

Abstract
The clinical course of polycythemia vera and essential thrombocythemia is potentially associated with long-term severe complications, such as evolution to myelofibrosis or acute myeloid leukemia. Allogeneic stem cell transplantation is currently the only potentially curative treatment for advanced polycythemia vera or essential thrombocythemia. We analyzed 250 consecutive patients with an initial diagnosis of polycythemia vera (n=120) or essential thrombocythemia (n=130), who underwent transplantation due to progression to myelofibrosis (n=193) or acute myeloid leukemia (n=57) and who were reported to the European Group for Blood and Marrow Transplantation registry between 1994 and 2010. Their median age was 56 years (range, 22–75) and in 52% of cases the interval between diagnosis and transplantation was 10 years or more. With a median follow-up from transplantation of 13 months, the 3-year overall survival rate and relapse incidence were 55% and 32%, respectively. In univariate analysis, the main parameters that negatively affected post-transplantation outcomes were older age (>55 years), a diagnosis at transplant of acute myeloid leukemia and the use of an unrelated donor. The overall 3-year cumulative incidence of non-relapse mortality was 28%, but was significantly higher in older patients than in younger ones (>55 years, 35% versus 20%, P=0.032), in those transplanted from an unrelated donor rather than a related donor (34% versus 18%, P=0.034) and in patients with a diagnosis of acute myeloid leukemia compared to myelofibrosis (29% versus 27%, P=0.045). This large retrospective study confirms that transplantation is potentially curative for patients with end-stage polycythemia vera/essential thrombocythemia progressing to myelofibrosis or acute myeloid leukemia. Relapse and non-relapse mortality remain unsolved problems for which innovative treatment approaches need to be assessed.
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET) are BCR-ABL-negative myeloproliferative neoplasms characterized by clonal proliferation of multiple lineage progenitor cells in bone marrow and by a relatively long median survival. The clinical course of both diseases is characterized by thrombosis as a major cause of morbidity and mortality1 and by long-term severe complications, such as evolution to myelofibrosis (MF) or acute myeloid leukemia (AML), which may occur in 2–25% of cases.2–5 Evolution to MF or AML is almost universally characterized by the development of cytopenias, due to progressive bone marrow failure, symptomatic splenomegaly and severe constitutional symptoms and is associated with early death.6 Once transformation develops, current medical treatments, which mainly include supportive therapy and/or cytoreductive treatment, are of limited efficacy and scanty prospective. Allogeneic hematopoietic stem cell transplantation (HSCT) is currently the only potentially curative treatment for advanced PV or ET, with the 3-year overall survival rate of patients so treated ranging from 39% to 67%.7–12
However, allogeneic HSCT is associated with a significant mortality (approximately 30%)10,13,14 and morbidity, mainly due to graft-versus-host disease (GVHD)8,15,16 with a clinical outcome that is particularly poor in patients of advanced age or in those with medical comorbidities.17 At present, the need for and timing of allogeneic HSCT remain under debate18 since its inherent risks are difficult to justify in patients with myeloproliferative neoplasms who are usually elderly, often have associated comorbidities and given that the existing literature is significantly under-powered for definite conclusions.
With this background, we carried out a retrospective study analyzing post-HSCT survival outcome, response rates and the relative contributions of different risk factors on clinical outcome in a large number of patients with an initial diagnosis of PV or ET who underwent allogeneic HSCT due to progression to MF or AML.
Methods
Patients and transplant characteristics
The individuals eligible for this study were 250 consecutive patients with an initial diagnosis of PV or ET, who underwent allogeneic HSCT between 1994 and 2010 due to progression to MF or AML and who were reported to the European Group for Blood and Marrow Transplantation (EBMT) registry by 89 European centers from 20 different countries. The EBMT database uses PV and ET sub-classifications at diagnosis. Transformation before transplantation to MF or AML is foreseen by the registry, and centers are asked to register the initial and the transformation diagnosis before transplantation. The study included data on: any pre-transplant status of disease; transplants performed after a standard myeloablative or a reduced intensity conditioning regimen; the use of either peripheral blood or marrow (thus excluding cord blood cells) as the source of stem cells; related or unrelated donors; and any GVHD prophylaxis that varied locally and according to the study period. The median follow-up from the time of allogeneic HSCT was 13 months (range, 0.03–123 months).
Institutional review board approval was obtained from all participating institutions.
Outcomes
The endpoints of the study were defined according to the Statistical Guidelines for the EBMT.19 The overall survival was defined as the probability of survival irrespective of disease state at any point in time from transplantation. Patients alive at their last follow-up were censored. The cumulative incidence of relapse was calculated as the time from transplantation to the first evidence of recurrence or progression of disease, with death with no prior relapse or progression as a competing risk. Similarly, non-relapse mortality was defined as the probability of dying without a previous relapse or progression, considering relapse or progression as a competing risk. GVHD was diagnosed according to previously established criteria.9,20,21
Statistical methods
Continuous variables are shown as medians with ranges and categorical variables as numbers with percentages. Kaplan-Meier curves were used to analyze survival figures (overall survival, relapse incidence), while relapse and non-relapse mortality were considered as competing events and were analyzed by means of cumulative incidence curves. The log-rank test was used to assess differences among variable categories.
We considered differences to be statistically significant if P values were ≤0.05. The analyses were carried out using SPSS software, version 19.0 (SPSS, Chicago, IL, USA).
Results
Patients and transplant characteristics
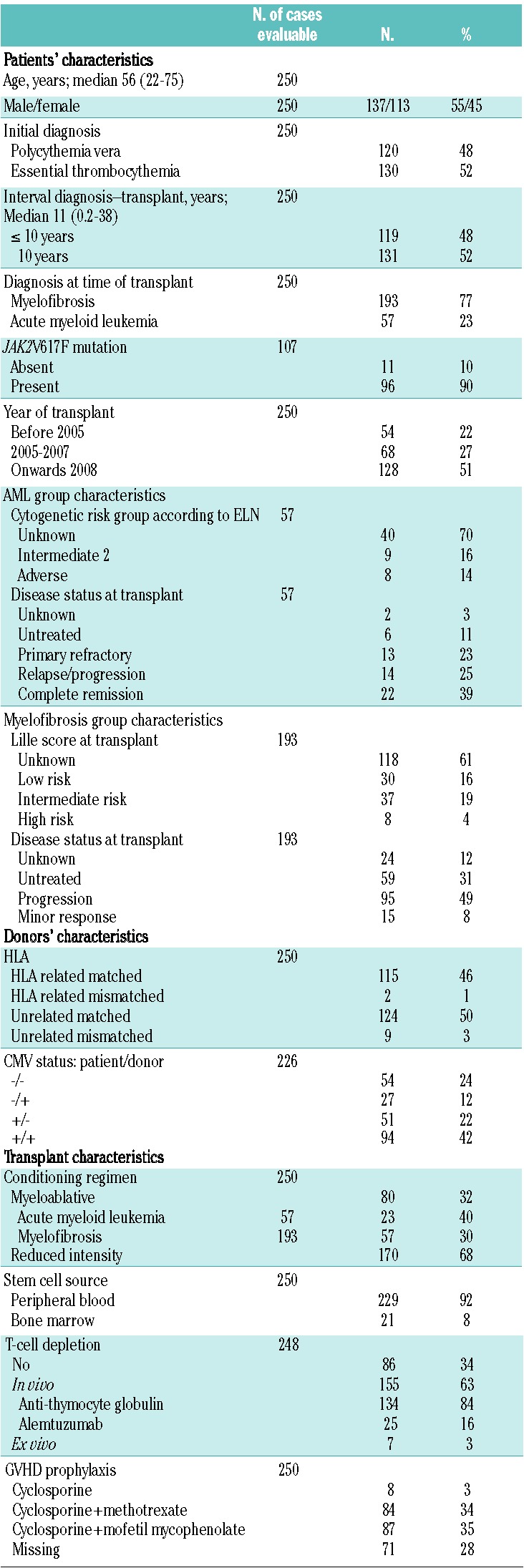
The characteristics of the patients and donors as well as transplantation and post-transplantation variables for the entire group of 250 patients are summarized in Table 1. This study included 250 consecutive patients with an initial diagnosis of PV (n=120) or ET (n=130), who underwent allogeneic HSCT due to progression to MF (n=193) or AML (n=57). The cohort consisted of 137 males and 113 females with a median age at transplantation of 56 years (range, 22–75 years) and in 52% of cases the interval between diagnosis and transplantation was 10 years or more. JAK2V617F mutational status was known for 107 patients (43%). Of all 250 transplants, 80 were performed after standard myeloablative conditioning and 170 after reduced intensity conditioning. The preferred source of stem cells was peripheral blood which was used in 229 patients (92%). Donors were HLA related matched (n=115) or mismatched (n=2), or unrelated matched (n=124) or mismatched (n=9). Fifty-four (22%) pairs were both seronegative for cytomegalovirus. GVHD prophylaxis was based on cyclosporine A in 179 cases either alone (n=8) or combined with methotrexate (n=84) or mofetil mycophenolate (n=87). T-cell-depletion was performed in vivo with antithymocyte globulin in 134 patients (54%) and with alemtuzumab in 25 (10%) or ex vivo in 7 (3%). In the AML group, cytogenetic data were available for 17 of 57 patients (30%) and all karyotypes were attributable, according to the European LeukemiaNet classification22, to an intermediate II or adverse prognostic category of risk. At the time of allogeneic HSCT, in the same group of patients 22 (39%) were considered in complete remission, 13 (23%) had refractory disease, 14 (25%) had relapsed/progressive disease, 6 (11%) were untreated, while the hematologic status was unknown for 2 (3%). A reduced intensity conditioning regimen was used in 34 patients (58%) (Table 1). In the MF group, the Lille score23 at the time of transplantation was available for 75 patients (49%). According to this prognostic scoring system, 30 (16%) patients belonged to a low risk group, 37 (19%) to an intermediate risk one, 8 (4%) to a high risk one; for 118 (61%) patients this information was not known. Among the whole group of patients with MF at the time of transplantation, 59 (31%) were untreated, 95 (49%) had progressive disease and 15 (8%) had a minor response; hematologic status was unknown for 24 (12%) (Table 1).
Table 1.
Characteristics of the 250 patients, donors and transplantation modalities.
Main clinical outcomes
With a median follow-up of 13 months (range, 0.03–123 months), 3-year overall survival and relapse incidences were 55% and 32%, respectively (Figure 1A,B). In the univariate analysis, overall survival was significantly longer in younger patients (<55 years, 65% versus 47%, P=0.015) and in patients with a diagnosis of MF compared to AML (62% versus 28%, P<0.001) (Figure 1C and Table 2). The overall survival rate was lower in patients receiving hematopoietic stem cells from an HLA unrelated donor than in those receiving a graft from HLA-matched related donors (50% versus 65%, P=0.085), although the difference was not statistically significant (Table 2). Older age and a diagnosis of AML were also risk factors associated with a higher relapse incidence rate (Table 2). Relapse incidence rate was lower in patients receiving myeloablative conditioning than in those receiving reduced intensity conditioning (16% versus 40%, P=0.09), although again this difference was not statistically significant (Table 2). Other potential risk factors, such as the initial diagnosis (PV versus ET), the time from initial diagnosis to transplantation (<10 years versus >10 years), the presence of JAK2V617F mutation, the disease status at transplantation, the stem cell source, T-cell depletion and the patient/donor cytomegalovirus status, had no impact on clinical outcomes (Table 2).
Figure 1.
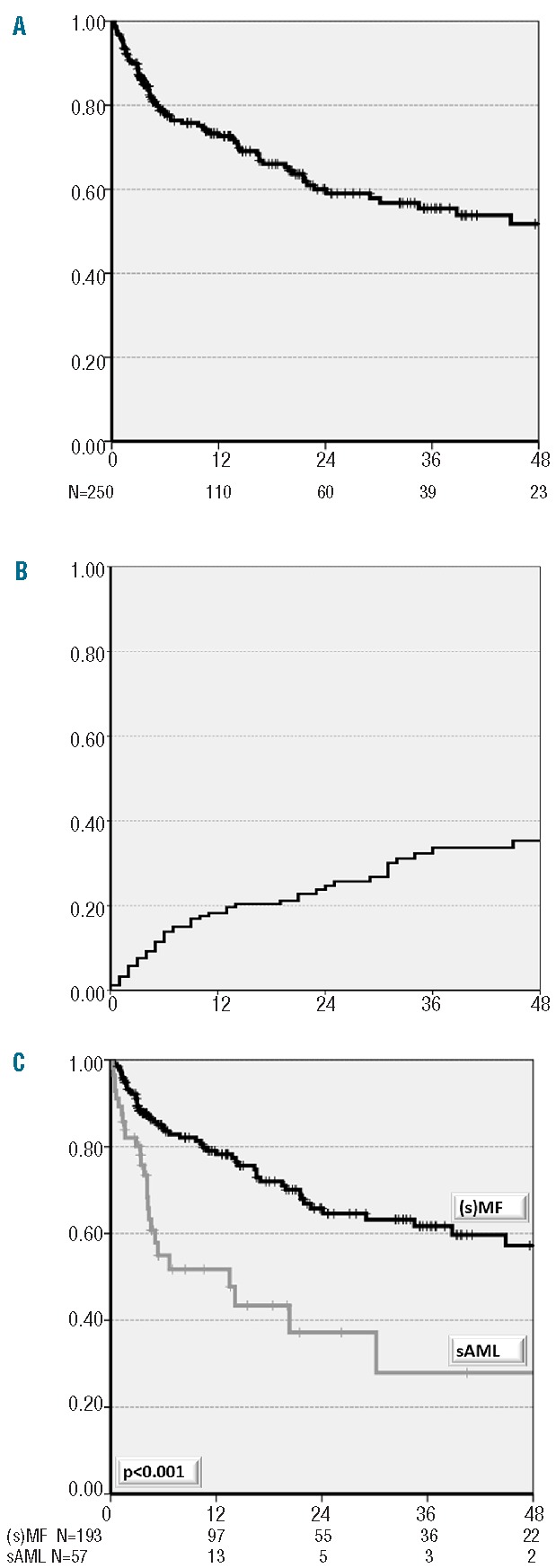
(A) Cumulative overall survival of allogeneic transplanted patients with transformed ET/PV. (B) Cumulative incidence of relapse of allogeneic transplanted patients with transformed ET/PV. (c) Cumulative overall survival of allogeneic transplanted patients with transformed ET/PV according to diagnosis at transplant.
Table 2.
Univariate analysis for the main clinical outcomes evaluated at 36 months after transplant.
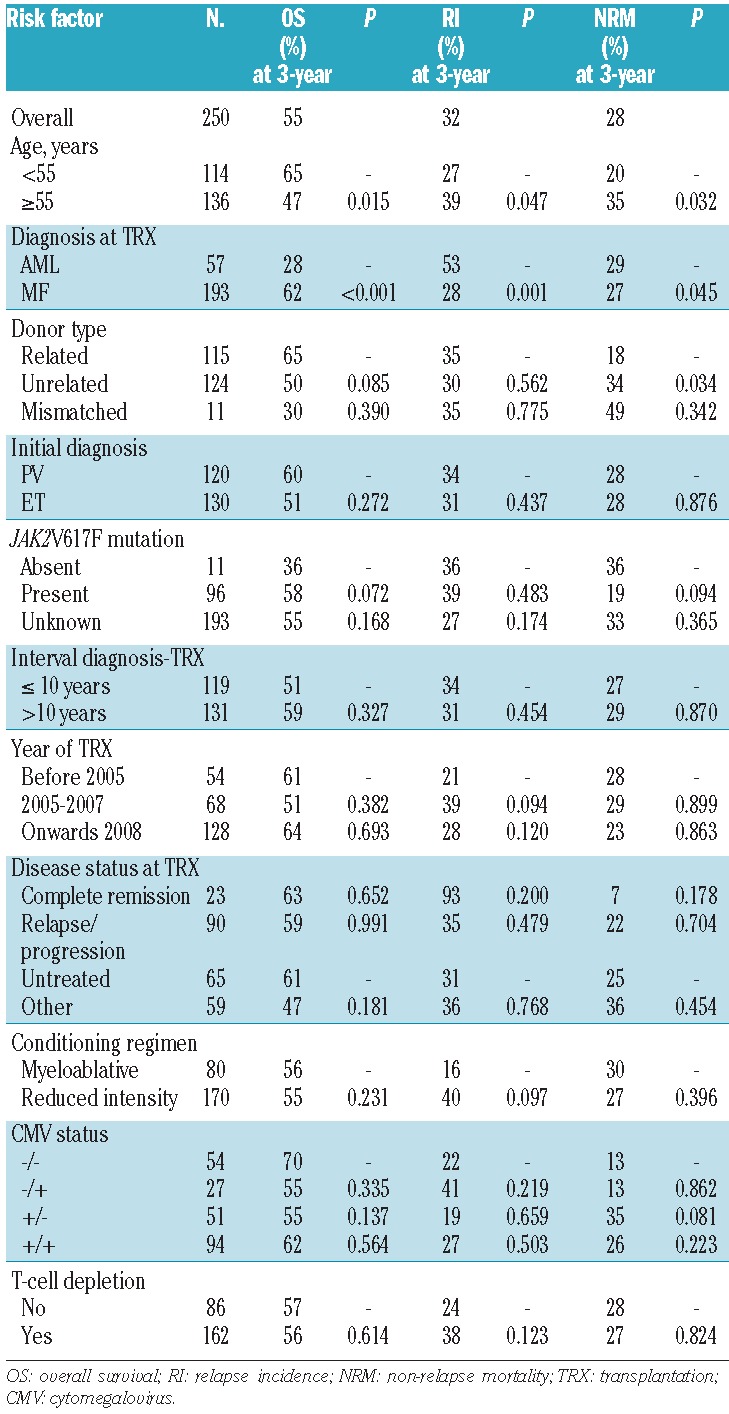
OS: overall survival; RI: relapse incidence; NRM: non-relapse mortality; TRX: transplantation; CMV: cytomegalovirus.
Mortality and causes of death
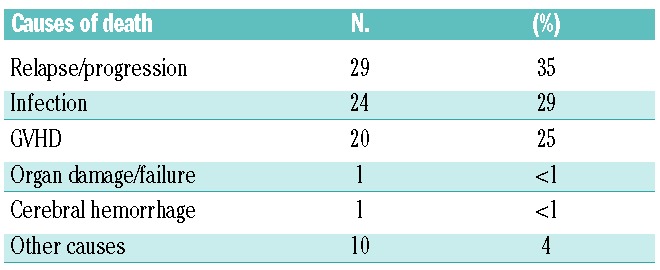
Of the 250 patients studied, 82 (33%) have died. The most frequent causes of death were relapse or progression (n=29), infections (n= 24) and GVHD (n=20), resulting in a non-relapse mortality rate of 28% (Table 3). The 3-year cumulative incidence of non-relapse mortality was significantly higher in older patients (>55 years, 35% versus 20%, P=0.032), in those transplanted from an unrelated donor rather than a related donor (34% versus 18%, P=0.034) and in patients with a diagnosis of AML compared to MF (29% versus 27%, P=0.045) (Table 2). The higher non-relapse mortality rate observed in patients with AML compared to those with MF was mainly due to a higher frequency of events occurring early after transplantation (e.g. at 6 months) in the patients with AML.
Table 3.
Causes of death (n=82).
Graft-versus-host disease
Grade 0–1 acute GVHD occurred in 68% of patients and grade 2–4 in 27%. In the evaluable patients the rate of chronic GVHD was 60%, with the extensive form occurring in 37% of patients and the limited form in 23% of patients. Data about acute and chronic GVHD were unknown for 10 (4%) and 67 (27%) patients, respectively.
Discussion
In keeping with previous reports our results support the potential curative effect of allogeneic HSCT in patients with PV or ET who have progressed to MF or AML.7,8,10–13,24 The main parameters that negatively affected post-transplant outcomes were older age (>55 years), a diagnosis at transplant of AML and donor type (unrelated versus related). Patients younger than 55 years had significantly better overall and event-free survival compared to those older than 55 years, because of the significantly higher non-relapse mortality observed in elderly patients. The reduced overall survival observed in patients with secondary AML was due to a higher incidence of relapse in AML (53%) compared to MF (28%) patients. Patients transplanted earlier in the course of the disease are those who benefit most from the procedure,8,10,25,26 suggesting that an appropriate timing for allogeneic HSCT is crucial to obtain better results. In this large series of patients we did not find that a longer interval between initial diagnosis and transplantation had a negative impact on survival, suggesting that the main parameter in terms of better outcome is probably the type of transformation before transplantation.
Among transplant-related factors, transplantation from matched unrelated donors showed an adverse impact on non-relapse mortality. These results are in keeping with those recently reported by Rondelli et al., who showed that after allogeneic HSCT with reduced intensity conditioning with fludarabine/melphalan, the unrelated donor transplants were associated with a higher risk incidence of graft rejection and failure and lower survival.27 It is worth noting that stem cells were obtained from a mismatched donor for only 11 patients and this should be carefully considered when evaluating the clinical outcome of this cohort of patients. Since a large proportion of patients (63%) received a graft subjected to in vivo T-cell depletion, we observed a relatively low incidence of acute GVHD grades 2–4, as compared to the incidence that would usually be expected in patients of such an advanced age. While intensive GVHD prophylaxis with in vivo T-cell depletion probably had a positive impact on the incidence and severity of acute GVHD, this strategy was associated with a high incidence of severe and fatal infections.
We observed only a tendency, which did not reach statistical significance, to a different relapse incidence rate when a standard myeloablative or reduced intensity conditioning regimen was used. On this topic, conflicting results have been reported and this uncertainty may be partly due to the retrospective design of most studies which have been highly heterogeneous regarding selection of patients, drugs used in the conditioning, donor type and stem cell source.8,11,12,25,28 Therefore, although many results would suggest the use of a busulfan-based conditioning regimen10,25,29 as most appropriate, this issue remains a matter of debate and of intense clinical investigation. Other factors, including initial diagnosis (PV versus ET), presence or absence of the JAK2V617F mutation, patient/donor cytomegalovirus status, disease status at transplantation, stem cell source and T-cell depletion, had no influence on clinical outcome in our cohort of patients. We found a significantly higher (90%) than expected proportion of patients with mutated JAK2V617F (70%) considering the cohort composition: this discrepancy is likely due to a reporting bias in the registry in favor of patients carrying the mutation. Unfortunately, the data required for calculating the Lille score were not available in the database for a large proportion of patients and similarly the data to calculate the more recent prognostic scoring systems, created to identify higher risk patients with primary M F,23,30–32 were also missing. Although this lack of information clearly represents a weakness of our study, it should be considered that none of these prognostic scoring systems has been validated for MF secondary to a previous PV and ET.
As for all multicenter retrospective studies some limitations need to be considered also for our study. First, this retrospective analysis collected data from many different European centers performing transplants over a long period of time with marked heterogeneity in terms of the patients’ age, co-morbidities, pre-transplant transfusion dependency, use of conditioning regimens and GVHD prophylaxis. Moreover, although to our knowledge it is the largest analysis published, it still does not allow an in-depth evaluation of the different risk factors that may characterize these patients, such as previous cytoreductive treatments or cytogenetics at transplantation. Similarly, it was not possible to get an accurate estimate of progression-free survival. The lack of this information in the database probably reflects the difficulties in assessing response in these diseases. Finally, the follow-up of the patients reported in the registry was relatively short, although we consider that this follow-up was probably sufficient to enable a correct evaluation of the main clinical outcomes. However, even if definitive conclusions cannot be drawn due to these limitations, the clinical implications of our findings are potentially very important: indeed, the potential curative effect of allogeneic HSCT is more consistently supported, with a similar 3-year survival rate compared to that of high-risk patients with MF who did not undergo allogeneic HSCT,33 with the advantage that transplanted patients may be definitely cured. These EBMT data are consistent with those recently reported by other cooperative study groups,8,28 indicating that overall survival following allogeneic HSCT may reach approximately 50%.
Nonetheless, non-relapse mortality and relapse remain unsolved problems for which future studies should explore the potential benefits of the use of new molecular therapies, such as Jak2 inhibitors, before and/or after allogeneic HSCT. In fact, these drugs have been shown to reduce spleen size and constitutional symptoms significantly, despite being unable to cure MF.34 The effect on spleen size could reduce the number of patients undergoing splenectomy, which cannot be recommended as a standard procedure before transplantation. Indeed, despite a favorable impact on engraftment,8,35 splenectomy is associated with significant surgery-related mortality (often exceeding 10%) and it may also be associated with an increased risk of relapse.25
In conclusion, this large retrospective study confirms that allogeneic HSCT is potentially curative for end-stage PV/ET patients progressing to MF or AML. Innovative treatment approaches with new molecular targeted therapies may increase the number of patients eligible for transplantation and reduce the risk of relapse and non-relapse mortality, but they need to be assessed in prospective clinical trials.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Marchioli R, Finazzi G, Landolfi R, Kutti J, Gisslinger H, Patrono C, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23(10):2224–32 [DOI] [PubMed] [Google Scholar]
- 2.Sterkers Y, Preudhomme C, Lai JL, Demory JL, Caulier MT, Wattel E, et al. Acute myeloid leukemia and myelodysplastic syndromes following essential thrombocythemia treated with hydroxyurea: high proportion of cases with 17p deletion. Blood. 1998;91(2):616–22 [PubMed] [Google Scholar]
- 3.Finazzi G, Ruggeri M, Rodeghiero F, Barbui T. Second malignancies in patients with essential thrombocythaemia treated with busulphan and hydroxyurea: long-term follow-up of a randomized clinical trial. Br J Haematol. 2000;110(3):577–83 [DOI] [PubMed] [Google Scholar]
- 4.Passamonti F, Rumi E, Pungolino E, Malabarba L, Bertazzoni P, Valentini M, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117(10):755–61 [DOI] [PubMed] [Google Scholar]
- 5.Bjorkholm M, Derolf AR, Hultcrantz M, Kristinsson SY, Ekstrand C, Goldin LR, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol. 2011;29(17):2410–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105(3):973–7 [DOI] [PubMed] [Google Scholar]
- 7.Kroger N, Alchalby H, Klyuchnikov E, Badbaran A, Hildebrandt Y, Ayuk F, et al. JAK2-V617F-triggered preemptive and salvage adoptive immunotherapy with donor-lymphocyte infusion in patients with myelofibrosis after allogeneic stem cell transplantation. Blood. 2009;113(8):1866–8 [DOI] [PubMed] [Google Scholar]
- 8.Robin M, Tabrizi R, Mohty M, Furst S, Michallet M, Bay J-O, et al. Allogeneic haematopoietic stem cell transplantation for myelofibrosis: a report of the Société Française de Greffe de Moelle et de Thérapie Cellulaire (SFGM-TC). B J Haematol. 2011; 152(3):331–9 [DOI] [PubMed] [Google Scholar]
- 9.Deeg HJ, Lin D, Leisenring W, Boeckh M, Anasetti C, Appelbaum FR, et al. Cyclosporine or cyclosporine plus methylprednisolone for prophylaxis of graft-versus-host disease: a prospective, randomized trial. Blood. 1997;89(10):3880–7 [PubMed] [Google Scholar]
- 10.Kerbauy DM, Gooley TA, Sale GE, Flowers ME, Doney KC, Georges GE, et al. Hematopoietic cell transplantation as curative therapy for idiopathic myelofibrosis, advanced polycythemia vera, and essential thrombocythemia. Biol Blood Marrow Transplant. 2007;13(3):355–65 [DOI] [PubMed] [Google Scholar]
- 11.Ballen KK, Woolfrey AE, Zhu X, Ahn KW, Wirk B, Arora M, et al. Allogeneic hematopoietic cell transplantation for advanced polycythemia vera and essential thrombocythemia. Biol Blood Marrow Transplant. 2012;18(9):1446–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patriarca F, Bacigalupo A, Sperotto A, Isola M, Soldano F, Bruno B, et al. Allogeneic hematopoietic stem cell transplantation in myelofibrosis: the 20-year experience of the Gruppo Italiano Trapianto di Midollo Osseo (GITMO). Haematologica. 2008;93(10):1514–22 [DOI] [PubMed] [Google Scholar]
- 13.Deeg HJ, Gooley TA, Flowers ME, Sale GE, Slattery JT, Anasetti C, et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood. 2003;102(12):3912–8 [DOI] [PubMed] [Google Scholar]
- 14.Guardiola P, Anderson JE, Bandini G, Cervantes F, Runde V, Arcese W, et al. Allogeneic stem cell transplantation for agnogenic myeloid metaplasia: a European Group for Blood and Marrow Transplantation, Societe Francaise de Greffe de Moelle, Gruppo Italiano per il Trapianto del Midollo Osseo, and Fred Hutchinson Cancer Research Center Collaborative Study. Blood. 1999;93(9):2831–8 [PubMed] [Google Scholar]
- 15.Stewart WA, Pearce R, Kirkland KE, Bloor A, Thomson K, Apperley J, et al. The role of allogeneic SCT in primary myelofibrosis: a British Society for Blood and Marrow Transplantation study. Bone Marrow Transplant. 2010;45(11):1587–93 [DOI] [PubMed] [Google Scholar]
- 16.Lissandre S, Bay JO, Cahn JY, Porcher R, Cacheux V, Cabrespine A, et al. Retrospective study of allogeneic haematopoietic stem-cell transplantation for myelofibrosis. Bone Marrow Transplant. 2011;46(4):557–61 [DOI] [PubMed] [Google Scholar]
- 17.Sorror ML, Maris MB, Storb R, Baron F, Sandmaier BM, Maloney DG, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood. 2005;106(8):2912–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, Griesshammer M, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29(6):761–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iacobelli S. Suggestions on the use of statistical methodologies in studies of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 2013;48(Suppl 1):S1–37 [DOI] [PubMed] [Google Scholar]
- 20.Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation. 1974;18(4):295–304 [DOI] [PubMed] [Google Scholar]
- 21.Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE, et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med. 1980;69(2):204–17 [DOI] [PubMed] [Google Scholar]
- 22.Mrozek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol. 2012;30(36):4515–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dupriez B, Morel P, Demory JL, Lai JL, Simon M, Plantier I, et al. Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood. 1996;88(3):1013–8 [PubMed] [Google Scholar]
- 24.Ditschkowski M, Elmaagacli AH, Trenschel R, Gromke T, Steckel NK, Koldehoff M, et al. Dynamic International Prognostic Scoring System scores, pre-transplant therapy and chronic graft-versus-host disease determine outcome after allogeneic hematopoietic stem cell transplantation for myelofibrosis. Haematologica. 2012;97(10):1574–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kroger N, Holler E, Kobbe G, Bornhäuser M, Schwerdtfeger R, Baurmann H, et al. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2009;114(26):5264–70 [DOI] [PubMed] [Google Scholar]
- 26.Platzbecker U, Gooley T, Anasetti C, Appelbaum FR, Clurman B, Doney K, et al. Curative therapy of advanced essential thrombocythemia or polycythemia vera by hemopoietic stem cell transplantation. Leuk Lymphoma. 2002;43(7):1409–14 [DOI] [PubMed] [Google Scholar]
- 27.Rondelli D, Goldberg JD, Marchioli R, Isola L, Shore TB, Prchal JT, et al. Results of phase II clinical trial MPD-RC 101: allogeneic hematopoietic stem cell transplantation conditioned with fludarabine/melphalan in patients with myelofibrosis. ASH Annual Meeting Abstracts 2011;118(21):1750 [Google Scholar]
- 28.McLornan DP, Mead AJ, Jackson G, Harrison CN. Allogeneic stem cell transplantation for Myelofibrosis in 2012. Br J Haematol. 2012; 157(4):413–25 [DOI] [PubMed] [Google Scholar]
- 29.Kroger N, Zabelina T, Schieder H, Panse J, Ayuk F, Stute N, et al. Pilot study of reduced-intensity conditioning followed by allogeneic stem cell transplantation from related and unrelated donors in patients with myelofibrosis. Br J Haematol. 2005;128(5):690–7 [DOI] [PubMed] [Google Scholar]
- 30.Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–7 [DOI] [PubMed] [Google Scholar]
- 31.Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–901 [DOI] [PubMed] [Google Scholar]
- 32.Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Cazzola M, et al. Dynamic International Prognostic Scoring System (DIPSS) predicts progression to acute myeloid leukemia in primary myelofibrosis. Blood. 2010;116(15):2857–8 [DOI] [PubMed] [Google Scholar]
- 33.Siragusa S, Passamonti F, Cervantes F, Tefferi A. Survival in young patients with intermediate-/high-risk myelofibrosis: estimates derived from databases for non transplant patients. Am J Hematol. 2009;84(3):140–3 [DOI] [PubMed] [Google Scholar]
- 34.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bacigalupo A, Soraru M, Dominietto A, Pozzi S, Geroldi S, Van Lint MT, et al. Allogeneic hemopoietic SCT for patients with primary myelofibrosis: a predictive transplant score based on transfusion requirement, spleen size and donor type. Bone Marrow Transplant. 2010;45(3):458–63 [DOI] [PubMed] [Google Scholar]