

Abstract
It is now widely recognized that cancer development is a protracted process requiring the stepwise acquisition of multiple oncogenic events. In humans, this process can take decades, if not a lifetime, blurring the notion of ‘healthy’ individuals. Follicular lymphoma exemplifies this multistep pathway of oncogenesis. In recent years, variants of follicular lymphoma have been recognized that appear to represent clonal B-cell expansions at an early stage of follicular lymphoma lymphomagenesis. These include follicular lymphoma in situ, duodenal follicular lymphoma, partial involvement by follicular lymphoma, and in the blood circulating follicular lymphoma-like B cells. Recent genetic studies have identified similarities and differences between the early lesions and overt follicular lymphoma, providing important information for understanding their biological evolution. The data indicate that there is already genomic instability at these early stages, even in instances with a low risk for clinical progression. The overexpression of BCL2 in t(14;18)-positive B cells puts them at risk for subsequent genetic aberrations when they re-enter the germinal center and are exposed to the influences of activation-induced cytidine deaminase and somatic hypermutations. The emerging data provide a rationale for clinical management and, in the future, may identify genetic risk factors that warrant early therapeutic intervention.
Nature and importance of follicular lymphoma precursors: a new challenge
Innovative approaches in cancer therapy have demonstrated the benefit of early treatment in lymphoma. This point is particularly well illustrated by the association between gastric MALT lymphomas and Helicobacter pylori infection, in which antibiotic therapy allows eradication of both the infectious agent and the clonal B-cell expansion, leading to long-term complete remission (CR).1 The success of this approach, however, is restricted to early phases of MALT lymphoma, as accumulation of genomic alterations in more advanced stages is associated with resistance to antibiotic therapy. The list of lymphomas evolving in response to antigen (bacterial or viral) has been growing rapidly in recent years, associated in some cases with similar therapeutic success, and with hopefully more to come.2 Although no such association with infectious agents or other early specific therapeutic target has yet been identified for follicular lymphoma (FL), this concept seems ideally suited to such an indolent disease.
Follicular lymphoma is the second most common form of non-Hodgkin lymphoma, accounting for approximately 30% of cases. The median survival is currently approximately 14 years, with most patients displaying an indolent form of the disease, slowly progressing over many years.3 In a minority of subjects, the disease might progress rapidly and aggressively in less than one year.4 In all cases, due to the insidious and often asymptomatic features of FL growth, patients are frequently diagnosed at an advanced stage (III/IV). The available therapeutic strategies thus have to cope with a largely disseminated tumor, and one resistant to long-term CR. Despite the indisputable progress in patient management, partly due to combination regimens including semi-targeted agents such as rituximab,5,6 there is no conclusive evidence that any of these approaches can fully eradicate the tumor cells. Therefore, for the moment, FL remains virtually incurable, or at best will be therapeutically converted into a chronic disease.5
In line with the clinical course, the molecular analysis of the tumor at different time points of disease progression has demonstrated that lymphomagenesis follows a complex multi-hit process that requires time for transformation to overt disease through active Darwinian-like selection.7 However, the clinical course likely represents the tip of the iceberg, as a large part of this very complex process might occur years, if not decades, before diagnosis.
One of the more remarkable illustrations of this long preclinical phase recently came from the detailed molecular description of a donor-recipient pair who synchronously developed FL grade 2/3A nine and seven years after allogeneic transplantation and donor lymphocyte infusion, respectively. Both donor and recipient harbored the same malignant FL clone, with over 90% of shared mutations, demonstrating acquisition at least seven years before clinical presentation.8 This report also revealed the capacity of the precursor cells to develop once transplanted in an allogeneic host. The study threw light on central aspects of the FL pathogenesis enigma and provided direct proof of principle that a ‘committed’ FL precursor can be present in the bone marrow (BM) and/or blood long before diagnosis. There is still no precise phenotypic, molecular, or functional definition of such committed precursors and this might range from so-called cancer stem cells (CSC), implying, among other features, self-renewing capacity at the apex of a hierarchical order,9,10 to a relatively advanced (if not already malignant) FL clone lying in wait for the opportunity to escape from immune surveillance. Further characterization of this committed entity, and the mechanisms involved in triggering its progression to FL, represent a formidable scientific and clinically significant challenge for the coming years.
Committed follicular lymphoma precursors in healthy individuals?
Follicular lymphoma results from the malignant transformation of mature B cells, and involves the aberrant proliferation of germinal center (GC)-like B cells in lymphoid organs.11,12 The hallmark and most recurrent feature of FL is the t(14;18)(q32;q21) translocation (>85% cases), which involves the B-cell lymphoma 2 (BCL2) proto-oncogene (on chromosome 18) and the non-expressed IGH allele (on chromosome 14).13,14 As a consequence, the BCL2 gene comes under the control of IGH enhancers, causing constitutive expression of the anti-apoptotic BCL-2 protein.15,16 t(14;18) is assumed to represent the earliest oncogenic event of FL. Remarkably, the translocation occurs early on in B-cell development in BM pre-B cells, due to a repair error during the V(D)J recombination process,17–19 seemingly without transforming consequence for immature B cells. This delayed malignant transformation represents the archetype of the uncoupling between the molecular oncogenic event and its oncogenic activity. One of the expected reasons underlying this uncoupling is that BCL2 is already expressed in virtually all normal B-cell subsets, with the notable exception of the two major GC-B cell subsets (centroblasts and centrocytes). The GC reaction aims to increase B-cell receptor (BCR) affinity to encountered antigens, through a random mutagenic process targeted to the IGH/L loci called somatic hypermutation (SHM). Germinal center B cells undergoing SHM will randomly increase or decrease their BCR affinity, and only those with the most affine BCR will be selected to survive and further differentiate.20 BCL2 is down-regulated in GC-B cells to ‘sensitize’ B cells to death by neglect as a means to eliminate useless and potentially dangerous B cells with decreased or modified BCR affinity. When t(14;18)-carrying B cells enter the GC through standard antigenic challenge, sustained BCL2 expression disrupts this selection process, by allowing the survival of t(14;18)-positive B cells irrespective of their BCR affinity.21,22 Although this scenario seems a plausible start of the pathogenesis and natural history of FL, this is undoubtedly far from the whole story. Indeed, cells carrying the t(14;18) can be detected (at low levels) in more than 50% of the ‘healthy’ adult population (here meaning devoid of clinical manifestation of lymphoma), a figure that obviously does not match FL prevalence (<0.03%) in adults. This indicates that most t(14;18)+ healthy individuals will never develop FL; thus, although t(14;18) translocation and ectopic BCL2 expression are critical early events in the natural history of lymphoma pathogenesis, they remain as such benign events, clearly insufficient to drive efficient FL lymphomagenesis. Could the primary antigen (Ag) encounter be the trigger¿ We and others have demonstrated that circulating t(14;18)+ B cells in healthy individuals are for the vast majority not naïve but rather Ag/GC-experienced B cells, indicating that the primary antigenic encounter and subsequent GC reaction did occur in most t(14;18)+ individuals, and consequently that primary antigenic stimulation and GC reaction are not the key factors driving in situ retention or the transformation of centroblasts/centrocytes.23 Most circulating t(14;18)+ cells in healthy individuals are therefore not ‘committed’ to FL development, and the mere detection of t(14;18) in the blood cannot constitute, as such, a predictive biomarker of this commitment.
Filling the gap with in situ FL precursors?
Follicular lymphoma cells are the transformed counterparts of centroblasts/centrocytes, blocked in their capacity to further differentiate into memory B cells, and largely addicted to the GC (or GC-like) microenvironment to survive and proliferate. The mechanisms responsible for the differentiation arrest of t(14;18)+ cells as GC-B cells are still unknown, although some secondary alteration candidates with matching functions are starting to emerge.24 One key step in the further characterization of circulating t(14;18)+ FL-like-B cells is their isolation and purification, which remains a yet unmet challenge due to their very low frequency in healthy individuals.
As the malignant counterpart of the GC-B cells, t(14;18)+ FL cells display an unusual phenotype: the expression of molecules/markers normally restricted to GC B cells (notably CD10, BCL6, and activation-induced cytidine deaminase (AID)) and translocation-induced constitutive BCL2 expression. This phenotypic combination, normally mutually exclusive in B cells at all differentiation stages, confers to FLs and precursors an easily recognizable histological GC pattern, largely used for diagnosis.25 To date, three histological lesions are recognized candidates to represent early/precursor stages of FL: intrafollicular neoplasia/in situ follicular lymphoma (FLIS), follicular lymphoma with partial involvement (PFL), and duodenal follicular lymphoma (DFL). FLIS, described in 2002 by the Jaffe group, is generally identified as reactive follicular hyperplasia in which some of the hyperplastic GCs are colonized by double positive BCL2+/CD10+ B cells.26 As the overall architecture and cytology of such lymph nodes (LN) are otherwise normal, BCL2 staining is mandatory for the diagnosis, and is usually manifested as one or more, often few, scattered germinal centers populated by BCL2+ centrocytes. The affected germinal centers may contain only a few such cells, or may be almost totally replaced. The mantle cuff is well preserved and the overall diameter of the follicle is usually not increased (Figure 1). Surprisingly, BCL2 (and CD10) staining is abnormally intense, higher than the positivity of any cells of the adjacent marginal zone and interfollicular areas, and notably higher than in most overt FL.25 Mechanisms regulating the peculiar BCL2 and CD10 staining intensities have still not been identified but might involve epigenetic processes, such as the regulation to chromatin access through or for given molecules.27 In FLIS, cells strongly positive for BCL2 are exclusively localized in the GCs of an otherwise reactive lymph node, without infiltration outside the GC, hence the term in situ to designate a condition in which the t(14;18)+ cells are restricted to an area normally occupied by their physiological counterparts. FLIS is generally an incidental finding in a lymph node excised because of enlargement or for another reason (e.g. another B-cell lymphoma or resection of an unrelated carcinoma) and appears to be very rare. Henopp et al. performed systematic BCL2 staining in unselected prospective specimens from 132 patients, and found 3 patients with an FLIS (approx. 2.3%). None of them had a history of FL, but interestingly one already had FLIS (derived from the same t(14;18) clone) in a LN excised two years before, attesting to early systemic dissemination.28 PCR assays on microdissected FLIS follicles generally show clonal rearrangement and FISH demonstrates the IGH/BCL2 rearrangement in the vast majority of the cases.26,29 The clinical significance of FLIS is still not clear. It can be associated with overt FL synchronously or metachronoulsy,26,28 as well with unrelated tumoral or inflammatory conditions,30,31 in keeping with the role of immune stimulation and FL’s opportunistic behavior and GC microenvironment dependency. In patients with concomitant FLIS and overt FL, the two entities were clonally related.29,30,32 The rate of progression from FLIS to overt FL appears to be very low: in the largest series of 34 FLIS patients,31 6 had prior or concomitant FL, 5 had FLIS composite with another lymphoma;33 in the 21 remaining FLIS-only patients with available follow up, only one (5%) developed overt FL at 29 months (median follow up 41 months).
Figure 1.

Histological and immunohistochemical (CD20, BCL2, CD10 and CD3) features of follicular lymphoma and its precursors. (A) Follicular lymphoma in situ (FLIS). Lymph node architecture is preserved, and BCL2+CD10+ cells occupy only selected GC. (B) Partial involvement by FL (PFL). Lymph node architecture is still partially preserved with open sinuses and intact paracortex. However, affected follicles are expanded and partially displace normal nodal elements. (C) Duodenal FL. A well circumscribed nodular accumulation of centrocytes expands in the mucosa. (D) Follicular lymphoma, Grade 1–2. Nodal architecture is replaced by the neoplastic follicular proliferation.
One of the burning questions concerning FLIS is what it takes mechanistically and functionally for a BCL2-expressing B cell to display such behavior.
- Is this merely a histological snapshot of a naïve t(14;18)+ B cell undergoing a primary GC reaction following antigenic challenge, or are these antigen-experienced GC B cells re-entering the GC¿ And in this case, how can we explain the simultaneous invasion of multiple GCs in the LN, or the occurrence in some cases of antecedent and clonally-related FL or FLIS at another site¿
- Does FLIS correspond to a secondary antigenic response, whereby cells from a previously expanded t(14;18)+ memory B-cell clone re-enter GCs in the attempt to produce a new generation of memory B cells34,35¿
- Is this phenomenon linked to or enhanced by the lack of GC selection provided by BCL2¿ Is BCL2 ectopic expression necessary and sufficient to generate a FLIS, and t(14;18) the only (functional) chromosomal lesion present in FLIS as previously proposed30¿
- Are FLIS t(14;18)+ cells already blocked in their capacity to further differentiate into memory B cells¿ Are FLIS t(14;18)+ cells already ‘committed’ to FL development or are they ahead of commitment¿
- Is FLIS a homogeneous entity, or could it possibly represent a multitude of stages (e.g. all of the above)¿
Part of the answer to these many questions was recently provided by comparative large genomic hybridization array (aCGH) analysis of a series of well-defined FLIS samples collected with or without concurrent FL or FL history, and which demonstrated the occurrence of genomic alterations.36,37 Together with the finding that FLIS display a lower proliferation index than normal reactive GCs and the additional presence of mutations in EZH2, CREBBP, TNFRSF14, these studies indicated that FLIS is not merely the BCL2-labeled counterpart of otherwise normal (14;18)+ memory B cells re-entering GC reactions during secondary challenge. Clearly, genomic instability is already at work in these cells, and might have further modified the functional properties of the BCL2-expressing cells. However, the low frequency of relevant and/or recurrent FL hits among the observed alterations does not support the idea that strong selection pressure towards FL pathogenesis already took place in most FLIS. Interestingly, a large fraction of the genomic alterations observed were functionally related to the biology of the GC, indicating that selective forces might initially converge on events contributing to the GC reaction and/or GC retention. Thus, FLIS likely correspond to an early FL precursor with increased genomic instability and preferential GC homing, but in which some of the specific events required for full lymphoma transformation did not yet occur (and might never transpire). Yet, it still remains to be determined whether FLIS t(14;18)+ cells can further differentiate into memory B cells, or whether they have already sustained the FL-characteristic block of differentiation at the stage of centrocytes/centroblasts. The recent evidence that such a block indeed occurred in some healthy individuals, and in particular in those with high t(14;18) frequencies, together with the detection at high frequency of the clonal blood counterpart of one FLIS case, seem to argue for this possibility.38 However, there is to date no firm evidence for a defined hierarchy between t(14;18)+ cells in FLIS patients and t(14;18)high healthy individuals. In the end, it seems very likely that t(14;18)+ cells from both FLIS and healthy individuals are very heterogeneous entities and represent a whole spectrum of overlapping stages (Figure 1).
Concerning the critical question of whether FLIS cells are already committed or not to FL development, available follow-up data of FLIS patients are currently limited and the cohorts too small to draw firm conclusions. Nevertheless, the current figure (5% at 7 years) tends to indicate that most FLIS would remain uncommitted. This might be different for the related PFL entity. Indeed, its clinical significance seems to be more important than the FLIS as it is associated with a higher risk of progression to FL; in the largest follow-up cohort to date, 53% (9 of 17) of untreated PFL patients developed overt FL in a 14-year follow-up period. PFL, however, remains a difficult differential diagnosis of FL. Histological criteria were recently proposed to reliably distinguish PFL from FLIS.26,31 Unlike FLIS, PFLs show altered architecture. The affected follicles are often larger than those in FLIS, and often grouped together in an area of the lymph node (versus scattered in FLIS). The margin of the PFL follicles may be ill-defined with attenuated mantle cuffs. Cytologically the follicles might contain admixed centroblasts (versus only centrocytes in FLIS), and show variable intensity of BCL2 and CD10. In some cases, BCL2+CD10+ cells may be outside the PFL follicles. Thus, histologically PFLs exhibit features closer to ‘true’ FL than those observed in FLIS (Figure 1).
In line with such criteria, a-CGH analysis of one series of PFL samples revealed the presence of significantly more genomic alterations per sample than in FLIS, even if still not reaching the level seen in low-grade FL.36 Notably, and in contrast to FLIS, a large fraction of the gains were shared with low-grade FL, suggesting significant selective pressure in line with PFLs higher progression rate. Overall, the existing data suggest that PFL generally do not constitute partial colonization of the LN by overt FL, but rather represent an earlier stage of tumor evolution.26,31 In keeping with these data, one study found that patients with PFL more often have low-stage disease.39
Note that the definition of PFL used for case selection in the genetic analysis mentioned above excluded patients with known FL at another site.36 In the presence of concurrent FL and/or FL history, partial colonization of lymph nodes by overt FL might histologically resemble/be undistinguishable from PFL. The presence of BCL2+CD10+ cells outside the PFL follicles suggests that t(14;18)+ PFL cells have already sustained the secondary oncogenic hits leading to the characteristic FL maturation arrest. Considering the progression rate, it is also likely that some (if not most) PFLs represent committed precursors. In this respect, the report that none from a small series of treated PFL patients (rituximab or localized radiotherapy) developed overt FL in a follow up of 14 years (compared to 53% of untreated patients) is of clinical significance. Longer follow up on a larger series will be of prime importance to validate or not the role that therapy might play in early stages of the disease.31
Duodenal follicular lymphoma (DFL) is a very peculiar form of FL, the study of which might provide further insights into the immunological side of FL development. Like FLIS, it is a rare condition (one per 3000–7000 gastroduodenoscopies). Despite its histological similarity with FLIS, i.e. BCL2+ immunostained follicles mimicking GCs colonized by t(14;18)+ cells, it exhibits peculiar clinical characteristics. While classical FL is a disseminated disease in the vast majority of cases, DFL is almost always localized and restricted to the mucosa/submucosa of the small intestine (most often the second part of the duodenum) and behaves as a very indolent disease, without dissemination outside the intestinal wall and with only rare transformation. Histologically, this lymphoma presents as mucosal warty polyps, each of them containing large neoplastic FL-like follicles made of centrocytes with few centroblasts. The atypical follicles show low-grade histology, and strong immunohistochemical expression of CD20, CD10 and BCL2, in addition to the t(14;18) translocation (Figure 1). Notably, the cells commonly express IgA, rather than IgG or IgM, as seen in nodal FL.5 In addition, despite a predominantly follicular growth pattern, the BCL2+ CD10+ cells may infiltrate the lamina propria, and extend into the villi, in contrast to FLIS, in which the cells remain confined to the follicular structure. DFL might be derived from a t(14;18)+ cell that homes to the intestine and encounters cognate Ag in the mucosal environment. The site of Ag encounter may explain the homing properties of DFL versus FLIS. In the largest reported series (63 patients),40 monoclonality and BCL2 rearrangements were found in almost all cases. Only 2 untreated patients developed nodal disease five years after diagnosis and no aggressive transformation was observed (median follow up 77 months). Seven spontaneous regressions were observed and no patient died from lymphoma. Interestingly, aCGH analysis revealed genomic alterations with similar frequencies to those observed in PFL. Unlike PFL, however, and in agreement with the rare progression of these lesions, only a small fraction of the genomic alterations in DFL was found to be shared with FL. It has been reported that t(14;18)+ cells accumulate in the duodenal wall after an antigen-driven process, but do not further acquire aggressive/disseminating potential, possibly due to the extinction of AID expression.41 It is still unclear whether the genomic instability observed in DFL is AID-independent, or acquired elsewhere in presence of AID (potentially in the frame of a classical GC reaction) before the colonization of duodenal GCs. By limiting further genomic instability and selection potential, this scenario might partly explain why this entity rarely evolves to overt malignancy. Interestingly, the low propensity of DFL to progress to overt FL, despite the presence of several major oncogenic alterations, adds to the evidence that such hits are not sufficient for transformation, and that extrinsic-related factors (such as successive immunological challenges and GCs co-opting) might play a key role in this process.
Defining the factors involved in commitment
A number of laboratories are currently analyzing the sequence of events leading to FL pathogenesis,8,42,45 and the recent a-CGH data on entities representing proposed sequential intermediates to FL transformation add to the list of putative genes involved in the early steps of t(14;18)+ cell transformation (Table 1).36
Table 1.
Genomic alterations reported in FL (by UDS sequencing) compared with early FL precursors, including FLIS, PFL and DFL (by array CGH or Sanger sequencing), and respective frequencies.
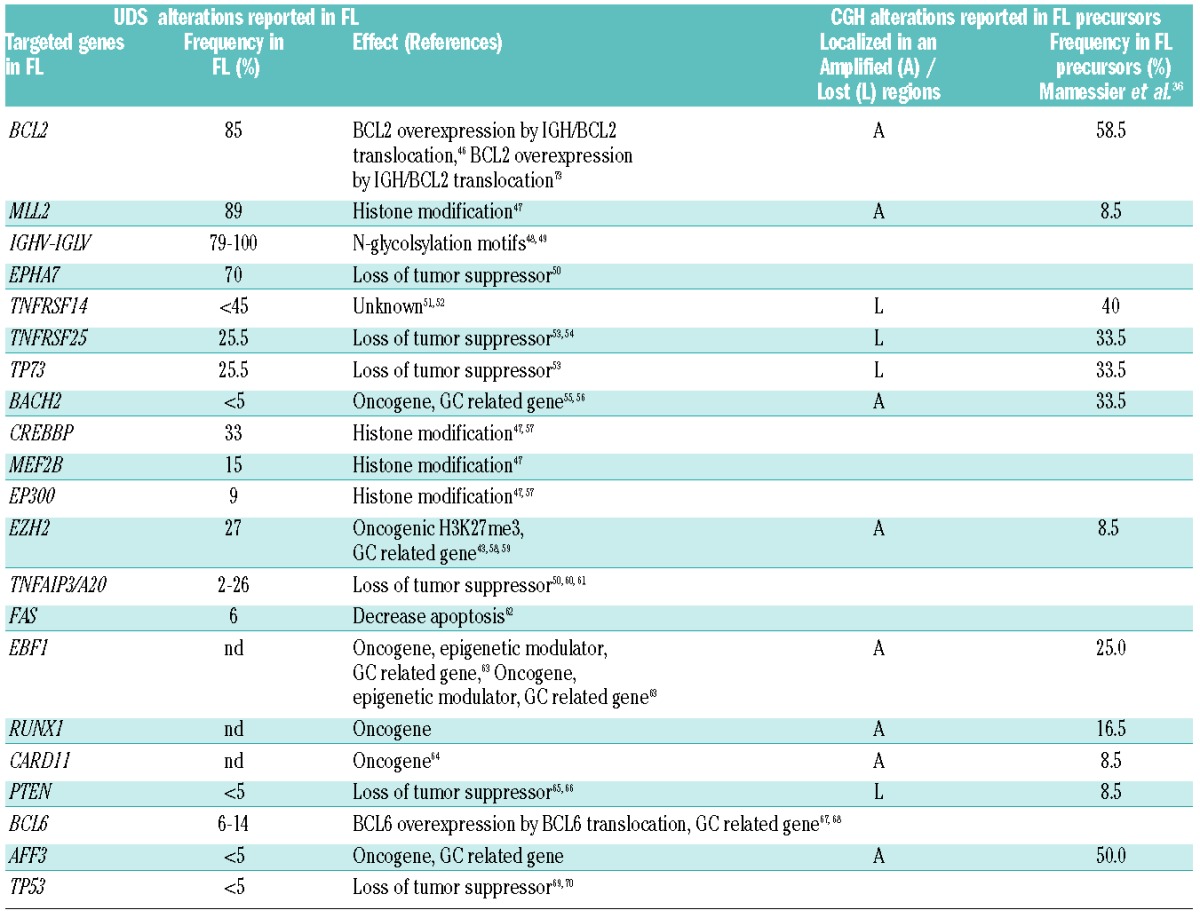
However, a major caveat in both approaches is that Darwinian evolution is complex, and likely involves multiple criss-crossing pathways, possible uncoupling between the time of oncogene activation and the time of oncogene activity, and genealogical dead ends, all of which lead to potentially extremely diverse kinetics. Among the numerous subclones and variants leading to dead ends, DFL and FLIS that rarely progress to FL might contain recurrent mutations that functionally contributed to the generation of these entities, but are not necessarily required for FL progression. On the other hand, the ‘root’ mutations uncovered in the genealogical trees of FL clones reconstituted a posteriori through ultra-deep sequencing (UDS) do indicate that such alterations occurred early in the given tree, but do not necessarily indicate that they are events required for early progression (i.e. an alteration can be acquired early as passenger mutation, and unleash its oncogenic potential later on, in a given context or a given tissue). Furthermore, some events might recurrently occur early because they are mechanistically constrained in B-cell development, but not necessarily required initially. For example, the hallmark t(14;18) is confined to the BM pre-B developmental stage due to the involvement of V(D)J recombination. However, it is still not clear whether BCL2 overexpression has functional consequences in the survival and/or differentiation potential of immature and naïve B cells, and consequently whether it enhances the risk of FL progression. The high prevalence of t(14;18) in the general adult population (>50%) and current risk estimates of FL development in the t(14;18)+ individuals compared to the t(14;18)− individuals, tend to argue against this possibility. Furthermore, oncogenic ‘hits’ are not limited to cell-intrinsic genomic alterations, and more elusive cell-extrinsic factors, such as changes in the microenvironment and/or immunological response, must be taken into account in the cascade of events triggering transformation.
Combined Next generation Sequencing (NGS) efforts on a large number and types of FL and FL intermediates will be needed to pull-out candidate genes, and the functional validation of these in innovative experimental systems/models will be key to reliably identify those responsible for driving the major pathways of stepwise FL progression, and among those, the ones responsible for commitment to FL.
Clinical perspectives
Asymptomatic pre-malignant conditions, which can progress to symptomatic disease states requiring therapy, have been identified in two other B-cell malignancies: chronic lymphocytic leukemia (CLL), with monoclonal B-cell lymphocytosis (MBL), and multiple myeloma (MM) with monoclonal gammopathy of undetermined significance (MGUS). The characterization of these precursors has helped to define the limits between non-progressive but clonal expansions, and those at higher risk to progress, requiring greater follow up and/or additional therapeutic management. For example, MBL is defined as a clonal B-cell expansion in the peripheral blood without symptoms or signs of a well-defined lymphoproliferative disorder. Based on the B-cell count, MBL is now divided into low-count MBL and clinical MBL. While low-count MBL seems to carry relevance mostly from an immunological perspective, clinical MBL and CLL appear to be overlapping entities and benefit from greater clinical surveillance until treatment may be required.71 In the case of MGUS, a score including elevated serum free light chain and M-spike was identified as the strongest risk factor for the subsequent development of MM. Studies of its pathogenesis led to the development of risk models and the estimation of the individual risk of progression, allowing individualized clinical management.72 For asymptomatic FL, one of the major challenges in the future will be to develop similar individual risk profiles allowing us to assess whether, when and how to treat with optimal long-term benefit for the patient.
Concerning FLIS and DFL, watchful waiting (WW) seems a reasonable option considering the relatively low progression rate (<5%). However, the hierarchical relationship between FLIS, DFL, FLLC and asymptomatic patients is currently unknown, and the clinical work up of a patient with FLIS still needs to be further defined. In some institutions, a computed tomography (CT) scan is performed, and in case of negativity, further clinical follow up may be obtained at regular intervals (e.g. every 6 months). In this regard, it is important to note that both FLIS and DFL are frequently incidental findings in the setting of histological assessment for other symptomatology, e.g. lymphadenopathy or unrelated gastrointestinal symptoms. The subsequent clinical management will thus often be based on other findings, not the presence of FLIS or DFL.
Concerning PFL, for which the progression rate is higher (approx. 50%), the management is often the same as for low-tumor burden. In FL, 10–15% of patients present with low tumor burden (GELF criteria: no involvement >7cm, no B-symptoms, no significant splenomegaly, no pleural effusion, no complications such as ascites/organ compression, normal LDH and β2-microglobulin levels). A number of such patients are relatively asymptomatic and thus detected fortuitously. Considering the indolent clinical course, it is reasonable to assume that a fraction of such patients remain undiagnosed, potentially for many years. Based on trials showing no benefit of immediate chemotherapy in patients with a low tumor burden,73,74 current guidelines recommend a WW approach, deferring treatment initiation until worsening of disease and/or clinical symptoms appear.75 Most patients with a low tumor burden under WW have an improved quality of life for an average period of 2.5 years by delaying exposure to the toxic side-effects of chemotherapy, the reduced number of hospital visits and related interventions (therapeutic agents administration, blood puncture, etc.). Yet 60–80% will eventually progress towards high tumor burden within a relatively short time, and will require radio/chemotherapy in a setting in which treatment may be not curative at that stage of disease. For some patients, WW may also be psychologically distressing because of a declared malignant diagnosis without administration of any treatment. Keeping in mind that any deviation from the WW therapeutic attitude must combine high response rate, improved survival, a good safety profile, and low impact on quality of life, a number of early therapeutic interventions are currently under investigation. For patients with localized disease, radiotherapy has been proposed to provide long-term benefit (50% freedom from treatment failure at 10 years).75–77 The role of rituximab-based therapy in the management of early stage FL is also under investigation. Although based on small numbers, it is notable that in a series of PFL treated with rituximab or radiotherapy none of the patients progressed to overt FL after seven years of follow up, compared to 53% when left untreated.31,40 Similarly, the use of both therapies has been very convincing in DFL. For the majority of patients with disseminated disease at diagnosis, rituximab is currently being explored as a promising alternative due to its low toxicity profile and its proven efficacy in symptomatic indolent FL.78 Intermediate data analysis from an ongoing phase III study in advanced stage FL patients with low tumor burden reported that induction/maintenance with rituximab as single-agent delayed the need for chemotherapy, and decreased the risk of worsening disease compared to the WW arm of the trial.79–81 Time-to-next-therapy and progression-free survival (PFS) were significantly increased (PFS at 3 years was of 33% in the WW arm vs. 81% in the rituximab arm), and the median time to initiation of chemotherapy was not reached at four years in patients on rituximab. In a second study (RESORT), patients with low tumor burden were treated with 4 weekly doses of rituximab.82 Patients achieving either partial or complete remissions were then randomized to maintenance treatment or observation with rituximab retreatment at the time of progression. Retreatment was as effective as maintenance treatment, and in the maintenance arm, patients received more rituximab than retreatment patients.83 There was no difference in quality of life. Collectively, these promising results led to a change of paradigm in the clinical practice, with only 20% of patients followed with WW at diagnosis in the US, and immunotherapy initiated in the large majority of cases.84 Longer-term follow up is clearly required to answer the many issues raised by the use of single agent rituximab treatment for patients with low tumor burden, including potential long-term toxicities and the possible emergence of rituximab-resistant relapses. Nevertheless, the pertinence of a standardized management is now frequently questioned, as patients with low tumor burden are likely a very heterogeneous group, with some potentially more at risk of swift FL progression than others. Thus, one of the current clinical challenges is to define reliable prognostic markers allowing for risk stratification of asymptomatic patients who might benefit from early therapy. Along these lines, the F2-study was designed as a prospective collection of data aiming to identify biological parameters for initiating treatment, and to evaluate whether an initial WW would have deleterious effects on treatment efficacy after progression or relapse in FL patients with low tumor burden.85 Although involvement of more than four nodal sites and decreased albumin levels were associated with a shorter time to lymphoma treatment, their prognostic value lacked robustness for application in clinical practice. Recently, FDG-PET has demonstrated a high predictive value for response to initial therapy in patients with advanced FL,86,87 and might be explored as a complementary prognostic tool for the stratification of low tumor burden patients.88,89 Other strategies have explored gene expression profiles to develop molecular predictors of response to treatment,90 revealing microenvironment signatures associated with survival.91,92 potentially transferable to clinical practice.93–96 The use of such signatures as a prognostic tool for the stratification of patients with low tumor burden also remains to be explored. The recent development of genome investigation techniques such as next-generation and ultra-deep sequencing, also offers the opportunity to further dissect early FL entities including FLIS, PFL, DFL and follicular lymphoma-like cells (FL-LC), and should bring new insights into molecular classifiers. Considering the biological complexity of FL lymphomagenesis, it seems clear that genomic, environmental and clinical investigations will need to identify a combination of risk factors in order to define reliable risk profiles and prognostic markers.
Many novel therapeutic agents are currently being investigated in pre-clinical and clinical studies, including monoclonal antibodies (modified anti-CD20, anti-CD19, anti-CD80, anti-CD22, anti-CD79b, anti-CTLA4, etc.) and small molecules that might alter the anti-apoptotic pathways (lenalidomide, ibrutinib, idealisib, BCL2 or BClx agonists, etc.).97–99 In this flourishing landscape of novel agents, the use of chemotherapy-free targeted therapy emerges as a promising change in the therapeutic paradigm for lymphoma, especially for early stage disease. To reach this goal, more than ever, a deep molecular and functional understanding of the oncogenic pathways involved in the various stages of FL progression is required. Among those, the identification of early biomarkers of commitment and of the factors responsible for this progression stage should be instrumental in the targeting of early and potentially less refractory forms of FL.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Wotherspoon AC, Doglioni C, de Boni M, Spencer J, Isaacson PG. Antibiotic treatment for low-grade gastric MALT lymphoma. Lancet. 1994;343(8911):1503. [PubMed] [Google Scholar]
- 2.Levine AM. Lymphomas and leukemias due to infectious organisms. Hematology. 2012;17(Suppl 1):S87–9 [DOI] [PubMed] [Google Scholar]
- 3.Swenson WT, Wooldridge JE, Lynch CF, Forman-Hoffman VL, Chrischilles E, Link BK. Improved survival of follicular lymphoma patients in the United States. J Clin Oncol. 2005;23(22):5019–26 [DOI] [PubMed] [Google Scholar]
- 4.Bastion Y, Sebban C, Berger F, Felman P, Salles G, Dumontet C, et al. Incidence, predictive factors, and outcome of lymphoma transformation in follicular lymphoma patients. J Clin Oncol. 1997; 15(4):1587–94 [DOI] [PubMed] [Google Scholar]
- 5.Bende RJ, Smit LA, Bossenbroek JG, Aarts WM, Spaargaren M, de Leval L, et al. Primary follicular lymphoma of the small intestine: alpha4beta7 expression and immunoglobulin configuration suggest an origin from local antigen-experienced B cells. Am J Pathol. 2003;162(1):105–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Czuczman MS, Leonard JP, Jung S, Johnson JL, Hsi ED, Byrd JC, Cheson BD. Phase II trial of galiximab (anti-CD80 monoclonal antibody) plus rituximab (CALGB 50402): Follicular Lymphoma International Prognostic Index (FLIPI) score is predictive of upfront immunotherapy responsiveness. Ann Oncol. 2012;23(9):2356–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossi D, Ciardullo C, Gaidano G. Genetic aberrations of signaling pathways in lymphomagenesis: Revelations from next generation sequencing studies. Semin Cancer Biol. 2013;23(6):422–30 [DOI] [PubMed] [Google Scholar]
- 8.Weigert O, Kopp N, Lane AA, Yoda A, Dahlberg SE, Neuberg D, et al. Molecular ontogeny of donor-derived follicular lymphomas occurring after hematopoietic cell transplantation. Cancer Discov. 2012;2(1):47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fox MF, Pontier A, Gurbuxani S, Sipkins DA. Stem cell factor expression in B cell malignancies is influenced by the niche. Leuk Lymphoma. 2013;54(10):2274–80 [DOI] [PubMed] [Google Scholar]
- 10.Weigert O, Weinstock DM. The evolving contribution of hematopoietic progenitor cells to lymphomagenesis. Blood. 2012;120(13):2553–61 [DOI] [PubMed] [Google Scholar]
- 11.Bende RJ, Smit LA, van Noesel CJ. Molecular pathways in follicular lymphoma. Leukemia. 2007;21(1):18–29 [DOI] [PubMed] [Google Scholar]
- 12.Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005; 5(4):251–62 [DOI] [PubMed] [Google Scholar]
- 13.Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985; 229(4720):1390–3 [DOI] [PubMed] [Google Scholar]
- 14.Weiss LM, Strickler JG, Medeiros LJ, Gerdes J, Stein H, Warnke RA. Proliferative rates of non-Hodgkin’s lymphomas as assessed by Ki-67 antibody. Hum Pathol. 1987;18(11):1155–9 [DOI] [PubMed] [Google Scholar]
- 15.Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986; 47(1):19–28 [DOI] [PubMed] [Google Scholar]
- 16.Seto M, Jaeger U, Hockett RD, Graninger W, Bennett S, Goldman P, et al. Alternative promoters and exons, somatic mutation and deregulation of the Bcl-2-Ig fusion gene in lymphoma. EMBO J. 1988;7(1):123–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jager U, Bocskor S, Le T, Mitterbauer G, Bolz I, Chott A, et al. Follicular lymphomas’ BCL-2/IgH junctions contain templated nucleotide insertions: novel insights into the mechanism of t(14;18) translocation. Blood. 2000;95(11):3520–9 [PubMed] [Google Scholar]
- 18.Marculescu R, Vanura K, Montpellier B, Roulland S, Le T, Navarro JM, et al. Recombinase, chromosomal translocations and lymphoid neoplasia: targeting mistakes and repair failures. DNA Repair (Amst). 2006;5(9–10):1246–58 [DOI] [PubMed] [Google Scholar]
- 19.Raghavan SC, Swanson PC, Wu X, Hsieh CL, Lieber MR. A non-B-DNA structure at the Bcl-2 major breakpoint region is cleaved by the RAG complex. Nature. 2004;428(6978):88–93 [DOI] [PubMed] [Google Scholar]
- 20.Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood. 2012;120(11):2240–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roulland S, Faroudi M, Mamessier E, Sungalee S, Salles G, Nadel B. Early steps of follicular lymphoma pathogenesis. Adv Immunol. 2011;111:1–46 [DOI] [PubMed] [Google Scholar]
- 22.Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8(1):22–33 [DOI] [PubMed] [Google Scholar]
- 23.Agopian J, Navarro JM, Gac AC, Lecluse Y, Briand M, Grenot P, et al. Agricultural pesticide exposure and the molecular connection to lymphomagenesis. J Exp Med. 2009;206(7):1473–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23(5):677–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed Bosman FT, Jaffe ES, Lakhani SR, Ohgaki H. editors. Lyon, France: International Agency for Research on Cancer; 2008 [Google Scholar]
- 26.Cong P, Raffeld M, Teruya-Feldstein J, Sorbara L, Pittaluga S, Jaffe ES. In situ localization of follicular lymphoma: description and analysis by laser capture microdissection. Blood. 2002;99(9):3376–82 [DOI] [PubMed] [Google Scholar]
- 27.Duan H, Heckman CA, Boxer LM. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol. 2005;25(5):1608–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henopp T, Quintanilla-Martinez L, Fend F, Adam P. Prevalence of follicular lymphoma in situ in consecutively analysed reactive lymph nodes. Histopathology. 2011;59(1):139–42 [DOI] [PubMed] [Google Scholar]
- 29.Lee JC, Hoehn D, Schecter J, Murty VV, Mansukhani MM, Alobeid B, et al. Lymphoid follicle colonization by Bcl-2CD10 B-cells ("follicular lymphoma in situ") at nodal and extranodal sites can be a manifestation of follicular homing of lymphoma. Hum Pathol. 2013;44(7):1328–40 [DOI] [PubMed] [Google Scholar]
- 30.Bonzheim I, Salaverria I, Haake A, Gastl G, Adam P, Siebert R, et al. A unique case of follicular lymphoma provides insights to the clonal evolution from follicular lymphoma in situ to manifest follicular lymphoma. Blood. 2011;118(12):3442–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jegalian AG, Eberle FC, Pack SD, Mirvis M, Raffeld M, Pittaluga S, et al. Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood. 2011;118(11):2976–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pillai RK, Surti U, Swerdlow SH. Follicular lymphoma-like B cells of uncertain significance (in situ follicular lymphoma) may infrequently progress, but precedes follicular lymphoma, is associated with other overt lymphomas and mimics follicular lymphoma in flow cytometric studies. Haematologica. 2013;98(10):1571–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carbone A, Tibiletti MG, Canzonieri V, Rossi D, Perin T, Bernasconi B, et al. In situ follicular lymphoma associated with non-lymphoid malignancies. Leuk Lymphoma. 2012;53(4):603–8 [DOI] [PubMed] [Google Scholar]
- 34.Bende RJ, van Maldegem F, Triesscheijn M, Wormhoudt TA, Guijt R, van Noesel CJ. Germinal centers in human lymph nodes contain reactivated memory B cells. J Exp Med. 2007;204(11):2655–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol. 2009;10(12):1292–9 [DOI] [PubMed] [Google Scholar]
- 36.Mamessier E, Song JY, Eberle FC, Pack S, Drevet C, Chetaille B, et al. Early lesions of follicular lymphoma: a genetic perspective. Haematologica. 2013;99(3):481–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmidt J, Salaverria I, Haake A, Bonzheim I, Adam P, Montes-Moreno S, et al. Increasing genomic and epigenomic complexity in the clonal evolution from in situ to manifest t(14;18) positive follicular lymphoma. Leukemia. 2013. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 38.Bretherick KL, Bu R, Gascoyne RD, Connors JM, Spinelli JJ, Brooks-Wilson AR. Elevated circulating t(14;18) translocation levels prior to diagnosis of follicular lymphoma. Blood. 2010;116(26):6146–7 [DOI] [PubMed] [Google Scholar]
- 39.Adam P, Katzenberger T, Eifert M, Ott MM, Rosenwald A, Muller-Hermelink HK, et al. Presence of preserved reactive germinal centers in follicular lymphoma is a strong histopathologic indicator of limited disease stage. Am J Surg Pathol. 2005;29(12):1661–4 [DOI] [PubMed] [Google Scholar]
- 40.Schmatz AI, Streubel B, Kretschmer-Chott E, Puspok A, Jager U, Mannhalter C, et al. Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol. 2011;29(11):1445–51 [DOI] [PubMed] [Google Scholar]
- 41.Takata K, Sato Y, Nakamura N, Tokunaka M, Miki Y, Yukie Kikuti Y, et al. Duodenal follicular lymphoma lacks AID but expresses BACH2 and has memory B-cell characteristics. Mod Pathol. 2013;26(1):22–31 [DOI] [PubMed] [Google Scholar]
- 42.Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL, et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;121(9):1604–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bodor C, Grossmann V, Popov N, Okosun J, O’Riain C, Tan K, et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood. 2013;122(18):3165–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okosun J, Bödör C, Wang J, Araf S, Yang CY, Pan C, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46(2):176–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6(1):130–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226(4678):1097–9 [DOI] [PubMed] [Google Scholar]
- 47.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coelho V, Krysov S, Ghaemmaghami AM, Emara M, Potter KN, Johnson P, et al. Glycosylation of surface Ig creates a functional bridge between human follicular lymphoma and microenvironmental lectins. Proc Natl Acad Sci USA. 2010;107(43):18587–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu D, McCarthy H, Ottensmeier CH, Johnson P, Hamblin TJ, Stevenson FK. Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood. 2002;99(7):2562–8 [DOI] [PubMed] [Google Scholar]
- 50.Oricchio E, Nanjangud G, Wolfe AL, Schatz JH, Mavrakis KJ, Jiang M, et al. The Ephreceptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell. 2011;147(3):554–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheung KJ, Johnson NA, Affleck JG, Severson T, Steidl C, Ben-Neriah S, et al. Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis. Cancer Res. 2010;70(22):9166–74 [DOI] [PubMed] [Google Scholar]
- 52.Launay E, Pangault C, Bertrand P, Jardin F, Lamy T, Tilly H, et al. High rate of TNFRSF14 gene alterations related to 1p36 region in de novo follicular lymphoma and impact on prognosis. Leukemia. 2012;26(3):559–62 [DOI] [PubMed] [Google Scholar]
- 53.Cheung KJ, Shah SP, Steidl C, Johnson N, Relander T, Telenius A, et al. Genome-wide profiling of follicular lymphoma by array comparative genomic hybridization reveals prognostically significant DNA copy number imbalances. Blood. 2009;113(1):137–48 [DOI] [PubMed] [Google Scholar]
- 54.Middendorp S, Xiao Y, Song JY, Peperzak V, Krijger PH, Jacobs H, et al. Mice deficient for CD137 ligand are predisposed to develop germinal center-derived B-cell lymphoma. Blood. 2009;114(11):2280–9 [DOI] [PubMed] [Google Scholar]
- 55.Green M, Gandhi MK, Camilleri E, Marlton P, Lea R, Griffiths L. High levels of BACH2 associated with lower levels of BCL2 transcript abundance in t(14;18)(q21;q34) translocation positive non-Hodgkin’s lymphoma. Leuk Res. 2009;33(5):731–4 [DOI] [PubMed] [Google Scholar]
- 56.Kobayashi S, Taki T, Chinen Y, Tsutsumi Y, Ohshiro M, Kobayashi T, et al. Identification of IGHCdelta-BACH2 fusion transcripts resulting from cryptic chromosomal rearrangements of 14q32 with 6q15 in aggressive B-cell lymphoma/leukemia. Genes Chromosomes Cancer. 2011;50(4):207–16 [DOI] [PubMed] [Google Scholar]
- 57.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyl-transferase genes in B-cell lymphoma. Nature. 2011;471(7337):189–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ryan RJ, Nitta M, Borger D, Zukerberg LR, Ferry JA, Harris NL, et al. EZH2 codon 641 mutations are common in BCL2-rearranged germinal center B cell lymphomas. PloS one. 2011;6(12):e28585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459(7247):712–6 [DOI] [PubMed] [Google Scholar]
- 61.Honma K, Tsuzuki S, Nakagawa M, Tagawa H, Nakamura S, Morishima Y, et al. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood. 2009;114(12):2467–75 [DOI] [PubMed] [Google Scholar]
- 62.Gronbaek K, Straten PT, Ralfkiaer E, Ahrenkiel V, Andersen MK, Hansen NE, et al. Somatic Fas mutations in non-Hodgkin’s lymphoma: association with extranodal disease and autoimmunity. Blood. 1998;92(9):3018–24 [PubMed] [Google Scholar]
- 63.Bouamar H, Abbas S, Lin AP, Wang L, Jiang D, Holder KN, et al. A capture-sequencing strategy identifies IRF8, EBF1, and APRIL as novel IGH fusion partners in B-cell lymphoma. Blood. 2013;122(5):726–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 2013; 110(4):1398–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miletic AV, Anzelon-Mills AN, Mills DM, Omori SA, Pedersen IM, Shin DM, et al. Coordinate suppression of B cell lymphoma by PTEN and SHIP phosphatases. J Exp Med. 2010;207(11):2407–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Butler MP, Wang SI, Chaganti RS, Parsons R, Dalla-Favera R. Analysis of PTEN mutations and deletions in B-cell non-Hodgkin’s lymphomas. Genes Chromosomes Cancer. 1999;24(4):322–7 [PubMed] [Google Scholar]
- 67.Lo Coco F, Ye BH, Lista F, Corradini P, Offit K, Knowles DM, et al. Rearrangements of the BCL6 gene in diffuse large cell non-Hodgkin’s lymphoma. Blood. 1994;83(7):1757–9 [PubMed] [Google Scholar]
- 68.Bastard C, Deweindt C, Kerckaert JP, Lenormand B, Rossi A, Pezzella F, et al. LAZ3 rearrangements in non-Hodgkin’s lymphoma: correlation with histology, immunophenotype, karyotype, and clinical outcome in 217 patients. Blood. 1994;83(9):2423–7 [PubMed] [Google Scholar]
- 69.Johnson NA, Savage KJ, Ludkovski O, Ben-Neriah S, Woods R, Steidl C, et al. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood. 2009;114(11):2273–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akasaka T, Lossos IS, Levy R. BCL6 gene translocation in follicular lymphoma: a harbinger of eventual transformation to diffuse aggressive lymphoma. Blood. 2003;102(4):1443–8 [DOI] [PubMed] [Google Scholar]
- 71.Vardi A, Dagklis A, Scarfo L, Jelinek D, Newton D, Bennett F, et al. Immunogenetics shows that not all MBL are equal: the larger the clone, the more similar to CLL. Blood. 2013;121(22):4521–8 [DOI] [PubMed] [Google Scholar]
- 72.Dhodapkar MV, Sexton R, Waheed S, Usmani S, Papanikolaou X, Nair B, et al. Clinical, genomic and imaging predictors of myeloma progression from asymptomatic monoclonal gammopathies (SWOG S0120). Blood. 2014;123(1):78–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ardeshna KM, Smith P, Norton A, Hancock BW, Hoskin PJ, MacLennan KA, et al. Long-term effect of a watch and wait policy versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma: a randomised controlled trial. Lancet. 2003;362(9383):516–22 [DOI] [PubMed] [Google Scholar]
- 74.Michallet AS, Lebras LL, Bauwens DD, Bouafia-Sauvy FF, Berger FF, Tychyj-Pinel CC, et al. Early stage follicular lymphoma: what is the clinical impact of the first-line treatment strategy¿ J Hematol Oncol. 2013;6:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McNamara C, Davies J, Dyer M, Hoskin P, Illidge T, Lyttelton M, et al. Guidelines on the investigation and management of follicular lymphoma. Br J Haematol. 2012;156(4):446–67 [DOI] [PubMed] [Google Scholar]
- 76.Mac Manus MP, Hoppe RT. Is radiotherapy curative for stage I and II low-grade follicular lymphoma¿ Results of a long-term followup study of patients treated at Stanford University. J Clin Oncol. 1996; 14(4):1282–90 [DOI] [PubMed] [Google Scholar]
- 77.Montoto S. Management of localized-stage follicular lymphoma: changing the paradigm¿ J Clin Oncol. 2012;30(27):3328–9 [DOI] [PubMed] [Google Scholar]
- 78.Vitolo U, Ladetto M, Boccomini C, Baldini L, De Angelis F, Tucci A, et al. Rituximab Maintenance Compared With Observation After Brief First-Line R-FND Chemoimmunotherapy With Rituximab Consolidation in Patients Age Older Than 60 Years With Advanced Follicular Lymphoma: A Phase III Randomized Study by the Fondazione Italiana Linfomi. J Clin Oncol. 2013;31(27):3351–9 [DOI] [PubMed] [Google Scholar]
- 79.Ardeshna KM, Smith P, Qian W, Warden J, Stevens L, Pocock CFE, et al. An Intergroup Randomised Trial of Rituximab Versus a Watch and Wait Strategy In Patients with Stage II, III, IV, Asymptomatic, Non-Bulky Follicular Lymphoma (Grades 1, 2 and 3a). A Preliminary Analysis. ASH Annual Meeting [Abstract]. 2010;116:6 [Google Scholar]
- 80.Ansell SM. Follicular lymphoma: watch and wait is watch and worry. Lancet Oncol. 2014;[Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 81.Ardeshna KM, Qian W, Smith P, Braganca N, Lowry L, Patrick P, et al. Rituximab versus a watch-and-wait approach in patients with advanced-stage, asymptomatic, non-bulky follicular lymphoma: an open-label randomised phase 3 trial. Lancet Oncol. 2014;[Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 82.Kahl BS, Hong F, Williams ME, (RESORT) ECOGPE , editors. A randomized phase III study comparing two different rituximab dosing strategies for low tumor burden follicular lymphoma. ASH Annual Meeting Abstracts. [Google Scholar]
- 83.Salles G, Seymour JF, Offner F, Lopez-Guillermo A, Belada D, Xerri L, et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet. 2011. 1;377(9759):42–51 [DOI] [PubMed] [Google Scholar]
- 84.Friedberg JW, Taylor MD, Cerhan JR, Flowers CR, Dillon H, Farber CM, et al. Follicular lymphoma in the United States: first report of the national LymphoCare study. J Clin Oncol. 2009;27(8):1202–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Solal-Celigny P, Bellei M, Marcheselli L, Pesce EA, Pileri S, McLaughlin P, et al. Watchful waiting in low-tumor burden follicular lymphoma in the rituximab era: results of an F2-study database. J Clin Oncol. 2012;30(31):3848–53 [DOI] [PubMed] [Google Scholar]
- 86.Dupuis J, Berriolo-Riedinger A, Julian A, Brice P, Tychyj-Pinel C, Tilly H, et al. Impact of [(18)F]fluorodeoxyglucose positron emission tomography response evaluation in patients with high-tumor burden follicular lymphoma treated with immunochemotherapy: a prospective study from the Groupe d’Etudes des Lymphomes de l’Adulte and GOELAMS. J Clin Oncol. 2012;30(35):4317–22 [DOI] [PubMed] [Google Scholar]
- 87.Trotman J, Fournier M, Lamy T, Seymour JF, Sonet A, Janikova A, et al. Positron emission tomography-computed tomography (PET-CT) after induction therapy is highly predictive of patient outcome in follicular lymphoma: analysis of PET-CT in a subset of PRIMA trial participants. J Clin Oncol. 2011;29(23):3194–200 [DOI] [PubMed] [Google Scholar]
- 88.Seymour JF, Trotman J, Hofman MS. Evaluating the place of FDG PET scanning in primary staging and beyond in patients with follicular lymphoma. Leuk Lymphoma. 2013;54(10):2093–5 [DOI] [PubMed] [Google Scholar]
- 89.Luminari S, Biasoli I, Arcaini L, Versari A, Rusconi C, Merli F, et al. The use of FDG-PET in the initial staging of 142 patients with follicular lymphoma: a retrospective study from the FOLL05 randomized trial of the Fondazione Italiana Linfomi. Ann Oncol. 2013;24(8):2108–12 [DOI] [PubMed] [Google Scholar]
- 90.Glas AM, Knoops L, Delahaye L, Kersten MJ, Kibbelaar RE, Wessels LA, et al. Gene-expression and immunohistochemical study of specific T-cell subsets and accessory cell types in the transformation and prognosis of follicular lymphoma. J Clin Oncol. 2007;25(4):390–8 [DOI] [PubMed] [Google Scholar]
- 91.Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351(21):2159–69 [DOI] [PubMed] [Google Scholar]
- 92.Cerhan JR, Wang S, Maurer MJ, Ansell SM, Geyer SM, Cozen W, et al. Prognostic significance of host immune gene polymorphisms in follicular lymphoma survival. Blood. 2007;109(12):5439–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Alvaro T, Lejeune M, Salvado MT, Lopez C, Jaen J, Bosch R, et al. Immunohistochemical patterns of reactive microenvironment are associated with clinicobiologic behavior in follicular lymphoma patients. J Clin Oncol. 2006;24(34):5350–7 [DOI] [PubMed] [Google Scholar]
- 94.Alvaro T, Lejeune M, Camacho FI, Salvado MT, Sanchez L, Garcia JF, et al. The presence of STAT1-positive tumor-associated macrophages and their relation to outcome in patients with follicular lymphoma. Haematologica. 2006;91(12):1605–12 [PubMed] [Google Scholar]
- 95.Canioni D, Salles G, Mounier N, Brousse N, Keuppens M, Morchhauser F, et al. High numbers of tumor-associated macrophages have an adverse prognostic value that can be circumvented by rituximab in patients with follicular lymphoma enrolled onto the GELA-GOELAMS FL-2000 trial. J Clin Oncol. 2008;26(3):440–6 [DOI] [PubMed] [Google Scholar]
- 96.de Jong D, Rosenwald A, Chhanabhai M, Gaulard P, Klapper W, Lee A, et al. Immunohistochemical prognostic markers in diffuse large B-cell lymphoma: validation of tissue microarray as a prerequisite for broad clinical applications–a study from the Lunenburg Lymphoma Biomarker Consortium. J Clin Oncol. 2007;25(7):805–12 [DOI] [PubMed] [Google Scholar]
- 97.Blanc V, Bousseau A, Caron A, Carrez C, Lutz RJ, Lambert JM. SAR3419: an anti-CD19-Maytansinoid Immunoconjugate for the treatment of B-cell malignancies. Clin Cancer Res. 2011;17(20):6448–58 [DOI] [PubMed] [Google Scholar]
- 98.Sosin AM, Burger AM, Siddiqi A, Abrams J, Mohammad RM, Al-Katib AM. HDM2 antagonist MI-219 (spiro-oxindole), but not Nutlin-3 (cis-imidazoline), regulates p53 through enhanced HDM2 autoubiquitination and degradation in human malignant B-cell lymphomas. J Hematol Oncol. 2012; 5:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Seiler TM, Hiddemann W. Advances in the management of follicular lymphoma. Curr Opin Oncol. 2012;24(6):742–7 [DOI] [PubMed] [Google Scholar]