGF‐A and CTGF expression in diabetic nephropathy is associated with podocyte loss. Kidney Int 2007; 71: 637–645 [DOI] [PubMed] [Google Scholar]
- 157.Shulman K, Rosen S, Tognazzi K, Manseau EJ, Brown LF. Expression of vascular permeability factor (VPF/VEGF) is altered in many glomerular diseases. J Am Soc Nephrol 1996; 7: 661–666 [DOI] [PubMed] [Google Scholar]
- 158.Baelde HJ, Eikmans M, Doran PP, Lappin DW, de Heer E, Bruijn JA. Gene expression profiling in glomeruli from human kidneys with diabetic nephropathy. Am J Kidney Dis 2004; 43: 636–650 [DOI] [PubMed] [Google Scholar]
- 159.Bortoloso E, Del Prete D, Dalla Vestra M, et al. Quantitave and qualitative changes in vascular endothelial growth factor gene expression in glomeruli of patients with type 2 diabetes. Eur J Endocrinol 2004; 150: 799–807 [DOI] [PubMed] [Google Scholar]
- 160.Zhu X, Wu S, Dahut WL, Parikh CR. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: systematic review and meta‐analysis. Am J Kidney Dis 2007; 49: 186–193 [DOI] [PubMed] [Google Scholar]
- 161.Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms‐like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 2003; 111: 649–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Eremina V, Sood M, Haigh J, et al. Glomerular‐specific alterations of VEGF‐A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 2003; 111: 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Eremina V, Baelde HJ, Quaggin SE. Role of the VEGF – a signaling pathway in the glomerulus: evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol 2007; 106: p32–p37 [DOI] [PubMed] [Google Scholar]
- 164.Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 2008; 358: 1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kang DH, Hughes J, Mazzali M, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model: II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. J Am Soc Nephrol 2001; 12: 1448–1457 [DOI] [PubMed] [Google Scholar]
- 166.Kang DH, Kim YG, Andoh TF, et al. Post‐cyclosporine‐mediated hypertension and nephropathy: amelioration by vascular endothelial growth factor. Am J Physiol Renal Physiol 2001; 280: F727–F736 [DOI] [PubMed] [Google Scholar]
- 167.Masuda Y, Shimizu A, Mori T, et al. Vascular endothelial growth factor enhances glomerular capillary repair and accelerates resolution of experimentally induced glomerulonephritis. Am J Pathol 2001; 159: 599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ostendorf T, Kunter U, Eitner F, et al. VEGF(165) mediates glomerular endothelial repair. J Clin Invest 1999; 104: 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood 2007; 109: 3161–3172 [DOI] [PubMed] [Google Scholar]
- 170.Aso Y, Inukai T, Takemura Y. Mechanisms of elevation of serum and urinary concentrations of soluble thrombomodulin in diabetic patients: possible application as a marker for vascular endothelial injury. Metabolism 1998; 47: 362–365 [DOI] [PubMed] [Google Scholar]
- 171.Hafer‐Macko CE, Ivey FM, Gyure KA, Sorkin JD, Macko RF. Thrombomodulin deficiency in human diabetic nerve microvasculature. Diabetes 2002; 51: 1957–1963 [DOI] [PubMed] [Google Scholar]
- 172.Isermann B, Vinnikov IA, Madhusudhan T, et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med 2007; 13: 1349–1358 [DOI] [PubMed] [Google Scholar]