

Abstract
Purpose.
To quantify fundus autofluorescence (qAF) in patients with recessive Stargardt disease (STGD1).
Methods.
A total of 42 STGD1 patients (ages: 7–52 years) with at least one confirmed disease-associated ABCA4 mutation were studied. Fundus AF images (488-nm excitation) were acquired with a confocal scanning laser ophthalmoscope equipped with an internal fluorescent reference to account for variable laser power and detector sensitivity. The gray levels (GLs) of each image were calibrated to the reference, zero GL, magnification, and normative optical media density to yield qAF. Texture factor (TF) was calculated to characterize inhomogeneities in the AF image and patients were assigned to the phenotypes of Fishman I through III.
Results.
Quantified fundus autofluorescence in 36 of 42 patients and TF in 27 of 42 patients were above normal limits for age. Young patients exhibited the relatively highest qAF, with levels up to 8-fold higher than healthy eyes. Quantified fundus autofluorescence and TF were higher in Fishman II and III than Fishman I, who had higher qAF and TF than healthy eyes. Patients carrying the G1916E mutation had lower qAF and TF than most other patients, even in the presence of a second allele associated with severe disease.
Conclusions.
Quantified fundus autofluorescence is an indirect approach to measuring RPE lipofuscin in vivo. We report that ABCA4 mutations cause significantly elevated qAF, consistent with previous reports indicating that increased RPE lipofuscin is a hallmark of STGD1. Even when qualitative differences in fundus AF images are not evident, qAF can elucidate phenotypic variation. Quantified fundus autofluorescence will serve to establish genotype-phenotype correlations and as an outcome measure in clinical trials.
Keywords: ABCA4, lipofuscin, retinal pigment epithelium, scanning laser ophthalmoscope, quantitative fundus autofluorescence, recessive Stargardt disease
Quantitative fundus autofluorescence (qAF) is significantly increased in Stargardt disease, consistent with previous reports of increased RPE lipofuscin. QAF will help to establish genotype-phenotype correlations and may serve as an outcome measure in clinical trials.
Introduction
Stargardt disease (STGD1) is the most common form of juvenile macular degeneration. The onset of visual symptoms usually occurs in the teenage years with progressive loss of central vision.1,2 The prevalence of STGD1 has been estimated at between 1 in 8000 and 1 in 10,000.3 A number of therapeutic trials for this disease, including those involving small molecule–, gene– and stem cell–based therapies, have recently commenced enrollment, although debate still exists on how to best document disease progression and which endpoints should be used to monitor treatment effect.
Stargardt disease is caused by mutations in the ABCA4 gene located on the short arm of chromosome 1.4 The protein plays an important role in the recycling of vitamin A in the visual cycle. It is located in the outer segment (OS) disc membranes of both rod and cone photoreceptors.5–9 N-retinylidene-phosphatidylethanolamine (NRPE), formed by the binding of all-trans-retinal to the phospholipid phosphatidylethanolamine (PE), is removed from the outer segment disc membranes by normally functioning ABCA4 protein.7,10,11 ABCA4 insufficiency, as occurs in STGD1, augments the build-up of NRPE in the disc space and thus allows binding of a second molecule of all-trans-retinal to NRPE, leading to increased formation of A2E and related bisretinoid molecules,12 which are the only known components of lipofuscin. Lipofuscin subsequently accumulates in the RPE due to OS disc shedding and phagocytosis and can be toxic to the RPE through a number of mechanisms.13 Histopathological examination of eyes from patients with STGD1 has demonstrated large RPE cells densely packed with granules that exhibit the structural and biochemical (periodic acid-Schiff positive) characteristics of lipofuscin.14–17 Studies of fundus autofluorescence (AF), an intrinsic signal originating from RPE lipofuscin,18 have corroborated these histopathological findings, suggesting a greater accumulation of lipofuscin in ABCA4 retinopathy in comparison with controls.19–21 In a previous study, fundus AF was measured spectrophotometrically at a position 7° temporal to the fovea using an excitation wavelength of 510 nm.19 Autofluorescence intensity was found to be ∼3-fold higher in STGD1 patients than in control subjects of the same age. The emission spectra of fundus AF in STGD1 patients was similar in shape to normal subjects, there being a broad emission maximum in the 620- to 640-nm range.18
Detection and assessment of abnormal AF patterns by confocal scanning laser ophthalmoscopy (cSLO) have proven helpful for diagnosing ABCA4 disease and allow progression to be monitored.22,23 Several investigators have used cSLO images to assess AF intensities in retinal dystrophies and have shown that patients with ABCA4 disease have elevated AF levels.20,21,24 However, because of inherent variability's in acquired AF images, even with a strict and standardized imaging protocol, comparisons amongst patients can be challenging.25
We recently reported a novel method, quantitative autofluorescence (qAF), for quantifying AF in images acquired with a cSLO.26 This methodology incorporates a fluorescence reference internal to the imaging device in such a way that the reference is part of the AF image. Analysis then consists of comparing the gray levels (GLs) in the AF image with the GL of the internal reference, accounting thereby for changes in laser power and detector sensitivity. Furthermore, the methodology includes corrections for magnification and optical media density from normative data on lens transmission spectra. This approach when used together with a standardized image acquisition protocol allows reproducible quantification of AF levels in individual patients, interpatient comparison, and monitoring of AF levels longitudinally. It would also permit the comparison of data acquired on different devices at several centers. Here we apply qAF to a cohort of genetically confirmed STGD1 patients and compare the qAF values with normal control subjects.
Materials and Methods
Patients
Patients were prospectively recruited at the Edward S. Harkness Eye Institute, Columbia University. The study cohort consisted of 42 patients (23 females) from 37 families. All patients had a complete dilated eye exam and the clinical diagnosis of STGD1 was confirmed by a retinal specialist (SHT, RTS). Clinical, demographic, and genetic data for all patients are presented in Table 1. All patients had at least one known disease-causing mutation in the ABCA4 gene and 85% had mutations on both chromosomes. Patients were aged between 7 and 52 years (median age: 28.8 years), and had eyes with refractions between −9 and +6 diopters ([D] ranges corresponding to control group described below). Thirty-nine patients were of European ancestry and there was one each of African American, Hispanic, and Indian origin. Duration of disease at the time of examination (time since first diagnosis) was 1 to 31 years (median: 6.8 years) and the age at first diagnosis was 4 to 51 years (median: 15.5 years). Control values consisted of previously published data from 277 healthy subjects (374 eyes; age range, 5–60 years).27
Table 1.
Clinical, Demographic, and Genetic Data for All STDG1 Patients. Results for qAF and Texture Factor
|
Patient Number |
Sex* |
Age at Exam, y |
Duration, y |
Visual Acuity, logMAR |
Fishman Grade |
ABCA4 Mutations |
qAF†, qAF Units |
TF†, qAF Units0.5 |
||||
|
OD |
OS |
OD |
OS |
OD |
OS |
OD |
OS |
|||||
| 1.1 | M | 11 | 1 | 0.54 | 0.60 | II | II | p.[L541P; A1038V] | 639 | 627 | 1.9 | 2.0 |
| 1.2 | M | 7 | 1 | 0.30 | 0.18 | – | I | p.[L541P; A1038V] | 413 | 1.9 | ||
| 2 | F | 25 | 11 | 0.80 | 0.80 | II | II | p.G863A; c.5898+1G > A | 710 | 675 | 3.4 | 3.3 |
| 3 | M | 11 | 6 | 0.80 | 0.70 | I | – | p.G1961E; p.P1380L | 267 | 2.3 | ||
| 4.1 | M | 35 | 10 | 0.30 | 0.18 | I | – | p.G1961E; p.[L541P; A1038V] | 426 | 2.1 | ||
| 4.2 | M | 35 | 10 | 0.40 | 0.48 | I | I | p.G1961E; p.[L541P; A1038V] | 356 | 354 | 1.7 | 1.8 |
| 5 | F | 14 | 1 | 0.60 | 0.60 | II | II | p.L2027F; p.T972N | 737 | 718 | 2.3 | 2.6 |
| 6 | M | 45 | 31 | 1.00 | 0.88 | I | I | p.G1961E; p.P1380L | 623 | 543 | 4.2 | 4.0 |
| 7 | F | 42 | 5 | 0.30 | CF | – | I | p.E1252* | 557 | 2.1 | ||
| 8 | M | 15 | 4 | 0.80 | 0.80 | II | II | p.L2027F; p.R2077W | 728 | 697 | 3.2 | 3.2 |
| 9 | F | 24 | 2 | 0.60 | 0.40 | II | II | p.R1161S | 571 | 647 | 3.8 | 3.5 |
| 10 | M | 46 | 15 | 1.30 | 1.30 | I | I | p.G1961E; p.Q636H | 394 | 351 | 2.3 | 2.4 |
| 11.1 | M | 12 | 2 | 1.00 | 1.00 | II | – | p.[L541P; A1038V]; p.R1640W | 911 | 3.3 | ||
| 11.2 | F | 10 | 4 | 1.00 | 1.00 | II | – | p.[L541P; A1038V]; p.R1640W | 850 | 4.4 | ||
| 12 | F | 27 | 9 | 1.30 | 1.00 | – | III | p.P1380L; p.P1380L | 577 | 4.8 | ||
| 13 | F | 39 | 8 | −0.12 | 0.00 | – | I | c.250_251insCAAA | 616 | 2.3 | ||
| 14 | M | 23 | 4 | 0.88 | 0.60 | – | II | p.C54Y | 535 | 5.1 | ||
| 15.1 | M | 49 | 17 | 1.00 | 0.88 | I | I | p.Y1557C | 646 | 604 | 4.1 | 3.9 |
| 15.2 | M | 46 | 7 | 0.10 | 0.48 | I | I | p.Y1557C | 456 | 508 | 2.6 | 2.3 |
| 16.1 | F | 27 | 14 | 0.88 | 0.88 | III | III | p.L2027F; p.G851D | 448 | 459 | 6.0 | 6.3 |
| 16.2 | F | 29 | 19 | 1.30 | 1.18 | III | III | p.L2027F; p.G851D | 538 | 569 | 7.4 | 7.9 |
| 17 | M | 22 | 18 | 1.30 | 1.00 | III | III | p.P1380L; p.R2030Q | 434 | 411 | 5.7 | 6.0 |
| 18 | M | 37 | 16 | 0.70 | 0.70 | I | I | p.G1961E; p.G1961E | 281 | 279 | 2.6 | 2.2 |
| 19 | F | 33 | 5 | 0.88 | 0.70 | I | I | p.G1961E; c.4540-2A > G | 412 | 420 | 2.5 | 2.8 |
| 20 | F | 26 | 12 | 0.60 | 0.60 | – | I | p.G1961E; p.[L541P; A1038V] | 398 | 2.4 | ||
| 21 | F | 45 | 31 | 0.88 | 0.88 | I | I | p.R1640W | 647 | 613 | 2.6 | 2.8 |
| 22 | M | 43 | 7 | 1.00 | 0.00 | – | III | p.A1773V; p.G1591G | 640 | 6.9 | ||
| 23 | F | 41 | 1 | 0.10 | CF | II | II | p.P1486L; p.A1598D | 613 | 572 | 6.0 | 6.5 |
| 24 | F | 19 | 4 | 0.60 | 0.70 | I | – | p.G1961E; p.P1380L | 368 | 2.4 | ||
| 25 | F | 23 | 4 | 0.88 | 0.80 | – | I | p.[A854T; A1038V]; p.C2150Y | 512 | 2.3 | ||
| 26 | F | 52 | 1 | 0.70 | 0.48 | I | – | p.R212C | 722 | 2.0 | ||
| 27 | F | 52 | 13 | 1.00 | 1.00 | – | I | p.A1038V; p.A848D | 459 | 4.1 | ||
| 28 | M | 20 | 5 | 0.30 | 0.40 | I | – | p.L2027F; p.R1108H | 507 | 2.3 | ||
| 29 | M | 23 | 7 | 1.00 | 1.00 | I | I | p.G1961E; p.R2030Q | 334 | 347 | 2.4 | 2.0 |
| 30 | M | 44 | 26 | 0.70 | 0.70 | – | II | p.P1380L; p.R1108H | 453 | 4.7 | ||
| 31 | F | 30 | 22 | 1.00 | 1.30 | – | I | p.G1961E; c.6005+1G > T | 428 | 2.3 | ||
| 32 | M | 12 | 8 | 0.40 | 0.40 | I | – | p.W821R; p.C2150Y | 306 | 2.0 | ||
| 33 | F | 20 | 9 | 0.88 | 0.88 | III | III | p.R602W; p.M1882I | 650 | 655 | 2.6 | 2.5 |
| 34 | F | 47 | 4 | 0.40 | 0.40 | I | – | p.G1961E; p.R1129C | 400 | 2.5 | ||
| 35 | F | 19 | 3 | 0.70 | 0.48 | II | II | p.[L541P; A1038V]; p.L2027F | 733 | 749 | 3.9 | 4.0 |
| 36 | F | 20 | 7 | 0.88 | 1.00 | II | II | p.R1640W | 571 | 552 | 3.4 | 3.8 |
| 37 | F | 12 | 3 | 0.80 | 0.80 | I | I | p.R1108C; p.Q1412* | 536 | 501 | 1.7 | 1.7 |
All subjects were white, except for patients 10, 22, and 36 who were Indian, Hispanic, and black, respectively.
Bolded data are higher than the upper 95% confidence interval for the mean of healthy subjects.
Based on color and fundus AF images, patients were assigned to one of three phenotype groups according to the classification of Fishman28: Fishman I, atrophy, and flecks predominantly restricted to the central region around the fovea; Fishman II, flecks throughout the posterior pole, which often extended anterior to the vascular arcades and/or nasal to the optic disc; and Fishman III, “resorbed” flecks with widespread atrophy of the RPE. The study population included only a few patients with advanced disease: 24 patients were classified as Fishman I, 12 as Fishman II, and six as Fishman III.
The study was carried out with the approval of the Institutional Review Board of Columbia University (IRB-AAAI9906), and all patients were enrolled in accordance with the tenets set out in the Declaration of Helsinki. Informed consent was obtained prior to enrollment.
Genotyping
The genotyping microarray (ABCR600; Asper Ophthalmics, Tartu, Estonia),29 which detects all known mutations in the ABCA4 gene (currently 632; Asper Biotech, Inc., www.asperbio.com, in the public domain), was used for initial screening of most patients. Mutations were confirmed by direct Sanger sequencing. In some cases where no mutations were detected by the array, or in more recently recruited patients, the next generation sequencing of the entire ABCA4 open reading frame and adjacent intronic sequences was performed on the Roche 454 platform.30
The four most common mutations found in six or more patients were: G1961E (12 patients from 11 families); L541P/A1038V (eight patients from five families); L2027F (six patients from five families); and P1380L (six patients from six families). Four other mutations were found in two to four patients: R1640W (four patients from three families); Y1557C (two patients from one family); G851D (two patients from one family); and R2030Q (two patients from two families). For the purposes of analyses reported below, we will refer to those eight mutations as the “more common mutations.” In two patients (two families), A1038V was present in a compound heterozygous state with other mutations while not as a complex allele with L541P (Table 1).
Image Acquisition
Autofluorescence images were acquired by three experienced operators (TRB, TD, JPG) using a cSLO device (Spectralis HRA+OCT; Heidelberg Engineering, Heidelberg, Germany) that had been modified by the insertion of an internal fluorescent reference. Pupils were dilated with 1% tropicamide and 2.5% phenylephrine to at least 7 mm in diameter. With room lights turned off, a near-infrared reflectance image (NIR-R; 787 nm) was recorded first. After switching to AF mode (488 nm excitation; beam power <260 μW), the camera was slowly moved toward the patient to allow the patient to adapt to the blue light. Patients were asked to focus on the central fixation light of the device. Alternatively, in cases of pronounced eccentric fixation, an external fixation light was used or the patient was asked to look “straight ahead,” and the orientation of the camera was adjusted to produce a field that was centered on the fovea and included the temporal edge of the disc. The fundus was exposed for 20 to 30 seconds to bleach rhodopsin,26 while the focus and alignment were refined to produce maximum and uniform signal over the whole field. The sensitivity (detector gain) was adjusted so that the GL did not exceed the linear range of the detector (GL < 175).26 Two or more images were then recorded (each of nine frames, in video format) in the high-speed mode (8.9 frames/s) within a 30 × 30° field (768 × 768 pixels). In addition, a horizontal scan through the fovea was recorded by spectral domain optical coherence tomography (SD-OCT).
Usually both eyes of patients were imaged. However, only one eye was imaged in five patients who proved to be challenging (due to photophobia or difficulties in alignment because of unstable or eccentric fixation) and for one patient who had high myopia in one eye (−12 D, study eye: −2 D).
After imaging, all videos were inspected for image quality and consistency in GLs by three of the authors (TRB, TD, JPG). For an imaging session, two videos were selected to generate the AF images for analysis. Only frames with no localized or generalized decreased AF signal (due to eyelid interference or iris obstruction) and no large misalignment of frames (causing double images after alignment) were considered. The frames then were aligned and averaged with the system software and saved in “non-normalized” mode (no histogram stretching). Fourteen out of 78 eyes were excluded after inspection of the videos because the image quality was not considered sufficient for reliable quantification. To evaluate possible selection bias (e.g., images of eyes with more advanced disease being excluded), we compared visual acuities, and found that there was no difference between those eyes that were included and those that were excluded (Mann-Whitney, z(83) = −0.66, P = 0.51).
To assess reproducibility, a second imaging session was performed only systematically in the second part of the study (21 patients, 32 eyes). The second session was performed after randomly changing the focus and the camera orientation and after repositioning the subject.
Image Analysis
Dedicated image analysis written in graphing and data analysis software (IGOR; WaveMetrics, Lake Oswego, OR, USA) was used to determine qAF. The software recorded the mean GLs of the internal reference and from eight circularly arranged segments positioned at an eccentricity of approximately 7 to 9° (Fig. 1). Segments were scaled to the horizontal distance between the fovea and the temporal edge of the optic disc. If the fovea could not be identified, then its position was estimated based on a SD-OCT scan that was registered to an AF or NIR-R image. When a myopic crescent obscured the true edge of the optic disc on AF, a NIR-R image aided in identifying the disc edge (crescent has high reflectance). The software accounted for the presence of vessels and marked atrophy in the segments (see Supplementary Material: “Histogram Analysis”). Segments were excluded, if geographic atrophy exceeded 50% of the area of the segment, or if the segment was partially positioned further than 15° from the center of the image.
Figure 1.

Fundus AF image analysis of a patient (#14) with widespread high AF flecks throughout the fundus. For qAF analysis, mean GLs are recorded from the internal reference (rectangular area outlined in white at the top of the image) and from eight circularly arranged segments (outlined in white). The horizontal distance, FD, between the temporal edge of the optic disc (white vertical line) and the center of the fovea (white cross) was used to define inner and outer radii of the ring of segments (0.58 × FD and 0.78 × FD, respectively).
For each segment, qAF was computed from the mean GL in that segment and the GL of the internal reference (after accounting for the zero level), media absorption (using normative data for a given age), image magnification, and internal reference calibration.26 For each image, a single measure, qAF8, was obtained from the mean qAFs of all segments. For each eye, the mean qAF8 from one or two imaging sessions (two or four images) was computed. Despite our efforts to center the fovea in the field, in some images, inferior segments were partially located outside the field, probably because the preferred locus of fixation in STGD1 patients is often situated superiorly.31,32 Thus, data were not available for the inferior segment in 11% of 192 images acquired in this study, compared with 4% for all other segments. After combining the data from all images of an eye, only five eyes had missing data in the inferior segment and in one of its neighboring segments. In such cases, qAF8 may be overestimated by 2% ± 2% on average (range, −2% to 10%) according to the average spatial distribution in healthy eyes.27
Texture Factor (TF)
Cideciyan et al.33 introduced a method to quantify small-scale heterogeneity of fundus AF (caused by flecks and small patches of partial atrophy) by defining a texture index as the coefficient of variation (SD/mean signal) when a small aperture was scanned in discrete areas of an AF image. In a photon noise–dominated system, the SD would be expected to increase with the square root of the mean signal, and the coefficient of variation to decrease. This was verified by quantifying images of uniform fluorescent targets using the cSLO device (Heidelberg Engineering; see Supplementary Material: “Texture Factor”). To obtain a metric for texture that would be independent of the mean signal (qAF), we defined a “texture factor” as TF = SD/
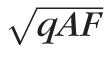 , where qAF is the mean value for the area of the segment that does not contain large vessels and SD is the standard deviation of qAF in the same area. To report the TF for one eye, the mean TF was calculated as TF =
, where qAF is the mean value for the area of the segment that does not contain large vessels and SD is the standard deviation of qAF in the same area. To report the TF for one eye, the mean TF was calculated as TF =
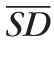 /
/
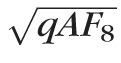 , where
, where
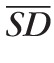 was derived from the mean variance (SD2) for the eight segments. We found that TF for the fluorescent targets exhibited little change with qAF8, which is consistent with photon noise limited detection. Thus, if TF is higher than the level observed in targets, the origin of this difference must be due to pathology or to variation in AF in healthy subjects. Since the cohort of STGD1 patients in this study was predominantly white, we compared their TF to those of 83 healthy white subjects (83 eyes; range, 5–58 years). For healthy eyes over 18 years of age, there was no effect of race on TF (P > 0.23).
was derived from the mean variance (SD2) for the eight segments. We found that TF for the fluorescent targets exhibited little change with qAF8, which is consistent with photon noise limited detection. Thus, if TF is higher than the level observed in targets, the origin of this difference must be due to pathology or to variation in AF in healthy subjects. Since the cohort of STGD1 patients in this study was predominantly white, we compared their TF to those of 83 healthy white subjects (83 eyes; range, 5–58 years). For healthy eyes over 18 years of age, there was no effect of race on TF (P > 0.23).
Statistical Analysis
Overall, images of 64 eyes (42 patients) were available for analysis. We used mixed-effects linear regression that accounts for within-subject correlations between eyes and between close family members (e.g., sibling, parent-child; Stata 12.1, College Station, TX, USA) to investigate the relationships between patient characteristics (age, genotype, etc.), qAF8, and TF. Models included binary and continuous factors. Continuous factors included age at examination, and disease duration (time since first diagnosis) in years. The binary factors that were examined included sex, eye, and mutation. Comparisons between specific mutations and healthy eyes were conducted for the four most common mutations (at least six patients and eight eyes). For between-genotype analyses, we only considered the eight more common mutations (see “Genotyping” section). The qAF8 data obtained from white STGD1 patients were compared with the confidence intervals of 87 white subjects with healthy eyes (125 eyes).27 For the Indian, Hispanic, and black STGD1 patients, qAF8 was compared with their respective confidence intervals.27
To evaluate the repeatability of the measurements between sessions, we used the method of Bland and Altman34 to compute the coefficient of repeatability (CR, 95% confidence interval) for the differences {log(qAFB) − log(qAFA)}, where qAFA and qAFB were the qAF8 in the two sessions. The coefficient of agreement (CA) between the qAF8 of right and left eyes was computed similarly.
Results
Autofluorescence images and color-coded qAF maps for three age-similar STGD1 patients and one healthy subject (Fig. 2) illustrate that the patients had higher qAF values. In these examples, the difference in qAF was greatest between the healthy eye (panel A) and the two STGD1 fundi on the right (patients #2 and #33, panels C and D). Conversely, patient #28 (panel B) had apparent disease confined to the central macula with a smaller elevation of qAF levels consistent with a milder phenotype.
Figure 2.

Quantified fundus autofluorescence images. Autofluorescence images (upper panels) with corresponding color-coded qAF images (lower panels) for the right eye of (A) a control (20 years) and for STGD1 patients: (B) #28 (20 years); (C) #2 (25 years); and (D) #33 (19 years). Images (B), (C), and (D) are examples of Fishman Stages I, II, and III, respectively. As shown in the qAF color-code scale (below), lower qAF levels are indicated as blue and higher qAF levels as red colors (see scale). Note that the images for all eyes had similar GLs after adjusting the sensitivity to optimize the dynamic range. Thus, the GL of the internal reference (rectangle in upper center of each image) is lower in the three STGD patients ([B–D], upper) than in the healthy eye ([A], upper), reflecting the higher AF levels of the patients. In the color-coded qAF images ([A–D], lower) the reference has the same color.
Spatial Distribution
The spatial distribution of qAFs within the ring of segments was calculated as before27 by averaging the normalized distribution measured in both eyes (accounting for mirror-image symmetry) to minimize instrumental nonuniformities. The spatial distribution for the 22 STGD1 patients for whom two eyes were imaged was similar to that in 97 healthy eyes (Fig. 3). Quantified fundus autofluorescence intensities were highest in the superotemporal quadrant and lowest in the inferonasal quadrant. Overall, there was no significant difference between STGD1 and control groups (mixed-effects linear regression, χ2(1) = 0.04, P = 0.84), but there was an interaction (χ2(7) = 18.9, P = 0.009), with STGD1 patients having higher qAF in the temporal segments (χ2(1) = 10.4, P = 0.001) and lower qAF in the inferior segment (χ2(1) = 3.8, P = 0.05). These small differences could reflect the natural history of STGD1 or be related to suboptimal area sampling due to eccentric fixation. Overall, the qAF8 in STGD1 patients can be compared with that in the healthy eyes because of the close similarity of their spatial distributions.
Figure 3.

Spatial distribution of qAF in individual segments of the ring (Fig. 1) for normal subjects (n = 97), and for STGD1 patients (n = 22). The segment positions are identified by S, superior; ST, superior temporal; T, temporal; IT, inferior temporal; I, inferior; IN, inferior nasal; N, nasal; and SN, superonasal. The segments' qAF were normalized to the average qAF8 (for all segments). Error bars are 95% confidence intervals.
Agreement Between Eyes
The coefficient of agreement for qAF8 between the two eyes of 22 STDG1 patients was ±13%, similar to that observed in healthy eyes (±15.3%).27 There was no difference between the qAF8 of left and right eyes (paired t = 1.3, P = 0.2).
Repeatability
Between-session repeatability of qAF8 (mean of two images per session) was evaluated from images of 32 eyes (21 patients). The coefficient of repeatability (for log(qAF2)-log(qAF1)) was ±0.042 log-qAF-units or ±10.3% of the mean qAF. This was slightly, but not significantly (Brown and Forsythe test, P = 0.51), higher than the ±9.4% observed in healthy subjects.27 The coefficient of repeatability for the TF was ±0.43 or ±12% of the mean TF.
qAF in STGD1 Patients Versus Healthy Subjects
Stargardt disease eyes exhibited elevated qAF8 relative to healthy eyes (P < 0.001) particularly at younger ages (Fig. 4). Between-subject variability in qAF8 was greater in STGD1 than in healthy eyes, again predominantly at young ages. Only nine eyes (six patients) had qAF below the upper 95% confidence limit of healthy eyes. For healthy eyes, qAF8 values increased with age (P < 0.001)27; but in STGD1 eyes, overall, there was no association with age (P = 0.88). As reported previously, amongst subjects with healthy eyes qAF8 was higher in females (P = 0.01)27; this was also the case for STGD1 patients (P = 0.009). For the five pairs of siblings (one twin pair), we found no significant difference in qAF8 (Wilcoxon paired; Z5 = 0.4, P = 0.7).
Figure 4.

qAF8 from one or two eyes of all STGD1 patients as a function of age. White STGD1 patients are shown as filled circles. For comparison, qAF8 of white healthy eyes (crosses) are plotted with mean (solid line) and 95% confidence intervals (dashed lines). The qAF8 of the black, Hispanic, and Indian subjects (open circles) can be compared with the corresponding upper 95% confidence limits for their different race/ethnicity group (short segments of solid line).
Genotype-Phenotype Relations (qAF8)
Figure 5 shows the qAF8 levels for patients with different ABCA4 mutations in our sample as a function of age. Comparing each of the four most common mutations separately with healthy eyes, G1961E (P = 0.001); L541P/A1038V (P < 0.001); L2027F (P < 0.001); and P1380L (P = 0.024) eyes had qAF8 that was significantly higher than in healthy eyes, when corrected for age. Autofluorescence images from these patients are presented in the Supplementary Material (“AF images of STDG1 patients”).
Figure 5.

qAF8 from one or two eyes of each STGD1 patients versus age with colored-symbol coding for mutations. The mutations were confirmed in six or more patients (G1961E, L541P/A1038V, L2027F, and P1380L) or in two to four patients (R1640W, Y1557C, G851D, and R2030Q). Also shown is mutation A1038V in a compound heterozygous state with other mutations while not as a complex allele with L541P. In this and the following figures, some points have been displaced slightly to avoid overlap.
A mixed-effects regression of all 64 STDG1 eyes was used to analyze the effects on qAF8 of the more common mutations in our cohort. When corrected for age (P = 0.04) and sex (P = 0.004), compared with all other patients who did not have that particular mutation, the mutations L2027F (P = 0.009) and L541P/A1038V (P = 0.015) were associated with higher qAF8, while A1038V (when not in conjunction with L541P, P = 0.06); G851D (P = 0.006); and G1961E (P < 0.001) were associated with lower qAF8 in this sample.
Texture Factor in STGD1 Patients and Healthy Subjects
As expected, TF of healthy eyes (n = 83) was higher than that of uniform fluorescent targets (Fig. 6), and increased with age (P = 0.001) when corrected for qAF8 (P = 0.15). This suggests that there is an increase in heterogeneity with age in the AF images of healthy subjects; this heterogeneity would be visualized as mottling. For eyes of STGD1 patients, the TFs in 42 eyes (27 patients) exhibited high TFs above the upper 95% confidence limit (Fig. 6). Among the 42 STGD1 patients, TF increased slightly with duration of disease (P = 0.05), but not with age (P = 0.77) when corrected for qAF8 (P = 0.22). For the five pairs of siblings, there was no significant difference in TF (Wilcoxon paired, Z5 = 0.9, P = 0.3).
Figure 6.

Texture factor in one or two eyes of each STGD1 patient (see legend) and healthy subjects (black crosses) as a function of age. Mean (solid black line) and 95% confidence intervals (interrupted line) for the healthy subjects are based on TF data from 83 white subjects (small +). The linear fit was TF = 1.58 + 0.014. Age (r2 = 0.42, P = 0.001). The horizontal (interrupted) line is the mean TF (1.1; 95% confidence interval: 0.8–1.4) associated with uniform fluorescent targets (see Supplementary Material: “Texture Factor”).
Genotype-Phenotype Relations (TF)
When compared separately with healthy eyes, three of the four most common mutations, L541P/A1038V (P < 0.001); L2027F (P < 0.001); and P1380L (P = 0.001), had TF that was significantly higher than in healthy eyes, when corrected for age. Conversely, G1961E did not have higher TF than healthy eyes (P = 0.61).
To investigate the effects of the eight more common mutations on the phenotype described by TF, we used a mixed-effects regression that included all STDG1 patients (64 eyes). When compared with all other patients who did not have that particular mutation, G851D (P < 0.001) and P1380L (P = 0.008) were associated with higher TF, while G1961E (P = 0.01) was associated with lower TF in this sample.
When all STDG1 eyes were considered, TF correlated with qAF8 (Spearman, P = 0.013). Forty eyes had both high qAF8 and high TF, seven had both normal qAF8 and TF, 15 eyes had high qAF8 and normal TF, and only two eyes had low qAF8 and high TF. The proportion of eyes with both high qAF8 and high TF was higher than expected by chance (Fisher's exact, P = 0.006).
Phenotype Classifications
Thirty-four eyes were classified as Fishman I, 20 as Fishman II, and 10 as Fishman III. All three groups had higher qAF8 than healthy eyes (P < 0.001). In Fishman II, qAF8 was higher (P < 0.001) than in Fishman I patients (Table 2), but qAF8 values of Fishman III patients were not significantly different than those of Fishman I (P = 0.07) and II (P = 0.10). qAF8 was within the normal range for nine of 34 Fishman I eyes, while no Fishman II (0/20) and no Fishman III (0/10) eyes had a qAF8 within the normal range (Fisher exact test, P < 0.009).
Table 2.
Fishman Grade: qAF and Texture Factor
|
Fishman Grade |
Eyes/Subjects, n |
Age, y |
Duration, y |
qAF, qAF Units |
Texture Factor, qAF Units0.5 |
| I | 34/24 | 33 ± 14* | 10 ± 8 | 458 ± 12* | 2.5 ± 0.7* |
| II | 20/12 | 21 ± 10* | 6 ± 7 | 684 ± 109* | 3.7 ± 1.2*† |
| III | 10/6 | 28 ± 8 | 9 ± 5 | 538 ± 94 | 5.6 ± 1.8† |
Mean ± SD. Same symbols (*, †) in each column indicated significant differences (P < 0.001), accounting for two eyes in some subjects and for family relationships.
Texture factor of Fishman II and III patients was higher than in healthy subjects (P < 0.001), but this effect was slightly less pronounced in Fishman I eyes (P = 0.001). Texture factor for Fishman III was higher than for Fishman II (P = 0.001), and the latter was higher than for Fishman I (P < 0.001). Texture factor was within the normal range for more than half the Fishman I eyes (20 out of 34 eyes), while few Fishman II (2/20) and no Fishman III (0/10) eyes had a TF within the normal range (Fisher exact test, P < 0.001).
The Fishman classification is based on the qualitative assessment of pathological changes in the entire posterior pole. Conversely, qAF8 and TF measures were restricted to the area of the segments (Fig. 1). Thus, to compare qAF8 and TF with the presence of flecks and atrophy in the segments, we applied (consensus of three observers: TRB, TD, SHT) an additional grading scheme that only took into consideration flecks and atrophy within the segments: grade A, no flecks in any segment; grade B, less than five flecks/segment on average; grade C, more than five flecks/segment on average, but the area covered by flecks smaller than that affected by atrophy; and grade D, area of resorbed flecks/atrophy larger than that occupied by flecks. This grading allowed us to better understand the relationship between TF and qAF8. Indeed, when these measures were plotted in a scatter diagram (Fig. 7), a pattern emerged in which grades A through D were well segregated. Grade A and B eyes tended to follow the same relationship as healthy eyes with an increase in qAF8 being accompanied by only a moderate increase in TF. The highest qAF8 values were seen for eyes with numerous flecks (grade C). For grades C and D, qAF8 decreased with increasing TF (Spearman, P = 0.01).
Figure 7.

Scatter diagram of qAF8 and TF for all STGD1 eyes. Symbols correspond to the four phenotype grades (assigned by three observers) for the ring of segments where the measurements are made: grade A if no flecks were seen in any segment, grade B is less than five flecks/segment on average were detected, grade C if more than five flecks were seen, but the area covered by flecks was smaller than that affected by atrophy, and grade D if the area of atrophy was larger than that occupied by flecks.
In all 25 grade A eyes (19 patients), the AF image appeared clinically normal in the ring of segments (Fig. 1), with no obvious high AF flecks nor other AF abnormalities. Even so, the qAF8 levels (P < 0.001) and texture (P = 0.02) in these eyes were significantly elevated compared with the healthy eyes (P < 0.001) when corrected for age.
Discussion
In keeping with the findings from previous histopathological14–17 and AF studies,19,22,35–38 qAF8 levels were markedly elevated in patients with STGD1. Consistent with the report that lipofuscin is a crucial and early component of the disease process in STGD1,33 we found that qAF8 levels were increased even in young patients with clinically diagnosed STDG1 (Fig. 4). This observation, together with the sizable differences in qAF8 observed between most STGD1 and control subjects, indicates that qAF may be a meaningful aid to identify patients for ABCA4 genetic screening.
The spatial distribution of the autofluorescence is similar in STDG1 patients and healthy subjects. Highest values are generally found in the superotemporal quadrant and lowest in the inferonasal quadrant. This corresponds with the well-established distribution of rod photoreceptors.39,40 The similarity of the spatial distributions facilitates comparisons of STDG1 and healthy eyes.
In this study, we classified all STDG1 patients according to the clinical phenotypes described by Fishman.28 This classification is based on an assessment of the presence of pigmentary changes, flecks, and atrophy over the entire posterior pole (and the spatial distribution of the flecks). Conversely, since qAF8 and TF only apply to the area defined by the ring of segments (Fig. 1), in addition to Fishman phenotypes we utilized a grading system (A–D) that was restricted to the segment areas. The A through D grading system, as shown in Figure 7, facilitated an interpretation of changes in qAF and TF and may contribute to a better understanding of the natural history of the disease. Despite the cross-sectional nature of the study, we observed an initial increase in qAF followed by a decline in qAF associated with RPE atrophy. This is also reflected in the lack of high qAF8 values at older ages and the decrease in between-subject variability with age. To further understand changes in AF levels and texture with disease progression, it will be necessary to conduct a larger study involving more genotyped patients and longitudinal follow-up. Such a study should also include patients without clinical changes and patients with advanced disease stages.
Only qAF8 for Fishman II patients was significantly elevated compared with the other Fishman groups (Table 2), but all groups had higher levels than healthy subjects. It has previously been noted33 that some cases of STGD1 can present with high AF in images, wherein structural abnormalities, such as flecks and/or atrophy, are not obvious outside the fovea. This was also the case in this study: all patients assessed as grade A in our classification (25 eyes, 19 patients) presented no flecks or other abnormalities in the ring of segments, but the qAF8 was significantly elevated in that group compared with healthy eyes (P < 0.001).
In addition to qAF8, we used TF to characterize the heterogeneity of fundus AF that depends upon the small-scale variations in lipofuscin distribution. The age-related increase in TF in healthy eyes may reflect RPE cell loss and monolayer thinning41,42 and RPE lipofuscin photodegradation.13 For two-thirds of the STDG1 eyes, TF was significantly higher than in healthy eyes. While more than half of the eyes classified as Fishman I had TFs within the normal range, Fishman II and III generally had levels outside normal limits. Interestingly, 42/64 STDG1 eyes had high TF, and all but two of those also had high qAF8. Conversely, Cideciyan and colleagues33 reported cases wherein mean AF intensity was within normal limits while AF texture index was elevated. In our cohort, the mutations G851D and P1380L had high AF texture while G1961E was associated with lower AF texture.
Given that STGD1 is a recessive disease and more than 600 disease-causing mutations have been described, compound heterozygosity is very common. Thus, it has been a challenge to determine the contribution of a single mutation to a phenotype and to understand interactions between alleles. In our current cross-sectional study, even though 42 subjects were included in the analysis, only limited genotype-phenotype correlations could be drawn from the data.
While in both the healthy eye43 and STGD1 patients lipofuscin accumulation begins at birth, our data suggest that the rate in STGD1 is determined, at least in part, by the specific ABCA4 mutation. For example, based on our cross-sectional data, the mutations L2027F and L541P/A1038V seem to confer a faster rate of accumulation, whereas G1961E and G851D seems to confer a slower increase (Fig. 5).
The complex allele L541P/A1038V involves missense mutations in both the exocytoplasmic domain-1 and nucleotide binding domain (NBD)-1 and confers severe STGD1 with relatively early onset of retina-wide disease.44–47 In our study, the L541P/A1038V complex allele was in some patients associated with high qAF8 even at young ages, while in other patients, particularly in those compound heterozygous for L541P/A1038V and G1961E, qAF8 values were relatively lower (Fig. 5). The latter group of patients, however, did not present features (e.g., atrophy) in the analyzed fundus segments that could explain a decrease in qAF8. Thus it is likely that in these cases, the lower qAF values do not reflect a decrease in qAF after an earlier rapid elevation, but rather a slower increase in qAF8 due to the presence of the G1961E allele (L541P/A1038V + G1961E). Additional missense mutations found to associate with elevated qAF8 compared with other mutations in our sample were L2027F and P1380L. The L2027F mutation causes an amino acid change in NBD-2 and confers reduced ATP binding.11,48 P1380L is also a severe mutation and is suggested to cause either impaired ATP binding11 or altered transport of ABCA4 protein across the outer segment membrane.46 When P1380L is carried in compound heterozygosity with R2077W, autosomal recessive cone-rod dystrophy results.49 When harbored as a homozygous mutation or as a compound heterozygous mutation with R2030Q or IVS40 + 5G>A, the mutation is associated with central atrophy and peripapillary disease, the latter being an uncommon phenotype.50
The missense mutation G1961E in exon 42 of the ABCA4 gene is the most common ABCA4 mutation.51 This sequence change results in a glycine to glutamate substitution within the NBD-2 of the protein.11,45 The G1961E allele always cosegregates with the disease in families.45,52 Nevertheless, the G1961E mutation in ABCA4 is perplexing since in an in vitro assay this mutation conferred a markedly aberrant decrease in all-trans-retinal stimulated ABCA4 ATPase activity,11 yet it is considered to be associated with mild disease. For instance, in Fishman's28 original classification of 29 STGD1 patients into three clinical phenotypes, patients with the G1961E variation on one ABCA4 allele were assigned to the mildest (Fishman I) of the three. These patients exhibited a generally small atrophic-appearing foveal lesion, visual acuity was slightly reduced, and normal cone and rod ERG amplitudes were observed. The G1961E variant was reported to confer a tendency toward later disease onset51 (though not in our sample), and relatively long delay before retina-wide changes.46 Also characteristic of this mutation is an absence of a dark choroid.28 A dark choroid in fluorescein angiography is generally attributed to high content and absorption by lipofuscin in the RPE, a feature that also confers a vermillion fundus appearance.19 Thus, absence of dark choroid is considered to reflect less pronounced lipofuscin accumulation; this interpretation is consistent with our finding that in the presence of the G1961E mutation, qAF levels outside the parafovea are lower when compared with other ABCA4 mutations. In fundus AF images, the G1961E allele in both the homozygous and compound heterozygous state is commonly associated with bull's eye maculopathy.53
Taken together, the clinical features of the disease associated with G1961E indicate a phenotype chiefly confined to cone-rich fovea and parafovea (central macula). The observation that in OCT images, foveal photoreceptor outer segment loss can occur in the presence of intact RPE,53 indicates that damage to central cones does not necessarily occur secondary to lipofuscin-related RPE damage. Since the bisretinoids that contribute to RPE lipofuscin originate in photoreceptor cells, one other possibility is direct lipofuscin-related damage to photoreceptor cells. In healthy eyes there is no appreciable accumulation of lipofuscin-related fluorophores in photoreceptor outer segments because of membrane turnover. However, histopathological studies14 have revealed that autofluorescent lipofuscin-like material can be detected in photoreceptor inner segment membrane in some patients with STGD1. Perhaps abnormal accretion of bisretinoid lipofuscin in photoreceptor inner and outer segments in the presence of the G1961E mutation is a feature deserving future investigation. A mechanism such as this could explain photoreceptor cell degeneration in the absence of marked lipofuscin accumulation in RPE.
On the operational side, difficulties in properly aligning the fovea in the center of the image caused data to be lost and introduced measurement errors because of image nonuniformities near the edges of the image.26,27 This resulted in part from the limited number of fixation targets available in the cSLO device (Heidelberg Engineering; one central and eight peripheral targets). Addition of intermediate targets in the current system or an improved external fixation (for the fellow eye) may substantially aid image acquisition and shorten the test duration. In this study, images/frames were not included in the analysis if they were of suboptimal quality. Going forward, it will be important to make qAF less operator-dependent. Software-driven feedback could assist image acquisition and standardized computer algorithms could be implemented to select which frames should be used to generate images for AF quantification.
Our approach to measuring texture was simple, yet provided a metric without requiring additional image analysis. In experiments using fluorescent targets we observed that qAF8 was independent of detector sensitivity as expected, while TF increased with decreasing sensitivity (see Supplementary Material: “Texture Factor”). Since fundus AF levels in STDG1 eyes are high, images are acquired using lower sensitivities than in healthy eyes. This sensitivity effect may explain why grade A STDG1 eyes with qualitatively normal-appearing segments had elevated TF (P = 0.02); eight of 25 of these eyes (seven patients, ages: 10–36 years) had TFs slightly higher (by 1%–12%) than the upper 95% confidence interval for healthy subjects (Fig. 6). Mean sensitivities were 80 and 87 for those STDG1 eyes and healthy eyes in the same age range, respectively. An estimate of the magnitude of the effect for all STDG1 eyes indicates that TF would be overestimated by 3 to 25% with a mean of 12% (see Supplementary Material, p. 8). However, this does not substantially alter our overall conclusion that higher than normal TF values are found in many STDG1 patients. Further efforts at developing metrics to describe pathological and age-related changes are warranted.
An inherent limitation of the qAF method is the necessity to bleach the photopigment for approximately 30 seconds with the 488-nm light of the Spectralis to minimize pigment absorption. This light is quite bright (5.1 log photopic Trolands) and causes discomfort for some patients (even when gradually implemented). The retinal irradiance during AF imaging is 330 μW/cm2 (488 nm, 30° field; 260 μW beam power), which is below the permissible exposure recommended by the ANSI standards for durations of up to 8 hours (permissible levels are approximately 10 times lower than damage threshold).54,55 Overall duration for bleaching and imaging for each eye was typically less than 2 minutes. However, the light exposure safety limits are based on data from healthy subjects and concerns have been raised about the long-term effects of intense retinal illuminations in patients with retinal disorders.56–58 Reducing the beam power of the instrument59 would alleviate patient discomfort but would not reduce the light exposure in our protocol, since duration of bleaching would have to be proportionally increased. Bleaching with an external light source centered on the rod absorption spectrum would reduce the needed exposure by only 15%. An external bleaching light that is gradually increased in intensity may alleviate patient discomfort and allow better control of the bleaching.
As expected for an inherited disease, qAF8 values for the right and left eyes of patients were found to be highly correlated. Similar right-left correspondence between eyes is generally observed clinically in STGD and the size of atrophic RPE lesions in the fellow eyes of patients with STGD1 are reported to be similar.60,61 Given the strength of this correlation, measurement of qAF in the eye contralateral to the treated eye, for example, in gene-based or stem cell therapies, could provide useful control data for future clinical trials.
The benefits of using qAF in diseases such as STGD1 are clear. Fundus AF imaging is quicker, easier to perform and cheaper than some other forms of clinical evaluation and provides a quantifiable parameter for assessment of disease status. While exact predictions of genotype-phenotype correlations are made difficult by the genetic and phenotypic heterogeneity observed in ABCA4 disease, qAF provides an additional parameter for the establishment of genotype-phenotype correlations. These correlations will aid both the selection of patients for ABCA4-related clinical trials, as well as the establishment of end-point measures for clinical studies.
Acknowledgments
Supported in part by Grants EY021163, EY019861, EY015520, and EY019007 (Core Support for Vision Research) from the National Eye Institute/National Institutes of Health; the Foundation Fighting Blindness (Owings Mills, Maryland, United States); and unrestricted funds from Research to Prevent Blindness (New York, New York) to the Department of Ophthalmology, Columbia University. The authors alone are responsible for the content and writing of the paper.
Disclosure: T.R. Burke, None; T. Duncker, None; R.L. Woods, None; J.P. Greenberg, None; J. Zernant, None; S.H. Tsang, None; R.T. Smith, None; R. Allikmets, None; J.R. Sparrow, None; F.C. Delori, None
References
- 1. Fishman GA, Farber M, Patel BS, Derlacki DJ. Visual acuity loss in patients with Stargardt's macular dystrophy. Ophthalmology. 1987; 94: 809–814 [DOI] [PubMed] [Google Scholar]
- 2. Rotenstreich Y, Fishman GA, Anderson RJ. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology. 2003; 110: 1151–1158 [DOI] [PubMed] [Google Scholar]
- 3. Blacharski PA. Fundus flavimaculatus. In: Newsome DA. ed Retinal Dystrophies and Degenerations. New York: Raven Press; 1988: 135–159 [Google Scholar]
- 4. Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997; 15: 236–246 [DOI] [PubMed] [Google Scholar]
- 5. Allikmets R, Shroyer NF, Singh N, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997; 277: 1805–1807 [DOI] [PubMed] [Google Scholar]
- 6. Azarian SM, Travis GH. The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt's disease (ABCR). FEBS Lett. 1997; 409: 247–252 [DOI] [PubMed] [Google Scholar]
- 7. Molday LL, Rabin AR, Molday RS. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet. 2000; 25: 257–258 [DOI] [PubMed] [Google Scholar]
- 8. Papermaster DS, Reilly P, Schneider BG. Cone lamellae and red and green rod outer segment disks contain a large intrinsic membrane protein on their margins: an ultrastructural immunocytochemical study of frog retinas. Vision Res. 1982; 22: 1417–1428 [DOI] [PubMed] [Google Scholar]
- 9. Sun H, Nathans J. Stargardt's ABCR is localized to the disc membrane of retinal rod outer segments. Nat Genet. 1997; 17: 15–16 [DOI] [PubMed] [Google Scholar]
- 10. Pawar AS, Qtaishat NM, Little DM, Pepperberg DR. Recovery of rod photoresponses in ABCR-deficient mice. Invest Ophthalmol Vis Sci. 2008; 49: 2743–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sun H, Smallwood PM, Nathans J. Biochemical defects in ABCR protein variants associated with human retinopathies. Nat Genetics. 2000; 26: 242–246 [DOI] [PubMed] [Google Scholar]
- 12. Quazi F, Lenevich S, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun. 2012; 3: 925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sparrow JR, Gregory-Roberts E, Yamamoto K, et al. The bisretinoids of retinal pigment epithelium. Prog Retin Eye Res. 2012; 31: 121–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Birnbach CD, Jarvelainen M, Possin DE, Milam AH. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology. 1994; 101: 1211–1219 [DOI] [PubMed] [Google Scholar]
- 15. Eagle RC Jr, Lucier AC, Bernardino VB Jr, Yanoff M. Retinal pigment epithelial abnormalities in fundus flavimaculatus: a light and electron microscopic study. Ophthalmology. 1980; 87: 1189–1200 [DOI] [PubMed] [Google Scholar]
- 16. McDonnell PJ, Kivlin JD, Maumenee IH, Green WR. Fundus flavimaculatus without maculopathy. A clinicopathologic study. Ophthalmology. 1986; 93: 116–119 [DOI] [PubMed] [Google Scholar]
- 17. Steinmetz RL, Garner A, Maguire JI, Bird AC. Histopathology of incipient fundus flavimaculatus. Ophthalmology. 1991; 98: 953–956 [DOI] [PubMed] [Google Scholar]
- 18. Delori FC, Dorey CK, Staurenghi G, Arend O, Goger DG, Weiter JJ. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci. 1995; 36: 718–729 [PubMed] [Google Scholar]
- 19. Delori FC, Staurenghi G, Arend O, Dorey CK, Goger DG, Weiter JJ. In vivo measurement of lipofuscin in Stargardt's disease--Fundus flavimaculatus. Invest Ophthalmol Vis Sci. 1995; 36: 2327–2331 [PubMed] [Google Scholar]
- 20. Cideciyan AV, Swider M, Aleman TS, et al. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci. 2005; 46: 4739–4746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lois N, Halfyard AS, Bird AC, Holder GE, Fitzke FW. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am J Ophthalmol. 2004; 138: 55–63 [DOI] [PubMed] [Google Scholar]
- 22. Boon CJ. Jeroen Klevering B, Keunen JE, Hoyng CB, Theelen T. Fundus autofluorescence imaging of retinal dystrophies. Vision Res. 2008; 48: 2569–2577 [DOI] [PubMed] [Google Scholar]
- 23. von Ruckmann A, Fitzke FW, Bird AC. In vivo fundus autofluorescence in macular dystrophies. Arch Ophthalmol. 1997; 115: 609–615 [DOI] [PubMed] [Google Scholar]
- 24. Lois N, Halfyard AS, Bird AC, Fitzke FW. Quantitative evaluation of fundus autofluorescence imaged “in vivo” in eyes with retinal disease. Br J Ophthalmol. 2000; 84: 741–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmitz-Valckenberg S, Holz FG, Fitzke FW. Perspectives in imaging technologies. In: Holz FG, Schmitz-Valckenberg S, Spaide RF, Bird A. eds Atlas of Fundus Imaging. Heidelberg: Springer-Verlag; 2007: 331–338 [Google Scholar]
- 26. Delori FC, Greenberg JP, Woods RL, et al. Quantitative measurements of autofluorescence with the scanning laser ophthalmoscope. Invest Ophthalmol Vis Sci. 2011; 52: 9379–9390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Greenberg JP, Duncker T, Woods RL, Smith RT, Sparrow JR, Delori FC. Quantitative fundus autofluorescence in healthy eyes. Invest Ophthalmol Vis Sci. 2013; 54: 5684–5693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fishman GA, Stone EM, Grover S, Derlacki DJ, Haines HL, Hockey RR. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol. 1999; 117: 504–510 [DOI] [PubMed] [Google Scholar]
- 29. Jaakson K, Zernant J, Kulm M, et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003; 22: 395–403 [DOI] [PubMed] [Google Scholar]
- 30. Zernant J, Schubert C, Im KM, et al. Analysis of the ABCA4 gene by next-generation sequencing. Invest Ophthalmol Vis Sci. 2011; 528479–8487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Messias A, Reinhard J. Velasco e Cruz AA, Dietz K, MacKeben M, Trauzettel-Klosinski S. Eccentric fixation in Stargardt's disease assessed by Tubingen perimetry. Invest Ophthalmol Vis Sci. 2007; 48: 5815–5822 [DOI] [PubMed] [Google Scholar]
- 32. Reinhard J, Messias A, Dietz K, et al. Quantifying fixation in patients with Stargardt disease. Vision Res. 2007; 47: 2076–2085 [DOI] [PubMed] [Google Scholar]
- 33. Cideciyan AV, Aleman TS, Swider M, et al. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum Mol Genet. 2004; 13: 525–534 [DOI] [PubMed] [Google Scholar]
- 34. Bland JM, Altman DG. Statistical method for assessing agreement between two methods of clinical measurement. Lancet. 1986; 1: 307–310 [PubMed] [Google Scholar]
- 35. Cideciyan AV, Swider M, Aleman TS, et al. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci. 2005; 46: 4739–4746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lois N, Halfyard AS, Bird AC, Fitzke FW. Quantitative evaluation of fundus autofluorescence imaged ‘in vivo' in eyes with retinal disease. Br J Ophthalmol. 2000; 84: 741–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lois N, Halfyard AS, Bird AC, Holder GE, Fitzke FW. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am J Ophthalmol. 2004; 138: 55–63 [DOI] [PubMed] [Google Scholar]
- 38. von Ruckmann A, Fitzke FW, Bird AC. In vivo fundus autofluorescence in macular dystrophies. Arch Ophthalmol. 1997; 115: 609–615 [DOI] [PubMed] [Google Scholar]
- 39. Curcio CA, Sloan KR, Kalina RE, Hendrickson AE. Human photoreceptor topography. J Comp Neurol. 1990; 292: 497–523 [DOI] [PubMed] [Google Scholar]
- 40. Delori FC, Goger DG, Dorey CK. Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest Ophthalmol Vis Sci. 2001; 42: 1855–1866 [PubMed] [Google Scholar]
- 41. Gao H, Hollyfield JG. Aging of the human retina. Differential loss of neurons and retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1992; 33: 1–17 [PubMed] [Google Scholar]
- 42. Del Priore LV, Kuo YH, Tezel TH. Age-related changes in human RPE cell density and apoptosis proportion in situ. Invest Ophthalmol Vis Sci. 2002; 43: 3312–3318 [PubMed] [Google Scholar]
- 43. Wing GL, Blanchard GC, Weiter JJ. The topography and age relationship of lipofuscin concentration in the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1978; 17: 601–607 [PubMed] [Google Scholar]
- 44. Rivera A, White K, Stohr H, et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet. 2000; 67: 800–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lewis RA, Shroyer NF, Singh N, et al. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet. 1999; 64: 422–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cideciyan AV, Swider M, Aleman TS, et al. ABCA4 disease progression and a proposed strategy for gene therapy. Hum Mol Genet. 2009; 18: 931–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wiszniewski W, Zaremba CM, Yatsenko AN, et al. ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum Mol Genet. 2005; 14: 2769–2778 [DOI] [PubMed] [Google Scholar]
- 48. Biswas EE. Nucleotide binding domain 1 of the human retinal ABC transporter functions as a general ribonucleotidase. Biochemistry. 2001; 40: 8181–8187 [DOI] [PubMed] [Google Scholar]
- 49. Kitiratschky VB, Grau T, Bernd A, et al. ABCA4 gene analysis in patients with autosomal recessive cone and cone rod dystrophies. Eur J Hum Genet. 2008; 16: 812–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hwang JC, Zernant J, Allikmets R, Barile GR, Chang S, Smith RT. Peripapillary atrophy in Stargardt disease. Retina. 2009; 29: 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Burke TR, Fishman GA, Zernant J, et al. Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2012; 53: 4458–4467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simonelli F, Testa F, de Crecchio G, et al. New ABCR mutations and clinical phenotype in Italian patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2000; 41: 892–897 [PubMed] [Google Scholar]
- 53. Cella W, Greenstein VC, Zernant-Rajang J, et al. G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull's eye maculopathy. Exp Eye Res. 2009; 89: 16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. ANSI. American National Standard for Safe Use of Lasers (ANSI 136.1). Orlando: The Laser Institute of America; 2007. [Google Scholar]
- 55. Delori FC, Webb RH, Sliney DH; American National Standards Institute. Maximum permissible exposures for ocular safety (ANSI 2000), with emphasis on ophthalmic devices. J Opt Soc Am A Opt Image Sci Vis. 2007; 24: 1250–1265 [DOI] [PubMed] [Google Scholar]
- 56. Cideciyan AV, Swider M, Aleman TS, et al. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J Opt Soc Am A Opt Image Sci Vis. 2007; 24: 1457–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Morgan JI, Hunter JJ, Masella B, et al. Light-induced retinal changes observed with high-resolution autofluorescence imaging of the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2008; 49: 3715–3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cideciyan AV, Jacobson SG, Aleman TS, et al. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc Natl Acad Sci U S A. 2005; 102: 5233–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cideciyan AV, Swider M, Aleman TS, et al. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J Opt Soc Am A Opt Image Sci Vis. 2007; 24: 1457–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen B, Tosha C, Gorin MB, Nusinowitz S. Analysis of autofluorescent retinal images and measurement of atrophic lesion growth in Stargardt disease. Exp Eye Res. 2010; 91: 143–152 [DOI] [PubMed] [Google Scholar]
- 61. Walia S, Fishman GA. Natural history of phenotypic changes in Stargardt macular dystrophy. Ophthalmic Genet. 2009; 30: 63–68 [DOI] [PubMed] [Google Scholar]