

Abstract
Oxidative stress is defined as excessive production of reactive oxygen species (ROS) in the presence of diminished anti‐oxidant substances. Increased oxidative stress could be one of the common pathogenic factors of diabetic complications. However, the mechanisms by which hyperglycemia increases oxidative stress are not fully understood. In this review, we focus on the impact of mitochondrial derived ROS (mtROS) on diabetic complications and suggest potential therapeutic approaches to suppress mtROS. It has been shown that hyperglycemia increases ROS production from mitochondrial electron transport chain and normalizing mitochondrial ROS ameliorates major pathways of hyperglycemic damage, such as activation of polyol pathway, activation of PKC and accumulation of advanced glycation end‐products (AGE). Additionally, in subjects with type 2 diabetes, we found a positive correlation between HbA1c and urinary excretion of 8‐hydroxydeoxyguanosine (8‐OHdG), which reflects mitochondrial oxidative damage, and further reported that 8‐OHdG was elevated in subjects with diabetic micro‐ and macro‐ vascular complications. We recently created vascular endothelial cell‐specific manganese superoxide dismutase (MnSOD) transgenic mice, and clarified that overexpression of MnSOD in endothelium could prevent diabetic retinopathy in vivo. Furthermore, we found that metformin and pioglitazone, both of which have the ability to reduce diabetic vascular complications, could ameliorate hyperglycemia‐induced mtROS production by the induction of PPARγ coactivator‐1α (PGC‐1α) and MnSOD and/or activation of adenosine monophosphate (AMP)‐activated protein kinase (AMPK). We also found that metformin and pioglitazone promote mitochondrial biogenesis through the same AMPK–PGC‐1α pathway. Taking these results, mtROS could be the key initiator of and a therapeutic target for diabetic vascular complications. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2010.00013.x, 2010)
Keywords: Adenosine monophosphate kinase, Mitochondria, Manganese superoxide dismutase
Introduction
Diabetic vascular complications are major causes of blindness, renal failure, myocardial infarction, stroke and limb amputation. Managing diabetes and its complications is not only a serious public health issue, but also a socioeconomic one in developed countries.
The Diabetes Control and Complications Trial (DCCT)1, Epidemiology of Diabetes Interventions and Complications (EDIC)2, UK Prospective Diabetes Study3,4, and our Kumamoto Study5–7 appear to have effectively resolved the long debate over whether chronic hyperglycemia is an important cause of diabetic vascular complications. However, the mechanisms through which hyperglycemia acts as a crucial risk factor for vascular complications are still unclear.
There are four main hypotheses about how hyperglycemia causes diabetic complications, which are: (i) increased glucose flux through polyol pathway8; (ii) glucose‐induced activation of protein kinase C (PKC) isoforms through de novo synthesis of the lipid second messenger diacylglycerol9; increased formation of glucose‐derived advanced glycation end‐products (AGE)10; and increased oxidative stress11,12. In particular, oxidative stress has recently emerged as an important mechanism for the development of diabetic complications. In the present review, we focus on proposed mechanisms for increasing oxidative stress in diabetes. In addition, we show that hyperglycemia‐induced mitochondrial reactive oxygen species (ROS) might be the key initiator and a potential therapeutic target of diabetic complications.
The concept and the role of oxidative stress
Oxidative stress is caused by an imbalance between the production of ROS and a biological system’s ability to readily detoxify ROS or easily repair the resulting damage (i.e. anti‐oxidant; Figure 1). ROS are free radicals that contain the oxygen atom and the biologically important ROS are superoxide (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (OH−). In contrast, common anti‐oxidants include vitamins A, C, and E, glutathione, α‐lipoic acid, mixed carotenoids, coenzyme Q10, several bioflavonoids, anti‐oxidant minerals (copper, zinc, manganese, and selenium), the cofactors (folic acid, vitamins B1, B2, B6, B12), and the enzymes superoxide dismutase (SOD), catalase, glutathione peroxidase and glutathione reductase. They work in synergy with each other and against different types of free radicals. Oxidative stress can damage all components of the cell, including cellular proteins, membrane lipids and nucleic acids.
Figure 1.
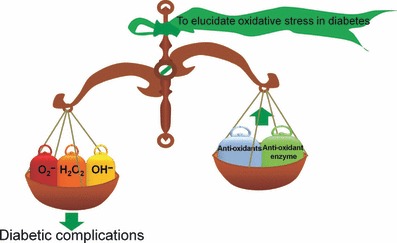
Oxidative stress. The relationship between excessive production of reactive oxygen species and the presence of reduced anti‐oxidant defense.
In patients with diabetes, especially in those with poor glycemic control, oxidative stress is increased11. In addition, it was reported that in aortic endothelial cells incubated with high glucose, ROS formation (measured as dichlorofluorescein [DCF]) was increased by 250% within 24 h, which resulted in lipid peroxidation (measured as malondialdehyde) by 330% within 168 h13. In accordance with these observations, animal studies have shown that the potent inhibition of oxidative stress with certain anti‐oxidants under experimental diabetic conditions showed preventive effects on the progression of diabetic complications14–16. However, human studies with various anti‐oxidants, including α‐tocopherol and ascorbic acid17,18, have been generally disappointing, although it was reported that supplementing with α‐lipoic acid was effective in reducing neuropathic symptoms of diabetic distal symmetric polyneuropathy19–21. Therefore, more targeted anti‐oxidant therapies based on the mechanisms of diabetes‐induced oxidative stress might be worth considering as part of the therapeutic strategy to prevent diabetic complications.
The mechanisms of oxidative stress in diabetes
Various mechanisms have been suggested to contribute to diabetes‐induced oxidative stress (Figure 2). First, it is one possibility that glucose oxidation is the source of ROS. In its enediol form, glucose is oxidized in a transition‐metal dependent reaction to an enediol radical anion that is converted into reactive ketoaldehydes and to superoxide anion radicals. The superoxide anion radicals undergo dismutation to hydrogen peroxide, which if not degraded by catalase or glutathione peroxidase and in the presence of transition metals, can lead to the production of extremely reactive hydroxyl radicals22. Second, the cytosolic enzyme aldose reductase converts high intracellular glucose concentrations to sorbitol using nicotinamide adenine dinucleotide phosphate (NADPH) derived from the pentose phosphate pathway as a cofactor. It is likely that during hyperglycemia, consumption of NADPH by this reaction inhibits replenishment of reduced glutathione, which is required to maintain glutathione peroxidase activity23. Third, hyperglycemia activates PKC in membrane fraction through an increase in de novo diacylglycerol synthesis in vascular cells, and hyperglycemia is reported to stimulate ROS production through PKC‐dependent activation of NAD(P)H oxidase24. NAD(P)H oxidase is a cytosolic enzyme complex initially discovered in neutrophils, where it plays a vital role in non‐specific host‐pathogen defense by producing superoxide. In addition to residing in phagocytic cells, NAD(P)H oxidase is present in non‐phagocytic cell types, such as endothelial cells, vascular smooth muscle cells, fibroblasts and mesangial cells25. Fourth, hyperglycemia increases non‐enzymatic glycation, characterized by the binding of reactive dicarbonyls and amino groups of proteins. This reaction leads to AGE. Glycation and oxidative stress are closely linked, and both phenomena are referred to as glycoxidation. All steps of glycoxidation generate ROS. In addition, plasma proteins modified by AGE precursors activate AGE receptors on macrophages and induce an intracellular oxidative stress by activating NAD(P)H oxidase26. Furthermore, it was reported that site specific and random fragmentation of copper, zinc SOD (Cu, ZnSOD), which is the key enzyme involved in the detoxification of superoxide radicals, was observed after the glycation reaction27. The last possible mechanism is mitochondrial ROS. Nishikawa et al. previously showed that hyperglycemia could increase ROS from the mitochondrial electron transport chain28 and proposed that this mitochondrial ROS production could be a key event in the development of diabetic complications29. However, it is still unclear whether mitochondrial ROS production is associated with the progression of diabetic complications in patients with type 2 diabetes.
Figure 2.
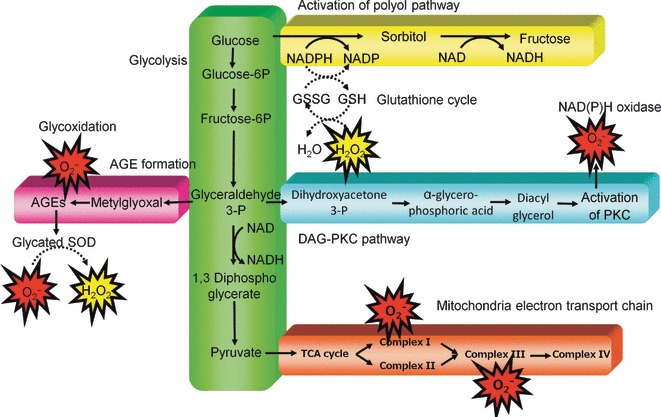
Proposed mechanism leading to diabetes‐induced oxidative stress in cells. AGE, advanced glycation end‐products; DAG, diacylglycerol; GSH, glutathione; GSSG, glutathione disulfide (oxidized glutathione); NADPH, nicotinamide adenine dinucleotide phosphate; PKC, protein kinase C; SOD, superoxide dismutase; TCA, tricarboxylic acid.
Mitochondrial ROS production in type 2 diabetic patients
To determine the role of mitochondrial ROS production in patients with type 2 diabetes, the relationship between urinary excretion of 8‐hydroxydeoxyguanosine (8‐OHdG) and the severity of diabetic complications were examined.
8‐OHdG is a product of oxidative DNA damage following specific enzymatic cleavage after 8‐hydroxylation of the guanine base. As intracellular ROS can cause strand breaks in DNA and base modifications, 8‐OHdG might serve as a sensitive biomarker of intracellular oxidative stress in vivo. There are several reasons to believe that mitochondrial DNA (mtDNA) is weak in oxidative stress. First, mitochondria are the major source of ROS under normal physiological conditions. Second, mtDNA lacks histone. Last, there is very little repair activity for mtDNA mutations. Because 8‐OHdG is a product of oxidative DNA damage and it has been reported that 8‐OHdG was 16‐fold higher in mtDNA than in nuclear DNA in rat liver30, 8‐OHdG might be a useful biomarker to evaluate ROS production from mitochondria.
We confirmed that a significant positive correlation existed between the amount of urinary 8‐OHdG excretion and HbA1c, suggesting that hyperglycemia could increase mitochondrial ROS production in patients with type 2 diabetes (Figure 3). In accordance with our results, it has been reported that the levels of 8‐OHdG in the mtDNA were significantly increased in the kidneys of streptozotosin‐induced diabetes rats, whereas those in nuclear DNA were not increased31. In addition, we found that the urinary 8‐OHdG excretion was significantly elevated in patients with diabetic retinopathy, in patients with increased urinary albumin excretion and in patients with increased mean intima‐media thickness (IMT) of the carotid arteries. Furthermore, we confirmed in the Kumamoto Study that urinary 8‐OHdG excretion was significantly lower in patients in the multiple insulin injection therapy group compared with those in the conventional insulin injection therapy group32. It was also reported that urinary 8‐OHdG excretion is a useful clinical marker to predict the development of diabetic nephropathy33. These results suggest that mitochondrial ROS production estimated by the urinary 8‐OHdG excretion could be associated with both micro‐angiopathy and macro‐angiopathy in patients with type 2 diabetes.
Figure 3.
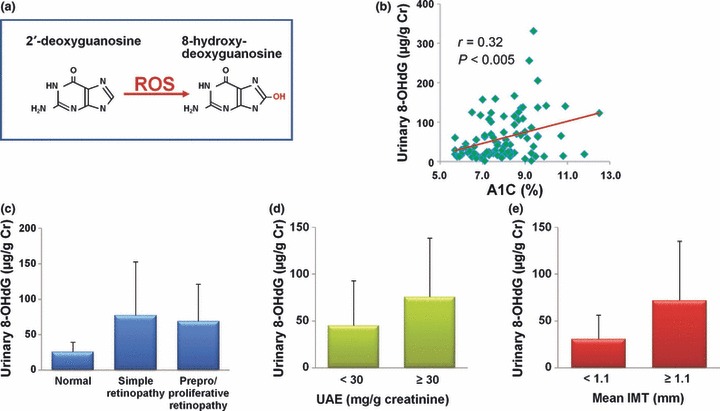
The relationship between urinary 8‐OHdG excretion and severity of diabetic complications. (a) 8‐ hydroxydeoxyguanosine (8‐OHdG) is an oxidized derivative of deoxyguanosine. (b) The relationship between urinary 8‐OHdG excretion and HbA1c. (c–e) The relationship between urinary 8‐OHdG excretion and severity of diabetic (c) retinopathy, (d) nephropathy and (e) macroangiopathy. The severity of diabetic nephropathy and macroangiopathy was evaluated by urinary albumin excretion (UAE) and mean intima‐media thickness (IMT) of the carotid arteries, respectively. ROS, reactive oxygen species.
Transgenic mice overexpressing manganese superoxide dismutase in the endothelium are resistant to diabetic retinopathy
The direct evidence of the relationship between mitochondrial ROS and the progression of diabetic vascular complications was clarified using transgenic mice, which specifically overexpressed manganese superoxide dismutase (MnSOD) in vascular endothelial cells, by using a Tie2 promoter and enhancer. To investigate the impact of mitochondrial ROS on diabetic retinopathy in vivo, diabetes was induced by an intraperitoneal injection of streptozotocin. After 4 weeks of injection, glycated hemoglobin was increased in both control and transgenic mice to similar degrees. In contrast, urinary 8‐OHdG excretion was suppressed in diabetic transgenic mice, although that was increased in control diabetic mice. Similarly, vascular endothelial growth factor (VEGF) mRNA and fibronectin mRNA in the retina, both of which are reported to be important in the pathogenesis of diabetic retinopathy34,35, were suppressed in diabetic transgenic mice but increased in control diabetic mice (Figure 4).
Figure 4.
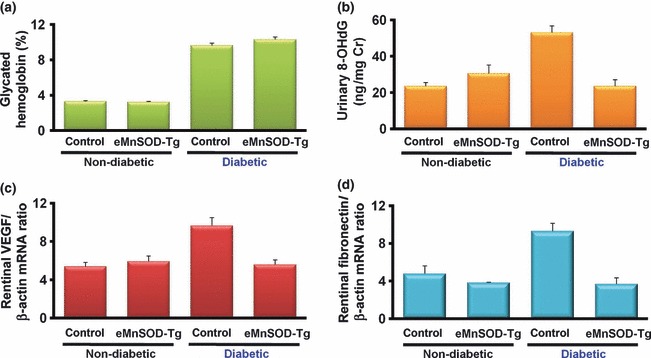
The effect of endothelial overexpression of manganese superoxide dismutase (MnSOD) on diabetic retinopathy in vivo. Diabetes was induced in endothelial cell‐specific manganese superoxide (eMnSOD‐Tg) or control C57Bl/6 male mice by an intraperitoneal injection of streptozotocin. Non‐diabetic mice were injected with buffer alone. After 4 weeks of injection, every parameter was measured in the groups of non‐diabetic control, non‐diabetic eMnSOD‐Tg, diabetic control and diabetic vascular eMnSOD‐Tg mice. (a) The level of glycated hemoglobin. (b) Urinary 8‐hydroxydeoxyguanosine (8‐OHdG) excretion. (c) The expression of vascular endothelial growth factor (VEGF) and (d) fibronectin mRNA in retinas.
Because commonly used diabetic animal models usually do not reach the proliferative retinopathy stage, we further applied relative hypoxia‐induced retinopathy to evaluate retinal neovascularization. The relative hypoxia‐induced retinopathy model is widely used in studies of retinal ischemic neovascular diseases, such as diabetic retinopathy and retinopathy of prematurity36,37. In the relative hypoxia‐induced retinopathy model, retinal flat‐mount pictures of control mice showed typical reactive central vaso‐obliteration, with the formation of peripheral tufts that indicate newly formed vasculature. In contrast, retinas of transgenic mice exposed to relative hypoxia showed normal‐appearing central vasculature and a lesser number of tufts. Taken together, these results suggest that the overexpression of mitochondrial‐specific SOD in endothelium could prevent diabetic retinopathy in vivo.
Effect of mitochondrial ROS amelioration by activation of an AMPK‐PGC‐1α‐pathway
The results from MnSOD transgenic mice suggest that mitochondrial ROS can be a target to develop new therapies to prevent diabetic complications. To explore the molecule to normalize hyperglycemia‐induce mitochondrial ROS, we focused on the results in the UKPDS38, PROACTIVE study39 and PERISCOPE study40. The UKPDS reported intensive glycemic control with metformin to decrease the risk of diabetes‐related end‐points in overweight patients with type 2 diabetes in comparison with sulfonylurea or insulin therapy. Given the equivalent HbA1c levels obtained by each therapy, the possible additional benefit of metformin to reduce cardiovascular events is not explicable on the basis of glycemic control. In contrast, the PROACTIVE study and PERISCOPE study have implied that pioglitazone also has the ability to reduce vascular complications in diabetic patients. Therefore, we investigated the impacts of metformin and pioglitazone on mitochondrial ROS production.
In order to clarify if metformin and pioglitazone can reduce hyperglycemia‐induced mitochondrial ROS production, we used the reduced MitoTracker Red probe (Molecular Probes, Eugene, OR, USA), which can specifically detect mitochondrial ROS production. The fluorescence of MitoTracker Red increased with 30 mM glucose, and this hyperglycemia‐induced fluorescence of MitoTracker Red was reduced by either metformin or piglitazone41,42. Therefore, these agents have an inhibitory effect on hyperglycemia‐induced mitochondrial ROS production. We also clarified that the inhibitory effect of these agents on hyperglycemia‐induced mitochondrial ROS production was, at least in part, dependent on the induction of PGC‐1α and MnSOD. Furthermore, the effect of metformin on PGC‐1α induction was dependent on the activation of AMPK.
Disturbance of the mitochondrial respiratory chain by ROS and the resulting reduced adenosine triphosphate production might be also a key factor in the development of diabetes and its complications43,44. Because PGC‐1α is a potent stimulator of mitochondrial biogenesis, we evaluated the effect of metformin and piglitazone on mitochondrial biogenesis. As expected, metformin, piglitazone and overexpression of PGC‐1α increased the expression of nuclear respiratory factor‐1 (NRF‐1) and mitochondrial DNA transcription factor A (mtTFA), both of which are regulators of mitochondrial DNA transcription and replication41,42. Metformin and pioglitazone also significantly increased the mitochondrial DNA content and mitochondria number in comparison with the incubation with high glucose alone. In addition, the effects of metformin were dependent on AMPK activity.
These observations suggested that metformin and pioglitazone could normalize hyperglycemia‐induced mitochondrial ROS production, at least in part, through the induction of PGC‐1α and MnSOD and/or activation of AMPK (Figure5). In addition, these agents also promote mitochondrial biogenesis through the same AMPK‐PGC‐1α pathway. Therefore, AMPK and PGC‐1α could be targets for the design of new pharmacological approaches to prevent diabetic complications.
Figure 5.
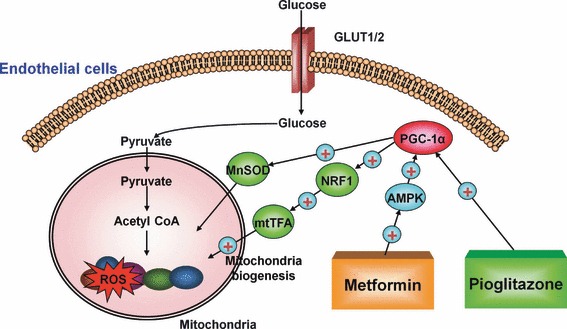
Proposed mechanism of metformin and pioglitazone induced suppression of mitochondrial reactive oxygen species (ROS) and promotion of mitochondrial biogenesis. Pioglitazone reduces mitochondrial ROS through the induction of PPARγ coactivator‐1α (PGC‐1α) and manganese superoxide dismutase (MnSOD). Metformin reduces mitochondrial ROS through the activation of AMP‐activated protein kinase (AMPK), and the resulting induction of PGC‐1α and MnSOD. Metformin and pioglitazone also promotes mitochondrial biogenesis through induction of PGC‐1α. These reactions might contribute, at least in part, to preventing diabetic complications. mtTFA, mitochondrial DNA transcription factor A; NRF‐1, nuclear respiratory factor‐1.
The main purpose of the treatment of diabetes is to prevent the onset and progression of chronic diabetic vascular complications. Oxidative stress, through the production of ROS, has been proposed as the root cause underlying the progression of long‐term diabetic complications. In the present study, we showed that hyperglycemia‐induced mitochondrial ROS production could be a key event in the development of diabetic complications (Figure 6) through the studies of subjects with type 2 diabetes, and in vascular endothelial cell‐specific MnSOD transgenic mice. In addition, we proposed that a blockade of mitochondrial ROS production through the induction of AMPK or PGC‐1α could, therefore, be useful in the design of new pharmacological approaches to prevent diabetic complications. Further clinical and basic studies will be required to clarify the benefits of reducing mitochondrial ROS production in the prevention of diabetic complications.
Figure 6.
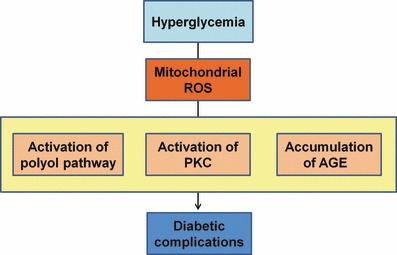
Proposed mechanism leading to diabetic vascular complications. Mitochondrial reactive oxygen species (ROS) could be the main cause of hyperglycemia‐induced oxidative stress. In addition, mitochondrial ROS is a causal link between hyperglycemia and each of the three main pathways responsible for hyperglycemic damage such as the activation of polyol pathway, the activation of protein kinase C (PKC) and the accumulation of advanced glycation end‐products (AGE).
Acknowledgments
This work was supported by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science, Japan (No. 20390259 to EA and No. 20591065 to TN).
References
- 1.The Diabetes Control and Complications Trial Research Group . The effect of intensive treatment of diabetes on the development and progression of long‐term complications in insulin‐dependent diabetes mellitus. N Engl J Med 1993; 329: 977–986 [DOI] [PubMed] [Google Scholar]
- 2.Nathan DM, Cleary PA, Backlund JY, et al.Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353: 2643–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.UK Prospective Diabetes Study (UKPDS) Group . Intensive blood‐glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352: 837–853 [PubMed] [Google Scholar]
- 4.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10‐year follow‐up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359: 1577–1589 [DOI] [PubMed] [Google Scholar]
- 5.Ohkubo Y, Kishikawa H, Araki E, et al.Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non‐insulin‐dependent diabetes mellitus: a randomized prospective 6‐year study. Diabetes Res Clin Pract 1995; 28: 103–117 [DOI] [PubMed] [Google Scholar]
- 6.Wake N, Hisashige A, Katayama T, et al.Cost‐effectiveness of intensive insulin therapy for type 2 diabetes: a 10‐year follow‐up of the Kumamoto study. Diabetes Res Clin Pract 2000; 48: 201–210 [DOI] [PubMed] [Google Scholar]
- 7.Shichiri M, Kishikawa H, Ohkubo Y, Wake N. Long‐term results of the Kumamoto Study on optimal diabetes control in type 2 diabetic patients. Diabetes Care 2000; 23(Suppl. 2): B21–B29 [PubMed] [Google Scholar]
- 8.Engerman RL, Kern TS, Larson ME. Nerve conduction and aldose reductase inhibition during 5 years of diabetes or galactosaemia in dogs. Diabetologia 1994; 37: 141–144 [DOI] [PubMed] [Google Scholar]
- 9.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes 1998; 47: 859–866 [DOI] [PubMed] [Google Scholar]
- 10.Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu Rev Med 1995; 46: 223–234 [DOI] [PubMed] [Google Scholar]
- 11.Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care 1996; 19: 257–267 [DOI] [PubMed] [Google Scholar]
- 12.Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008; 57: 1446–1454 [DOI] [PubMed] [Google Scholar]
- 13.Giardino I, Edelstein D, Brownlee M. BCL‐2 expression or antioxidants prevent hyperglycemia‐induced formation of intracellular advanced glycation endproducts in bovine endothelial cells. J Clin Invest 1996; 97: 1422–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunisaki M, Bursell SE, Clermont AC, et al.Vitamin E prevents diabetes‐induced abnormal retinal blood flow via the diacylglycerol‐protein kinase C pathway. Am J Physiol 1995; 269: E239–E246 [DOI] [PubMed] [Google Scholar]
- 15.Melhem MF, Craven PA, Liachenko J, DeRubertis FR. Alpha‐lipoic acid attenuates hyperglycemia and prevents glomerular mesangial matrix expansion in diabetes. J Am Soc Nephrol 2002; 13: 108–116 [DOI] [PubMed] [Google Scholar]
- 16.Odetti P, Pesce C, Traverso N, et al.Comparative trial of N‐acetyl‐cysteine, taurine, and oxerutin on skin and kidney damage in long‐term experimental diabetes. Diabetes 2003; 52: 499–505 [DOI] [PubMed] [Google Scholar]
- 17.Heart Outcomes Prevention Evaluation Study Investigators . Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO‐HOPE substudy. Lancet 2000; 355: 253–259 [PubMed] [Google Scholar]
- 18.Belch J, MacCuish A, Campbell I, et al.The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008; 337: a1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ziegler D, Hanefeld M, Ruhnau KJ, et al.Treatment of symptomatic diabetic peripheral neuropathy with the anti‐oxidant alpha‐lipoic acid. A 3‐week multicentre randomized controlled trial (ALADIN Study). Diabetologia 1995; 38: 1425–1433 [DOI] [PubMed] [Google Scholar]
- 20.Ametov AS, Barinov A, Dyck PJ, et al.The sensory symptoms of diabetic polyneuropathy are improved with alpha‐lipoic acid: the SYDNEY trial. Diabetes Care 2003; 26: 770–776 [DOI] [PubMed] [Google Scholar]
- 21.Tang J, Wingerchuk DM, Crum BA, Rubin DI, Demaerschalk BM. Alpha‐lipoic acid may improve symptomatic diabetic polyneuropathy. Neurologist 2007; 13: 164–167 [DOI] [PubMed] [Google Scholar]
- 22.Hunt JV, Smith CC, Wolff SP. Autoxidative glycosylation and possible involvement of peroxides and free radicals in LDL modification by glucose. Diabetes 1990; 39: 1420–1424 [DOI] [PubMed] [Google Scholar]
- 23.Williamson JR, Chang K, Frangos M, et al.Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 1993; 42: 801–813 [DOI] [PubMed] [Google Scholar]
- 24.Inoguchi T, Li P, Umeda F, et al.High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C‐‐dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000; 49: 1939–1945 [DOI] [PubMed] [Google Scholar]
- 25.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 1994; 74: 1141–1148 [DOI] [PubMed] [Google Scholar]
- 26.Yan SD, Schmidt AM, Anderson GM, et al.Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem 1994; 269: 9889–9897 [PubMed] [Google Scholar]
- 27.Ookawara T, Kawamura N, Kitagawa Y, Taniguchi N. Site‐specific and random fragmentation of Cu,Zn‐superoxide dismutase by glycation reaction. Implication of reactive oxygen species. J Biol Chem 1992; 267: 18505–18510 [PubMed] [Google Scholar]
- 28.Nishikawa T, Edelstein D, Du XL, et al.Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404: 787–790 [DOI] [PubMed] [Google Scholar]
- 29.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001; 414: 813–820 [DOI] [PubMed] [Google Scholar]
- 30.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev 1998; 78: 547–581 [DOI] [PubMed] [Google Scholar]
- 31.Kakimoto M, Inoguchi T, Sonta T, et al.Accumulation of 8‐hydroxy‐2′‐deoxyguanosine and mitochondrial DNA deletion in kidney of diabetic rats. Diabetes 2002; 51: 1588–1595 [DOI] [PubMed] [Google Scholar]
- 32.Nishikawa T, Sasahara T, Kiritoshi S, et al.Evaluation of urinary 8‐hydroxydeoxy‐guanosine as a novel biomarker of macrovascular complications in type 2 diabetes. Diabetes Care 2003; 26: 1507–1512 [DOI] [PubMed] [Google Scholar]
- 33.Hinokio Y, Suzuki S, Hirai M, Suzuki C, Suzuki M, Toyota T. Urinary excretion of 8‐oxo‐7, 8‐dihydro‐2′‐deoxyguanosine as a predictor of the development of diabetic nephropathy. Diabetologia 2002; 45: 877–882 [DOI] [PubMed] [Google Scholar]
- 34.Aiello LP, Avery RL, Arrigg PG, et al.Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 1994; 331: 1480–1487 [DOI] [PubMed] [Google Scholar]
- 35.Nishikawa T, Giardino I, Edelstein D, Brownlee M. Changes in diabetic retinal matrix protein mRNA levels in a common transgenic mouse strain. Curr Eye Res 2000; 21: 581–587 [PubMed] [Google Scholar]
- 36.Zhang SX, Sima J, Shao C, et al.Plasminogen kringle 5 reduces vascular leakage in the retina in rat models of oxygen‐induced retinopathy and diabetes. Diabetologia 2004; 47: 124–131 [DOI] [PubMed] [Google Scholar]
- 37.Kondo T, Vicent D, Suzuma K, et al.Knockout of insulin and IGF‐1 receptors on vascular endothelial cells protects against retinal neovascularization. J Clin Invest 2003; 111: 1835–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.UK Prospective Diabetes Study (UKPDS) Group . Effect of intensive blood‐glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998; 352: 854–865 [PubMed] [Google Scholar]
- 39.Dormandy JA, Charbonnel B, Eckland DJ, et al.Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet 2005; 366: 1279–1289 [DOI] [PubMed] [Google Scholar]
- 40.Nissen SE, Nicholls SJ, Wolski K, et al.Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. JAMA 2008; 299: 1561–1573 [DOI] [PubMed] [Google Scholar]
- 41.Kukidome D, Nishikawa T, Sonoda K, et al.Activation of AMP‐Activated Protein Kinase Reduces Hyperglycemia‐Induced Mitochondrial Reactive Oxygen Species Production and Promotes Mitochondrial Biogenesis in Human Umbilical Vein Endothelial Cells. Diabetes 2006; 55: 120–127 [PubMed] [Google Scholar]
- 42.Fujisawa K, Nishikawa T, Kukidome D, et al.TZDs reduce mitochondrial ROS production and enhance mitochondrial biogenesis. Biochem Biophys Res Commun 2009; 379: 43–48 [DOI] [PubMed] [Google Scholar]
- 43.Perez‐Campo R, Lopez‐Torres M, Cadenas S, Rojas C, Barja G. The rate of free radical production as a determinant of the rate of aging: evidence from the comparative approach. J Comp Physiol B 1998; 168: 149–158 [DOI] [PubMed] [Google Scholar]
- 44.Petersen KF, Befroy D, Dufour S, et al.Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 2003; 300: 1140–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]