

Abstract
Aims/Introduction: Endothelial lipase (EL) plays an important role in high‐density lipoprotein (HDL) metabolism and experimental data suggest that EL might be proatherogenic. We have investigated whether serum EL concentration is associated with changes in serum capacity to induce cholesterol efflux and arterial stiffness in type 2 diabetes.
Materials and Methods: Serum EL was assayed by ELISA in 172 diabetic patients and 175 controls. The ability of serum to induce cholesterol efflux was measured using a cell culture system and arterial stiffness was determined by measuring pulse wave velocity (PWV) between carotid and femoral arteries.
Results: Diabetic patients had significantly higher C‐reactive protein (CRP) and EL (27.7 ± 16.6 ng/mL vs 24.0 ± 11.3, P < 0.05). Cholesterol efflux to serum mediated through scavenger receptor class B type I was impaired (15.1 ± 2.5%vs 16.7 ± 3.1, respectively, P < 0.01). In controls, serum EL correlated with cholesterol efflux to serum (r = −0.16, P = 0.025), but only a trend was seen in the diabetic patients. Linear regression showed that in controls, HDL, serum EL and waist circumference were major independent determinants of cholesterol efflux; whereas in the diabetic cohort, the major independent determinants of cholesterol efflux were HDL, CRP and age. PWV was increased in the diabetic patients (P < 0.01), but no association between serum EL and PWV was seen in either groups.
Conclusions: Serum EL was increased in diabetic patients, but impaired serum capacity to induce cholesterol efflux in these patients was mainly related to low HDL and subclinical inflammation. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2010.00016.x, 2010)
Keywords: Endothelial lipase, Reverse cholesterol transport, Cholesterol efflux
Introduction
Endothelial lipase (EL) is a new member of the triacylglycerol lipase family. It is a phospholipase with little triacylglycerol lipase activity and plays a significant role in the metabolism of high‐density lipoprotein (HDL)1,2. Decreased HDL cholesterol and phospholipid levels have been shown in mouse models over‐expressing EL, whereas inhibition of EL activity by antibodies or gene knock‐out leads to an increase in HDL cholesterol and phospholipid levels2–5. In humans, increased plasma EL concentration has been associated with a less favorable lipid profile with elevated plasma triglyceride and apolipoprotein (APO) B as well as smaller low‐density lipoprotein (LDL) particles size, and not all studies have shown an association with HDL6–8. There are data suggesting that EL might be proatherogenic. An increased level of plasma EL has been associated with visceral obesity, metabolic syndrome, type 2 diabetes mellitus and inflammation6–9. It has been shown that EL is expressed in endothelial cells, macrophages and smooth muscle cells in atherosclerotic lesions of human coronary arteries10, and an association between plasma EL concentration and coronary artery calcification score has been reported in a large cross‐sectional study6.
EL is involved in the remodeling and catabolism of HDL particles in the plasma compartment and EL efficiently cleaves phospholipids in HDL and releases fatty acids. In addition to its lipase activity, Strauss et al. have shown that EL also facilitates HDL particle binding and uptake as well as selective uptake of HDL cholesteryl ester in HepG2 cells by its bridging function11. Modulation of HDL by EL might affect the capacity of HDL to act as extracellular acceptors of free cholesterol in reverse cholesterol transport. Gauster et al. have shown that EL modification alters chemical composition and physical properties of HDL, resulting in its decreased binding capacity to scavenger receptor class B type I (SR‐BI) and a diminished ability to mediate SR‐BI‐dependent cholesterol efflux12. In contrast, Qiu et al. have reported that EL promotes apolipoprotein (APO) A1‐mediated cholesterol efflux in THP‐1 macrophages13. Overexpression of EL in human APOA1 transgenic mice led to a large reduction in HDL cholesterol and in serum phospholipids/APOA1 ratio14. As a result, the efflux potential of serum through SR‐BI decreased by 90% and adenosine triphosphate (ATP)‐binding cassette transporter A1 (ABCA1)‐mediated efflux increased by 63%. ATP‐binding cassette transporter A1 mainly mediates cholesterol efflux to APOA1 and pre‐β‐HDL, whereas SR‐BI mainly mediates cholesterol efflux to mature HDL. Because serum EL concentration is increased in patients with type 2 diabetes, we have investigated whether increased EL is associated with changes in the serum capacity to induce cholesterol efflux and cardiovascular risk in diabetic patients. Cardiovascular risk was assessed non‐invasively by measuring arterial stiffness, which provides useful information on vascular health and serves as a surrogate for cardiovascular morbidity and mortality risk15,16.
Methods
Patients with type 2 diabetes mellitus according to World Health Organization criteria were recruited from the diabetes clinics at Queen Mary Hospital. Because we have previously shown that cellular cholesterol efflux to serum was impaired in diabetic patients with incipient or overt nephropathy17, only diabetic patients with normoalbuminuria (urinary albumin excretion rate <30 mg/day) were recruited for the present study to avoid the potential confounding effect of nephropathy. Patients on insulin therapy or lipid lowering agents, or had a history of cardiovascular disease were also excluded. All subjects must have had stable glycaemic control with no change in anti‐diabetic therapy for the preceding 3 months. Healthy age‐matched controls were recruited from the community. Fasting blood samples were taken for the measurement of glucose, glycated hemoglobin (HbA1c), lipids, high sensitivity C‐reactive protein (CRP), cholesterol efflux and EL. Arterial stiffness was assessed non‐invasively by measuring aortic pulse wave velocity (PWV) using SphygmoCor Vx PWV system (version 7.0; AtCor Medical, Sydney, Australia). Pulse wave velocity was determined by measuring the velocity of the blood pressure waveform between the carotid and femoral artery sites using a single‐lead electrocardiogram and a tonometer to measure the pressure pulse waveform sequentially in the two artery sites. The present study was approved by the Ethics Committee of the University of Hong Kong and informed consent was obtained from all subjects.
Plasma total cholesterol and triglyceride were determined enzymatically on a Hitachi 912 analyzer (Roche Diagnostics, Mannheim, Germany). HDL cholesterol was measured using a homogenous method with polyethylene glycol‐modified enzymes and alpha‐cyclodextrin. LDL cholesterol was calculated by the Friedewald equation or measured directly if plasma triglyceride was >4.5 mmol/L. Plasma APOA1 and APOB were measured by rate nephelometry using the Beckman Array System (Beckman Instruments, Fullerton, CA, USA). HbA1c was measured in whole blood using ion‐exchange high performance liquid chromatography with the Bio‐Rad Variant Haemoglobin Testing System (Bio‐Rad Laboratories, Hercules, CA, USA). Plasma high sensitivity CRP was measured by a particle‐enhanced immunoturbidimetric assay (Roche Diagnostics) using anti‐CRP mouse monoclonal antibodies coupled to latex microparticles.
Serum EL was measured by competitive ELISA using a rabbit polycloncal antibody specific to human EL (Novus Biologicals, Littleton, CO, USA) as described9. The immunogen used for raising the antibody was an N‐terminal synthetic peptide made to the human EL protein sequence. The anti‐EL antibody was highly specific and there was no cross‐reactivity with hepatic lipase or lipoprotein lipase. Briefly, a 96‐well EIA microtiter plate (Costar, Corning, NY, USA) was coated with antigen, the N‐terminal synthetic peptide of the human endothelial lipase (Novus Biologicals) in coating buffer overnight at 4°C. The wells were then washed extensively five times and incubated with blocking reagent at room temperature for 2 h. After rinsing five times, equal volume of one‐quarter diluted serum samples plus anti‐EL antibody (Novus Biologicals) was added. The plate was then incubated for 3 h at room temperature. After another five washes, horseradish peroxidase‐conjugated goat‐anti‐rabbit secondary antibody was added and incubated for 1 h. The plate was finally measured at 450 nm by an ELISA reader. The interassay precision was 7.3%.
Fu5AH rat hepatoma cells (a generous gift from Dr GH Rothblat) were used to measure the capacity of serum to induce cholesterol efflux by SR‐BI and ABCA1. Fu5AH rat hepatoma cells have a high expression level of SR‐BI, but they lack functional ATP‐binding cassette transporters. We have previously shown that ABCA1 expression can be specifically induced in Fu5AH cells by stimulating the cells with 22(R)‐hydroxycholesterol (22R‐HC)/9‐cis‐retinoic acid (9cRA) and can be used to assay ABCA1‐mediated cholesterol efflux, and both SR‐BI and ABCA1 expression was not affected by incubation with serum17. The magnitude of ABCA1‐mediated cholesterol efflux in our assay is similar to the magnitude of ABCA1‐mediated cholesterol efflux reported in efflux assays using RAW264.7 cells stimulated with cyclic adenosine monophosphate (cAMP) to induce ABCA1 expression18. To measure cellular cholesterol efflux to serum, Fu5AH cells were cultured in minimal essential medium (MEM) containing 5% calf serum and labeled with [3H]cholesterol for 48 h (1 μCi/well; GE Healthcare Biosciences, Piscataway, NJ, USA). ABCA1‐mediated cholesterol efflux was measured using [3H]cholesterol‐labeled Fu5AH cells treated with 22R‐HC (5 μg/mL) and 9cRA (10 μmol/L) and SR‐BI‐mediated cholesterol efflux was measured in untreated cells. 5% diluted serum in MEM was added and incubated at 37°C for 4 h. Radioactivity was measured in both medium and cells, and the radioactivity released to the medium was expressed as the fraction of the total radioactive cholesterol present in the well. Cholesterol efflux mediated by SR‐BI, expressed as percent, was calculated as the amount of label recovered in the medium divided by the total label in each well in untreated cells. Cholesterol efflux mediated by ABCA1 was calculated as fractional cholesterol efflux from 22R‐HC/9cRA‐treated cells minus fractional cholesterol efflux from untreated control cells. Each assay of cholesterol efflux was carried out in triplicate. The interassay coefficient of variations for SR‐BI mediated and ABCA1‐mediated cholesterol efflux were 8.0% and 4.5%, respectively.
Results are expressed as means ± SD or as median and interquartile range if the data were not normally distributed. Comparisons between two different groups were carried out using an independent samples t‐test, and skewed data were logarithmically transformed before analysis. Pearson’s correlations were used to test the relationship among variables. Multiple linear regression analysis was used to simultaneously assess the relationship between EL and various variables.
Results
The clinical characteristics of the subjects are shown in Table 1. Age, sex and smoking status were not significantly different between the two groups, but body mass index (BMI) and waist circumference were significantly higher in the diabetic patients than controls. In the present study, 32 patients were taking metformin, 23 were taking sulphonylurea and the rest were taking a combination of metformin and sulphonylurea. The mean duration of diabetes was 9.7 ± 4.9 years. Forty‐nine percent of patients were on anti‐hypertensive agents and 27% had evidence of retinopathy. Fasting glucose, HbA1c and CRP were significantly higher in the diabetic patients than the controls. Pulse wave velocity was also elevated in the diabetic patients.
Table 1. Clinical characteristic of controls and diabetic patients.
| Controls (n = 175) | DM (n = 172) | |
|---|---|---|
| Male/female (%) | 51/49 | 53/47 |
| Age (years) | 49.6 ± 6.6 | 51.3 ± 8.4 |
| Smokers (%) | 14 | 12.8 |
| BMI (kg/m2) | 24.5 ± 3.1 | 26.1 ± 4.2** |
| Waist circumference (cm) | 80.9 ± 9.2 | 87.6 ± 11.9** |
| Systolic BP (mmHg) | 114 ± 17 | 128 ± 18** |
| Diastolic BP (mmHg) | 74 ± 10 | 77 ± 9* |
| PWV (m/s) | 7.1 ± 1.1 | 9.0 ± 1.5** |
| Fasting glucose (mmol/L) | 4.84 ± 0.82 | 8.60 ± 2.22** |
| HbA1c (%) | 5.67 ± 0.80 | 8.21 ± 1.35** |
| CRP (mg/L) | 0.75 (0.41–1.70) | 1.35 (0.63–2.49)** |
Values are mean ± SD or median (interquartile range). *P < 0.05, **P < 0.01 vs controls. BMI, body mass index; BP, blood pressure; CRP, C‐reactive protein; DM, diabetes mellitus; HbA1c, glycated hemoglobin; PWV, pulse wave velocity.
Plasma levels of lipids and apolipoproteins are shown in Table 2. Plasma HDL cholesterol and APOA1 were reduced and triglyceride was higher in the diabetic group. SR‐BI mediated cholesterol efflux to serum was impaired in the diabetic patients, whereas no significant difference in ABCA1 mediated cholesterol efflux to serum was found. Because plasma HDL levels differed significantly between diabetic patients and controls, data were also analyzed after adjusting for HDL cholesterol level, and SR‐BI mediated cholesterol efflux to serum remained impaired in the diabetic patients (P < 0.01). Serum EL was significantly increased in the diabetic patients (Table 2). There was no significant correlation between serum EL and HDL in controls (r = −0.08) or diabetic patients (r = −0.05), whilst there was a weak trend between serum EL and logCRP (controls r = 0.13, P = 0.08; diabetics r = 0.14, P = 0.07). No association between serum EL and PWV was observed in either the diabetic patients or controls. In the diabetic patients, PWV correlated with logCRP (r = 0.20, P < 0.05) and SR‐BI mediated cholesterol efflux (r = −0.17, P < 0.05) in addition to its strong correlation with systolic BP (r = 0.54, P < 0.01). Neither the association between PWV and logCRP nor SR‐BI mediated cholesterol efflux remained significant after adjusting for blood pressure.
Table 2. Plasma lipids, endothelial lipase and serum cholesterol efflux capacity.
| Controls | DM | |
|---|---|---|
| Total cholesterol (mmol/L) | 5.06 ± 0.8 | 5.06 ± 0.97 |
| Triglyceride (mmol/L) | 1.00 (0.80–1.60) | 1.40 (0.90–2.10)** |
| LDL cholesterol (mmol/L) | 3.04 ± 0.76 | 3.06 ± 0.88 |
| HDL cholesterol (mmol/L) | 1.42 ± 0.38 | 1.14 ± 0.26** |
| APOA1(g/L) | 1.44 ± 0.27 | 1.29 ± 0.23** |
| APOB (g/L) | 0.88 ± 0.20 | 0.93 ± 0.23* |
| EL (ng/mL) | 24.0 ± 11.3 | 27.7 ± 16.6* |
| SR‐BI mediated cholesterol efflux to serum (%) | 16.68 ± 3.14 | 15.13 ± 2.46** |
| ABCA1 mediated cholesterol efflux to serum (%) | 1.60 ± 0.49 | 1.56 ± 0.50 |
Values are mean ± SD or median (interquartile range). *P < 0.05, **P < 0.01 vs controls. ABCA1, adenosine triphosphate‐binding cassette transporter A1; APOA1, apolipoprotein A1; APOB, apolipoprotein B; DM, diabetes mellitus EL, endothelial lipase; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; SR‐BI, scavenger receptor class B type I.
Univariate analysis was carried out to investigate which clinical parameters were associated with cholesterol efflux. In the control subjects, serum EL correlated inversely with SR‐BI mediated cholesterol efflux (Figure 1a) but not with ABCA1 mediated cholesterol efflux. SR‐BI mediated cholesterol efflux to serum also correlated with HDL (r = 0.59, P < 0.001), APOAI (r = 0.60, P < 0.001), waist circumference (r = −0.16, P < 0.05), but not with logCRP. However, in the diabetic patients, there was only a trend towards an association between serum EL and SR‐BI mediated cholesterol efflux (Figure 1b) and there was no correlation with ABCA1 mediated cholesterol efflux. In the diabetic cohort, SR‐BI mediated cholesterol efflux to serum correlated with HDL (r = 0.62, P < 0.001), APOAI (r = 0.51, P < 0.001), waist circumference (r = −0.19, P < 0.05) and logCRP (r = −0.32, P < 0.001), whereas ABCA1 mediated cholesterol efflux to serum correlated only with HDL (r = 0.23, P < 0.01).
Figure 1.
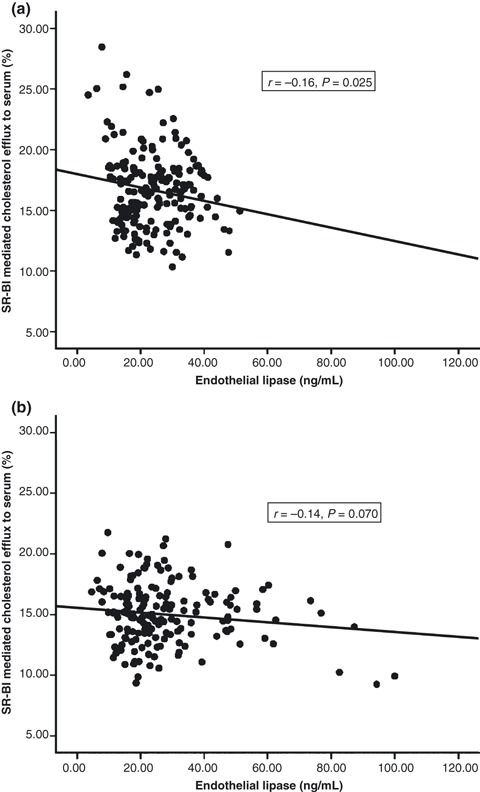
Correlation between serum endothelial lipase and scavenger receptor class B type I mediated cholesterol efflux to serum in (a) controls and (b) diabetic patients. SR‐BI, scavenger receptor class B type I.
To determine whether serum EL was an independent determinant of SR‐BI mediated cholesterol efflux to serum, multiple linear regression analysis was carried out including variables that had significant associations. In controls, age, sex, waist circumference, smoking status, HDL and serum EL were entered into the model, and the results are shown in Table 3. Plasma HDL, serum EL and waist circumference were the independent determinants of SR‐BI mediated cholesterol efflux to serum in controls, accounting for 24%, 10% and 2% of the variability of cholesterol efflux respectively. In contrast, the major independent determinants of SR‐BI mediated cholesterol efflux in the diabetic patients were HDL, logCRP and age (Table 4), accounting for 26%, 6% and 2% of the variability of cholesterol efflux, respectively. Forcing serum EL and/or HbA1c into the model did not change the results.
Table 3. Multiple regression analysis with scavenger receptor class B type I mediated cholesterol efflux to serum as the dependent variable in control subjects.
| Regression coefficient | SE of regression coefficient | P‐value | |
|---|---|---|---|
| Intercept | 6.103 | 3.142 | <0.001 |
| Age | 0.005 | 0.028 | 0.054 |
| Sex | 0.099 | 0.515 | 0.849 |
| Waist circumference | 0.063 | 0.029 | 0.031 |
| Smoker, yes/no | −0.021 | 0.409 | 0.960 |
| HDL | 5.222 | 0.647 | <0.001 |
| EL | −0.196 | 0.022 | <0.001 |
Adjusted R2 of model = 36%. EL, endothelial lipase; HDL, high‐density lipoprotein.
Table 4. Multiple regression analysis with scavenger receptor class B type I mediated cholesterol efflux to serum as the dependent variable in diabetic subjects.
| Regression coefficient | SE of regression coefficient | P‐value | |
|---|---|---|---|
| Intercept | 12.188 | 1.910 | <0.001 |
| Age | −0.049 | 0.019 | 0.012 |
| Sex | 0.295 | 0.353 | 0.404 |
| Waist circumference | 0.005 | 0.015 | 0.736 |
| Smoker, yes/no | 0.431 | 0.266 | 0.108 |
| HDL | 3.690 | 0.526 | <0.001 |
| Log (CRP) | −1.061 | 0.412 | 0.010 |
Adjusted R2 of model = 34%. CRP, C‐reactive protein; HDL, high‐density lipoprotein.
Discussion
Efflux of free cholesterol from cells is an early step of reverse cholesterol transport and the efficiency of cellular cholesterol efflux is influenced by the concentrations of lipoproteins that act as cholesterol acceptors and the activities of serum proteins, such as lipases and lipid transfer proteins involved in the remodeling of lipoproteins19. The capacity of whole serum or plasma to stimulate cholesterol efflux from cells has been used by a number of laboratories as a means to investigate reverse cholesterol transport, as whole serum provides integrated information on lipoproteins and serum components involved in promoting cholesterol efflux from cells20–24. The capacity of serum to induce cholesterol efflux has been shown to be an independent predictor of coronary artery atherosclerosis in clinical studies22,23. Serum capacity to induce SR‐BI‐mediated cellular cholesterol efflux is reduced in type 2 diabetic patients with or without diabetic complications17,24 and ABCA1‐mediated cholesterol efflux is also impaired in patients with nephropathy17. Similar to previous studies, we have shown that serum capacity to induce SR‐BI‐mediated cellular cholesterol efflux is decreased in diabetic patients, whereas no significant reduction was seen in serum capacity to induce ABCA1‐mediated cholesterol efflux.
In the present study, we have determined whether serum capacity to induce cellular cholesterol efflux was related to serum EL level. As expected, plasma HDL level was the major determinant of serum capacity to induce cellular cholesterol efflux, be it mediated by SR‐BI or ABCA1. In healthy control subjects, there was a weak but significant inverse correlation between EL and serum capacity to induce SR‐BI‐mediated cellular cholesterol efflux. This is consistent with animal studies showing that increased EL was associated with a decrease in efflux potential of serum through SR‐B112,14. EL can reduce SR‐BI‐dependent cholesterol efflux partly by altering the chemical composition and physical properties of HDL12. EL has both catalytic and non‐catalytic functions and can mediate binding of lipoproteins independent of its enzymatic activity25. Because the association between EL and serum capacity to induce SR‐BI‐mediated cellular cholesterol efflux remains independent of HDL on analysis, whether this might be related to the non‐catalytic function of EL warrants further investigation.
In contrast to the findings in healthy controls, only a trend towards an association between serum EL and serum capacity to induce cellular cholesterol efflux was seen in type 2 diabetic patients. In these patients, CRP turned out to be an important determinant of serum capacity to induce SR‐BI‐mediated cellular cholesterol efflux. Inflammation has a major effect on reverse cholesterol transport26 and our data would suggest that the influence of inflammation on serum capacity to induce SR‐BI‐mediated cellular cholesterol efflux might override that of EL in diabetic subjects. Chronic subclinical inflammation has been shown in patients with type 2 diabetes mellitus27. Although inflammation has been shown to activate EL28, this is unlikely to be the main mechanism whereby inflammation impairs serum capacity to induce cholesterol efflux in our diabetic patients, because we did not find any significant correlation between EL and cholesterol efflux. Inflammation can influence serum capacity to induce cholesterol efflux by a number of mechanisms independent of EL. Inflammation also activates secretory phospholipase A2 and inflammatory remodeling of HDL impairs its capacity to serve as cholesterol acceptor ex vivo and in vivo26,29,30. In addition, inflammation also affects serum proteins that are involved in promoting cholesterol efflux from cells. In human subjects, attenuation of lecithin cholesterol acyltransferase and cholesterol ester transfer protein activity has been reported during infection and inflammation31,32. Taken together, subclinical inflammation might play a role in reducing serum capacity to induce cellular cholesterol efflux in type 2 diabetes mellitus. However, the present study was limited by its cross‐sectional nature, and we could only show an association and not a causal relationship.
Because EL might be proatherogenic and plasma EL has been associated with coronary artery calcification score in healthy individuals with a family history of premature coronary heart disease6, we have also determined in the present study whether plasma EL is correlated with arterial stiffness in diabetic patients. Vascular stiffness reflects both functional and structural changes in the artery wall that precede and accompany atherosclerosis. Pulse wave velocity reflects arterial stiffness and is regarded as a surrogate marker of severity of atherosclerosis33. Experimental studies have shown that aortic PWV increases with the development of atherosclerosis in primates34 and there is a strong association between aortic PWV with intima‐media thickness and severity of plaques evaluated by ultrasonographic images15. We have shown that arterial stiffness is increased in diabetic patients without overt cardiovascular disease, but we did not find an association between EL and PWV in our diabetic patients.
In conclusion, in patients with type 2 diabetes, serum EL concentration was increased, but impaired serum capacity to induce cholesterol efflux in these patients was mainly related to low HDL and subclinical inflammation.
Acknowledgements
The authors declare no conflict of interest.
References
- 1.McCoy MG, Sun GS, Marchadier D, Maugeais C, Glick JM, Rader DJ. Characterization of the lipolytic activity of endothelial lipase. J Lipid Res 2002; 43: 921–929 [PubMed] [Google Scholar]
- 2.Jaye M, Lynch KJ, Krawiec J, et al. A novel endothelial‐derived lipase that modulates HDL metabolism. Nat Genet 1999; 21: 424–428 [DOI] [PubMed] [Google Scholar]
- 3.Ishida T, Choi S, Kundu RK, et al. Endothelial lipase is a major determinant of HDL level. J Clin Invest 2003; 111: 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin W, Millar JS, Broedl U, Glick JM, Rader DJ. Inhibition of endothelial lipase causes increased HDL cholesterol levels in vivo. J Clin Invest 2003; 111: 357–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma K, Cilingiroglu M, Otvos JD, Ballantyne CM, Marian AJ, Chan L. Endothelial lipase is a major genetic determinant for high‐density lipoprotein concentration, structure, and metabolism. Proc Natl Acad Sci U S A 2003; 100: 2748–2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Badellino K, Wolfe M, Reilly M, Rader D. Endothelial lipase concentrations are increased in metabolic syndrome and associated with coronary atherosclerosis. PLoS Med 2006; 3: e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paradis ME, Badellino KO, Rader DJ, et al. Visceral adiposity and endothelial lipase. J Clin Endocrinol Metab 2006; 91: 3538–3543 [DOI] [PubMed] [Google Scholar]
- 8.Paradis ME, Badellino KO, Rader DJ, et al. Endothelial lipase is associated with inflammation in humans. J Lipid Res 2006; 47: 2808–2813 [DOI] [PubMed] [Google Scholar]
- 9.Shiu SW, Tan KC, Huang Y, Wong Y. Type 2 diabetes mellitus and endothelial lipase. Atherosclerosis 2008; 198: 441–447 [DOI] [PubMed] [Google Scholar]
- 10.Azumi H, Hirata K, Ishida T, et al. Immunohistochemical localization of endothelial cell‐derived lipase in atherosclerotic human coronary arteries. Cardiovasc Res 2003; 58: 647–654 [DOI] [PubMed] [Google Scholar]
- 11.Strauss JG, Zimmermann R, Hrzenjak A, et al. Endothelial cell‐derived lipase mediates uptake and binding of high‐density lipoprotein (HDL) particles and the selective uptake of HDL‐associated cholesterol esters independent of its enzymic activity. Biochem J 2002; 368: 69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gauster M, Oskolkova OV, Innerlohinger J, Glatter O, Knipping G, Frank S. Endothelial lipase‐modified high‐density lipoprotein exhibits diminished ability to mediate SR‐BI (scavenger receptor B type I)‐dependent free‐cholesterol efflux. Biochem J 2004; 382: 75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiu G, Hill JS. Endothelial lipase promotes apolipoprotein AI‐mediated cholesterol efflux in THP‐1 macrophages. Arterioscler Thromb Vasc Biol 2009; 29: 84–91 [DOI] [PubMed] [Google Scholar]
- 14.Yancey PG, Kawashiri MA, Moore R, et al. In vivo modulation of HDL phospholipid has opposing effects on SR‐BI‐ and ABCA1‐mediated cholesterol efflux. J Lipid Res 2004; 45: 337–346 [DOI] [PubMed] [Google Scholar]
- 15.van Popele NM, Grobbee DE, Bots ML, et al. Association between arterial stiffness and atherosclerosis: the Rotterdam Study. Stroke 2001; 32: 454–460 [DOI] [PubMed] [Google Scholar]
- 16.Cruickshank K, Riste L, Anderson SG, Wright JS, Dunn G, Gosling RG. Aortic pulse‐wave velocity and its relationship to mortality in diabetes and glucose intolerance: an integrated index of vascular function? Circulation 2002; 106: 2085–2090 [DOI] [PubMed] [Google Scholar]
- 17.Zhou H, Tan KC, Shiu SW, Wong Y. Cellular cholesterol efflux to serum is impaired in diabetic nephropathy. Diabetes Metab Res Rev 2008; 24: 617–623 [DOI] [PubMed] [Google Scholar]
- 18.Catalano G, Julia Z, Frisdal E, et al. Torcetrapib differentially modulates the biological activities of HDL2 and HDL3 particles in the reverse cholesterol transport pathway. Arterioscler Thromb Vasc Biol 2009; 29: 268–275 [DOI] [PubMed] [Google Scholar]
- 19.von Eckardstein A, Nofer JR, Assmann G. High density lipoproteins and arteriosclerosis. Role of cholesterol efflux and reverse cholesterol transport. Arterioscler Thromb Vasc Biol 2001; 21: 13–27 [DOI] [PubMed] [Google Scholar]
- 20.Rothblat GH, de la Llera‐Moya M, Atger V, Kellner‐Weibel G, Williams DL, Phillips MC. Cell cholesterol efflux: integration of old and new observations provides new insights. J Lipid Res 1999; 40: 781–796 [PubMed] [Google Scholar]
- 21.Movva R, Rader DJ. Laboratory assessment of HDL heterogeneity and function. Clin Chem 2008; 54: 788–800 [DOI] [PubMed] [Google Scholar]
- 22.Pajunen P, Syvänne M, Castro G, Nieminen MS, Taskinen MR. Cholesterol efflux capacity in vitro predicts the severity and extent of coronary artery disease in patients with and without type 2 diabetes. Scand Cardiovasc J 2001; 35: 96–100 [DOI] [PubMed] [Google Scholar]
- 23.Mikkola TS, Anthony MS, Clarkson TB, St Clair RW. Serum cholesterol efflux potential is an independent predictor of coronary artery atherosclerosis. Atherosclerosis 2003; 170: 31–38 [DOI] [PubMed] [Google Scholar]
- 24.Syvanne M, Castro G, Dengremont C, et al. Cholesterol efflux from Fu5AH hepatoma cells induced by plasma of subjects with or without coronary artery disease and non‐insulin‐dependent diabetes: importance of LpA‐I:A‐II particles and phospholipid transfer protein. Atherosclerosis 1996; 127: 245–253 [DOI] [PubMed] [Google Scholar]
- 25.Fuki IV, Blanchard N, Jin W, et al. Endogenously produced endothelial lipase enhances binding and cellular processing of plasma lipoproteins via heparan sulfate proteoglycan‐mediated pathway. J Biol Chem 2003; 278: 34331–34338 [DOI] [PubMed] [Google Scholar]
- 26.McGillicuddy FC, de la Llera Moya M, Hinkle CC, et al. Inflammation impairs reverse cholesterol transport in vivo. Circulation 2009; 119: 1135–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alexandraki K, Piperi C, Kalofoutis C, Singh J, Alaveras A, Kalofoutis A. Inflammatory process in type 2 diabetes: The role of cytokines. Ann N Y Acad Sci 2006; 1084: 89–117 [DOI] [PubMed] [Google Scholar]
- 28.Badellino KO, Wolfe ML, Reilly MP, Rader DJ. Endothelial lipase is increased in vivo by inflammation in humans. Circulation 2008; 117: 678–685 [DOI] [PubMed] [Google Scholar]
- 29.Tietge UJ, Maugeais C, Lund‐Katz S, Grass D, deBeer FC, Rader DJ. Human secretory phospholipase A2 mediates decreased plasma levels of HDL cholesterol and apoA‐I in response to inflammation in human apoA‐I transgenic mice. Arterioscler Thromb Vasc Biol 2002; 22: 1213–1218 [DOI] [PubMed] [Google Scholar]
- 30.Ansell BJ, Fonarow GC, Navab M, Fogelman AM. Modifying the anti‐inflammatory effects of high‐density lipoprotein. Curr Atheroscler Rep 2007; 9: 57–63 [DOI] [PubMed] [Google Scholar]
- 31.Khovidhunkit W, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Cholesterol efflux by acute‐phase high density lipoprotein: role of lecithin: cholesterol acyltransferase. J Lipid Res 2001; 42: 967–975 [PubMed] [Google Scholar]
- 32.Levels JH, Pajkrt D, Schultz M, et al. Alterations in lipoprotein homeostasis during human experimental endotoxemia and clinical sepsis. Biochim Biophys Acta 2007; 1771: 1429–1438 [DOI] [PubMed] [Google Scholar]
- 33.Cohn JN, Quyyumi AA, Hollenberg NK, Jamerson KA. Surrogate markers for cardiovascular disease: functional markers. Circulation 2004; 25(Suppl. 1): IV31–IV46 [DOI] [PubMed] [Google Scholar]
- 34.Farrar DJ, Green HD, Bond MG, Wagner WD, Gobbeé RA. Aortic pulse wave velocity, elasticity, and composition in a nonhuman primate model of atherosclerosis. Circ Res 1978; 43: 52–62 [DOI] [PubMed] [Google Scholar]