

Abstract
Paroxysmal nocturnal hemoglobinuria is a rare, acquired disease associated with hemolytic anemia, bone marrow failure, thrombosis, and, frequently, poor quality of life. The International PNH Registry is a worldwide, observational, non-interventional study collecting safety, effectiveness, and quality-of-life data from patients with a confirmed paroxysmal nocturnal hemoglobinuria diagnosis or detectable paroxysmal nocturnal hemoglobinuria clone, irrespective of treatment. In addition to evaluating the long-term safety and effectiveness of eculizumab in a global population, the registry aims to improve diagnosis, optimize patient management and outcomes, and enhance the understanding of the natural history of paroxysmal nocturnal hemoglobinuria. Here we report the characteristics of the first 1610 patients enrolled. Median disease duration was 4.6 years. Median granulocyte paroxysmal nocturnal hemoglobinuria clone size was 68.1% (range 0.01–100%). Overall, 16% of patients had a history of thrombotic events and 14% a history of impaired renal function. Therapies included anticoagulation (31%), immunosuppression (19%), and eculizumab (25%). Frequently reported symptoms included fatigue (80%), dyspnea (64%), hemoglobinuria (62%), abdominal pain (44%), and chest pain (33%). Patients suffered from poor quality of life; 23% of patients had been hospitalized due to paroxysmal nocturnal hemoglobinuria-related complications and 17% stated that paroxysmal nocturnal hemoglobinuria was the reason they were not working or were working less. This international registry will provide an ongoing, valuable resource to further the clinical understanding of paroxysmal nocturnal hemoglobinuria.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired disease characterized by chronic intravascular hemolysis caused by uncontrolled complement activation.1 The cellular abnormality in this life-threatening disease originates from a mutation in the phosphatidylinositol glycan class A (PIGA) gene, resulting in a deficiency of glycosylphosphatidyl-inositol (GPI)-anchored complement regulatory proteins, including CD55 and CD59, on the surface of blood cells.1 Patients with chronic hemolysis experience a marked increased risk of thromboembolism (TE), which may ultimately lead to target organ damage and death.2,3 Retrospective analyses have reported that, despite best supportive care, the 10-year survival rate in patients with PNH ranged from 50% for patients diagnosed between 1940 and 19703–5 to 75% in a more recent series.6 TE is the leading cause of mortality in patients with PNH, accounting for between 40% and 67% of deaths with known causes.7 Patients with PNH also experience symptoms including fatigue, abdominal pain, headache, shortness of breath, dysphagia, and erectile dysfunction.1 These symptoms can be debilitating and significantly reduce the quality of life (QoL) of patients with PNH.8 PNH may develop in the absence of another bone marrow disorder (BMD), as a condition secondary to BMDs such as aplastic anemia (AA) or myelodysplastic syndrome, or as subclinical PNH.1
The only potentially curative therapy for PNH is allogeneic bone marrow transplantation; however, this procedure is associated with substantial morbidity and mortality9–11 and, consequently, is not an appropriate therapeutic option for most patients. Historically, management of PNH was limited to the use of supportive measures such as blood transfusions and anticoagulation therapy. However, it has been reported that the risk of TE in patients with PNH remains high even in patients who have no clinical evidence of TE or are receiving prophylactic anticoagulation,7,12 which is itself associated with an increased risk of bleeding complications.13 In one study, spontaneous, long-term remission was observed in approximately 15% of patients.4
In 2007, eculizumab (Soliris®, Alexion Pharmaceuticals, Inc., Cheshire, CT, USA), a humanized monoclonal antibody that inhibits terminal complement activation, was approved for the treatment of patients with PNH. A phase II study (PILOT)14 and two phase III studies (TRIUMPH and SHEPHERD)15,16 demonstrated that eculizumab was well tolerated and provided a rapid, sustained, and clinically meaningful reduction in hemolysis, fatigue, and transfusion requirements, along with improved QoL. Other studies have shown that eculizumab therapy in patients with PNH is also associated with improvements in pulmonary hypertension and renal function.2,17 Subsequent studies have demonstrated that eculizumab is associated with a 92% reduction in the risk of TE (P<0.001)7 and with a highly significant improvement in patient survival to a level comparable to that of age-matched healthy controls.3
The natural history of PNH is highly variable and has previously been investigated by retrospective analyses involving relatively small patient populations.4,6,18,19 The burden of disease from the patient’s perspective has not been previously documented. The International PNH Registry was implemented to evaluate the safety and effectiveness of eculizumab, to gather comprehensive data on the natural history of PNH and the management of patients with PNH, and to evaluate the clinical symptoms and outcomes of the disease in order to better understand its progression and variability on a global scale. The long-term aim of the registry is to improve diagnosis and therapeutic strategies, optimize patient management and outcomes, enhance knowledge of pregnancy and related issues, and better understand the natural history of the disease.
In this first publication of data from the International PNH Registry, we report on the cross-sectional analysis of demographic and clinical characteristics of patients enrolled through June 30, 2012, and describe disease-associated morbidities commonly experienced in patients with PNH. In addition, we also report initial findings from a subset of patients who had completed base-line study questionnaires relating to impacts of the disease on various measures, including QoL, symptomatology, and employment status.
Methods
Patient population
The International PNH Registry is a prospective, non-interventional, observational study. It collects information from patients monitored in current medical practice irrespective of past, present, or future treatment. The registry was approved by the institutional review boards (or equivalent) of participating centers and all patients provided written informed consent prior to inclusion. The registry is sponsored by Alexion Pharmaceuticals, Inc., and is overseen by an independent executive committee of international PNH experts.
Patients of any age with a clinical diagnosis of PNH (by any applicable diagnostic method) and/or detectable PNH clone of 0.01% or over were eligible for inclusion in the registry. A PNH clone was defined as a population of granulocytes and/or erythrocytes deficient in GPI.
Data collection
Data captured in the registry include patients’ demographics, medical and treatment history, comorbid conditions, PNH clone size, disease characteristics and outcomes, symptoms, PNH-specific treatments, PNH-related events, morbidity (including myeloproliferative disease, other malignancies, and infections), mortality, pregnancy (maternal and fetal outcomes), patient QoL, and health resource utilization. Clinical data captured include lactate dehydrogenase (LDH) levels, hemoglobin levels, transfusion requirements, thrombotic events (identified using major adverse vascular event categories), physician-reported renal dysfunction, and other laboratory data. Specific information collected for eculizumab-treated patients includes dosage and dose adjustments, meningococcal vaccination status, infusion reactions, reasons for treatment discontinuation, and outcomes associated with discontinuation.
Patient medical information and study questionnaire data are collected at study enrollment and every six months thereafter. The patients’ questionnaires are collected at study enrollment and during routine office visits close to the 6-month follow-up time window and include QoL data based on the validated Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue20 and European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire version 3.0 (EORTC QLQ-C30)21 instruments, common PNH symptoms, use of health care resources including hospitalizations, work status and time lost from work due to PNH, and treatment satisfaction.
Statistical analysis
Continuous variables were described using standard summary statistics; categorical variables were described using frequencies and percentages. Group differences were determined using analysis of variance or the Student’s t-test for continuous variables and χ2 tests for categorical variables. Non-parametric Wilcoxon and Kruskal-Wallis tests were used for laboratory results (e.g. granulocyte clone size and LDH) with non-normal distributions. P<0.05 was considered statistically significant.
Patient-reported symptoms, history of TE, and transfusion requirements were stratified by PNH clone size at enrollment (<10%, 10–49%, and ≥50%), diagnosis (current or prior) of AA or other BMD, and LDH level (<1.5 or ≥1.5 × upper limit of normal (ULN), the higher levels having been shown to be associated with significantly increased risks of TE and mortality22,23). Clone size categories were used for analyses, as their bimodal distribution limits their suitability for consideration as a continuous variable.
For the EORTC QoL and FACIT-Fatigue assessments, lower scores indicate a poorer QoL and increased levels of fatigue. Differences in average scores between groups of 5 points or over24 or 3 points or over,25 respectively, are considered clinically meaningful.
Results
Patients’ demographics and clinical characteristics
As of June 30, 2012, 1610 patients from 273 centers in 25 countries were enrolled in the International PNH Registry (Online Supplementary Table S1). Overall, 92.5% of patients were from Europe and North America and 87.5% of patients were Caucasian. The remaining patients were Asian/Pacific Islanders (5.0%), of African descent (3.5%), Native/Aboriginal (0.2%), or of other/unknown ethnicity/race (3.9%).
Patients’ demographics and clinical characteristics of the 1610 enrolled patients are provided in Table 1. Median patient age at enrollment was 42 years (range 3–99 years), and 53.2% of patients were female. Median PNH duration from disease start to enrollment was 4.6 years, with a minimum of less than 1 year and a maximum of 47 years. Overall, 774 (48.1%) of the patients had been diagnosed with one or more types of BMD, including AA or hypoplastic anemia (n=701; 43.5%), myelodysplastic syndromes (n=93; 5.8%), myelofibrosis (n=7; 0.4%), and/or acute myeloid leukemia (n=6; 0.4%). In the 900 of 1610 patients who had not received eculizumab in the 12 months prior to enrollment and for whom LDH values were available, the median LDH level at the time of enrollment was approximately twice the ULN. A history of any prior thrombotic event was reported in 250 patients (15.5%), with the majority of these patients (169 of 250, 67.6%) having experienced just one event. A similar number of patients (n=223,13.9%) had a history of impaired renal function. At enrollment, 31.1% of patients were receiving anticoagulation therapy, 18.7% immunosuppressive therapy, and 8.3% pain medication; approximately 25% of the patients were being treated with eculizumab (Table 1). At the time of analysis, completed base-line patients’ questionnaires relating to symptoms of PNH, QoL, and work were available for 856 of the 1610 (53%) enrolled patients.
Table 1.
Patients’ demographics and clinical characteristics at enrollment into the International PNH Registry.
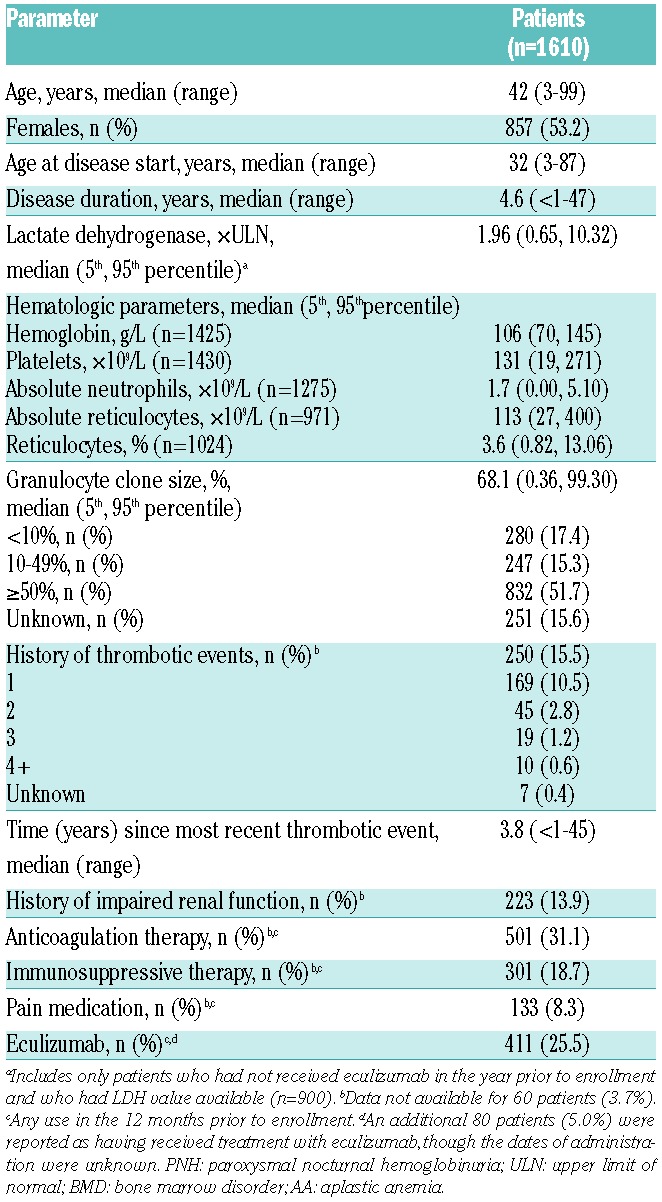
Due to the observational nature of this study, the number of patients contributing data to each assessment was not consistent. In addition, as eculizumab reduces hemolysis and prevents release of LDH, patients who had received eculizumab at any time in the 12 months prior to enrollment were excluded from all data summaries assessed by LDH levels. A graphical summary of the number of patients included in each assessment, along with the criteria defining each subpopulation, is provided in Online Supplementary Figure S1.
PNH clone size
The median granulocyte clone size at enrollment was 68.1% (Table 1), and the entire range of clone sizes was represented, from 0.01% (the minimum inclusion criterion for enrollment) through 100%. The median clone size was significantly larger in patients without a history of BMD than in patients who had at any point been diagnosed with AA (83% vs. 35%; P<0.001 by Wilcoxon test). The distribution of clone sizes varied with history of BMD: clone sizes of 50% or over and less than 10% were reported in, respectively, 34% and 40% of patients with a history of AA compared with 76% and 8% of patients without a diagnosis of BMD other than PNH.
In patients who had not received eculizumab in the 12 months prior to study enrollment, median LDH levels generally increased with PNH clone size category (P<0.001 by Kruskal-Wallis test) and in patients with no history of BMD other than PNH compared with those patients ever diagnosed with AA (Figure 1A). There was also a significant increase in the percentage of patients with LDH levels ≥1.5 × ULN as PNH clone size increased, going from 8% of patients with a clone size less than 10% to 55% of patients with a clone size of 10–49% and 83% of patients with a clone size 50% or over (Figure 1B).
Figure 1.

LDH concentration. (A) Median LDH concentration at enrollment by PNH clone size and diagnosis. (B) Percentage of patients with LDH <1.5×ULN or ≥1.5×ULN by PNH clone size. Only includes patients who had not received eculizumab in the 12 months prior to study enrollment.
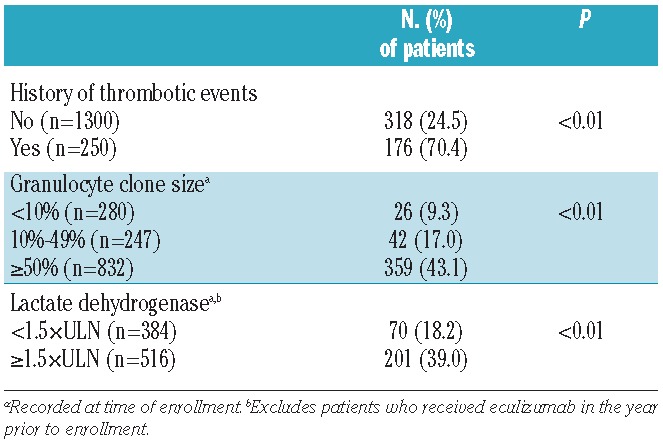
In patients who had not received eculizumab in the 12 months prior to enrollment, there was a positive correlation between history of thrombosis and clone size at enrollment: 5.3% of patients with clone size less than 10% had a history of thrombosis and 7.7% of patients with clone size 10–49% had such a history, whereas 15.4% of patients with clone size 50% or over had such a history (P<0.001). In addition, a larger percentage of patients with LDH ≥1.5 × ULN at enrollment, compared with LDH <1.5 × ULN at enrollment reported a history of thrombosis (15.6% vs. 8.4%; P<0.001) (Figure 2A).
Figure 2.

Thrombosis and red blood cell transfusion history. (A) Percentage of patients with a history of thrombosis by PNH clone size and LDH level at enrollment. (B) Percentage of patients receiving red blood cell transfusion in the year prior to enrollment by PNH clone size and diagnosis. Only includes patients who had not received eculizumab in the 12 months prior to study enrollment.
Concomitant therapies
The number of patients receiving anticoagulation therapy or immunosuppressive therapy in the 12 months prior to enrollment and the number of patients receiving red blood cell transfusions in the six months prior to enrollment are shown in Table 2. Compared with patients with no history of BMD, patients who had at some point been diagnosed with AA were less likely to have received treatment with anticoagulants (21.0% vs. 42.0%; P<0.001) or with eculizumab (18.7% vs. 33.8%; P<0.001), but they were more likely to have received immunosuppressive therapies such as cyclosporine and/or anti-thymocyte globulin (38.5% vs. 2.8%; P<0.001). There was no significant difference between these patient groups in the percentage who had received red blood cell transfusions in the six months before study enrollment (P=0.21), though patients who had at some point been diagnosed with AA received a significantly greater mean number of units of packed red blood cells during this time frame compared with patients with no history of BMD (9.0 vs. 6.8; P=0.001).
Table 2.
Treatment receiveda prior to enrollment into the International PNH Registry by history of AA.

Compared with patients with no history of BMD, a significantly greater percentage of patients who had at some point been diagnosed with AA had received treatment with immunosuppressive therapies plus anticoagulants, eculizumab, or red blood cell transfusions (all P<0.001) (Table 2). In contrast, significantly more patients with no history of BMD had received treatment with anticoagulation therapy plus eculizumab or red blood cell transfusions (both P<0.001) (Table 2).
Patients were significantly more likely to have received anticoagulation therapy in the 12 months prior to enrollment if they had a history of thrombosis, a granulocyte PNH clone size 50% or over, or an LDH level ≥1.5 × ULN (Table 3). Figure 2B shows the percentage of patients who received red blood cell transfusions by diagnosis and PNH clone size. Patients who had at some point been diagnosed with AA were more likely to have received a blood transfusion in the six months prior to enrollment. This difference reached statistical significance in patients with clone sizes over 50% (P=0.02).
Table 3.
Anticoagulant use (within 12 months prior to enrollment) by selected characteristics.
PNH symptoms
Of the 856 patients for whom self-reported symptom data were available (i.e. patients with a base-line questionnaire), a total of 799 patients (93.3%) reported at least one symptom associated with PNH. The percentage of patients reporting common symptoms associated with PNH in the past six months is shown in Online Supplementary Figure S2. Commonly reported symptoms included fatigue (80%), dyspnea (64%), headache (63%), and hemoglobinuria (62%), and 38% of male patients had experienced erectile dysfunction. Although fatigue was the most frequently reported symptom, 91.4% of patients (782 of 856) reported at least one symptom other than fatigue, whereas only 2.0% of patients (17 of 856) reported fatigue and no other symptoms.
Although each of the common PNH-related symptoms were reported in all categories of clone size, patients with clone sizes 50% or over reported significantly more hemoglobinuria, dyspnea, abdominal pain, scleral icterus, erectile dysfunction, and dysphagia (Figure 3A). There were no significant differences in the prevalence of fatigue, headache, dyspnea, or confusion between clone size categories. The most frequently reported symptom was fatigue, reported in over 75% of patients in each clone size category. However, 83.3% of patients with clone size less than 10%, 89.3% with clone size 10–49%, and 93.3% with clone size 50% or over reported at least one symptom other than fatigue, whereas only 4.0%, 3.6%, and 0.8% of patients, respectively, reported fatigue and no other symptoms. In patients who had not been treated with eculizumab, the prevalence of dyspnea, hemoglobinuria, abdominal pain, scleral icterus, chest pain, and dysphagia was significantly greater in patients with LDH ≥1.5 × ULN at enrollment than in patients with lower LDH levels (Figure 3B). There were no significant differences in the prevalence of confusion, headache, fatigue, or erectile dysfunction between patients with LDH levels ≥1.5 × ULN and those with LDH levels <1.5 × ULN. Overall, 95.9% of patients with LDH ≥1.5 × ULN reported at least one symptom other than fatigue, whereas only 0.6% of patients with elevated LDH levels reported fatigue and no other symptoms. Similarly, 88.7% of patients with LDH <1.5 × ULN had symptoms other than fatigue, while only 2.0% reported fatigue alone.
Figure 3.

Patient-reported PNH symptoms. (A) Patient-reported symptoms by PNH clone size. (B) Patient-reported symptoms by LDH level. Only includes patients who had not received eculizumab in the 12 months prior to study enrollment. *Statistically significant. †Male patients only; n=62, 68, and 222. ‡Male patients only; n=82 and 134.
For the majority of the patient-reported PNH-related symptoms, history of BMD was not associated with prevalence of the symptom. However, hemoglobinuria, scleral icterus, and dysphagia were significantly more common in patients never diagnosed with BMD (all P<0.001).
QoL, hospitalization, and employment
Patient-reported EORTC QLQ-C30 QoL and FACIT-Fatigue assessments indicated that, compared with patients without a history of thrombosis, patients with a prior TE had significantly lower global health status/QoL (P=0.01), physical functioning (P<0.01), and social functioning (P=0.02) and significantly greater fatigue (P=0.04) (Online Supplementary Figure S3).
Statistically significantly lower QoL scores for all EORTC domains were reported by patients who had reported a clinical symptom of abdominal pain, chest pain, confusion, dysphagia, dyspnea, erectile dysfunction, fatigue, headache, hemoglobinuria, or scleral icterus in the six months prior to completing the baseline questionnaire compared with patients who had not experienced the symptom. All results were highly significant (P<0.0001) with the exception of emotional functioning for patients with erectile dysfunction (P=0.039), physical functioning for patients with hemoglobinuria (P=0.003), and emotional and cognitive functioning in patients with scleral icterus (P=0.001 and P=0.007, respectively).
In the six months prior to completion of the base-line questionnaire, 194 of 856 patients (22.7%) reported being hospitalized due to their PNH. Patients were significantly more likely to be hospitalized if they had a history of thrombosis or had experienced self-reported PNH-related symptoms of scleral icterus, chest pain, dysphagia, abdominal pain, hemoglobinuria, dyspnea, or fatigue in the past six months (all P<0.01) (Online Supplementary Figure S4).
The impact of PNH on a patient’s employment was assessed in patients aged 18–59 years. Eighty-eight of 506 patients (17.4%) reported PNH as the reason they were either not working or working less (e.g. part time rather than full time). Of the 312 full-time or part-time workers, 82 (26.3%) had missed work in the past six months for reasons related to PNH.
Discussion
As of June 30, 2012, the International PNH Registry had enrolled 1610 patients from 5 continents, with completed patient questionnaires at enrollment available for 856 patients. Collection of outstanding data, as well as new recruitment into the registry, is ongoing. The current analysis evaluated the base-line characteristics of the enrolled population and investigated the burden of disease in terms of symptomatology and clinical outcomes. As the questionnaires completed at enrollment are largely concerned with the patients’ medical history, much of the data are retrospective. However, these base-line data are an important foundation for future analyses on prospectively collected data.
Assessing data from an extensive population is important, particularly for rare conditions, as the disease course can vary significantly among patients; at enrollment our population has an age range spanning almost 100 years, a disease duration of less than one to 47 years, and granulocyte PNH clone sizes ranging from less than 1% to 100%. Given these variations, associations between patients’ characteristics and outcomes and determination of robust therapeutic strategies can be achieved only via an international registry.
The level of morbidity was high in our patient population: at enrollment, 43.5% had a history of AA or hypoplastic anemia, 15.5% had experienced at least one TE, and 13.9% had a history of impaired renal function. The majority of patients not treated with eculizumab had LDH concentrations ≥1.5 × ULN, a level associated with significantly increased risks of TE and mortality.22,23 Our analyses suggested an association between PNH granulocyte clone size and LDH levels; in patients with a clone size less than 10%, median LDH levels were towards the ULN, with only 8% of patients having LDH ≥1.5 × ULN, whereas in patients with a clone size 50% or over, median levels were to >4.0 × ULN and 62% of patients had LDH ≥1.5 × ULN. Elevated LDH levels were also associated with higher prevalence of TE and symptoms such as abdominal pain, chest pain, and hemoglobinuria, all significant risk factors for TE.23
It is perhaps not surprising that a significantly larger percentage of patients who at some point have been diagnosed with AA had received immunosuppressive therapy but less often received anticoagulation compared with patients with no history of BMD. This latter group of patients was significantly more likely to have been treated with eculizumab; however, the percentage of patients requiring red blood cell transfusions appeared to be independent of underlying BMD.
Not only is thrombosis, the leading cause of death in PNH,7 associated with a significant risk for mortality, but patients experiencing a TE are 5-fold more likely to experience subsequent thrombotic events.5,6,26 Our findings demonstrate that PNH patients at all clone sizes may have a history of thrombosis, and patients with larger clones were more likely to have a history of thrombosis. These analyses suggest that all patients with PNH are at risk for thrombosis. This finding indicates that all patients should be routinely monitored for signs and symptoms of TE, including clinically evident hemolysis and abdominal pain.
Overall, 31.5% of patients with a PNH clone size less than 10% had required at least one red blood cell transfusion in the year prior to enrollment. As the majority of patients with this PNH clone size for whom LDH assessments at enrollment were available did not show signs of elevated hemolysis, the reason for transfusions is most likely related to an underlying BMD. These and other concomitant conditions may also be the cause of some of the symptoms classified as “PNH related”. It should be noted that not all of the transfusion, symptom and other data collected at enrollment may be directly related to hemolysis due to PNH, though collection and analysis of all such data are important in order to provide greater insight into the course of the disease and help to identify patients at risk of TE.
Our results showed that patients were significantly more likely to have been prescribed anticoagulant therapy if they had a history of TE, a larger granulocyte clone size, or elevated LDH concentrations. It must be remembered that these factors are not mutually exclusive but interrelated.
Common PNH-related symptoms were experienced by over 93% of patients for whom data was available. Although significantly more patients with a clone size 50% or over experienced hemoglobinuria, abdominal pain, scleral icterus, dysphagia, and erectile dysfunction, a substantial number of patients with a clone size less than 10% also experienced these symptoms, indicating that clone size alone is not a good indicator of burden of disease. The percentage of patients reporting PNH-related symptoms was greater in those with LDH levels ≥1.5 × ULN, with, for example, abdominal pain, a symptom associated with a 3.6-fold greater risk of thrombosis,27 being significantly more common in patients with elevated LDH levels. Overall, our results confirm that patients with PNH and elevated LDH levels are at increased risk of experiencing complications of PNH associated with increased risk of TE, mortality, and poor QoL.
Reference values for FACIT-Fatigue28 give a mean fatigue score of 43.6 in the general population and 40.0 in non-anemic cancer patients. The lower scores of 35.9 and 33.4 in patients without and with a history of thrombosis, respectively, indicate that PNH patients have a clinically meaningful greater level of fatigue29 than either of these other 2 populations. Similarly, the EORTC global health QoL assessments indicated that patients with PNH had a clinically meaningful worse QoL22 (with mean scores of 63.7 and 57.5 in patients without and with a history of thrombosis, respectively) than the general population (reference score 71.230) and similar QoL scores to those seen in cancer patients.28 These findings show that PNH has a clinically meaningful impact on QoL in all patients, with the occurrence of a thrombotic event having a significantly greater impact on patient levels of fatigue and overall QoL.
The EORTC scores also showed that global health status in patients with PNH is most affected by fatigue, confusion, and chest pain; these symptoms also significantly impact physical, role, and emotional functioning. Abdominal pain significantly affects global health status, particularly in terms of role, emotional, and cognitive functioning. This is not surprising given that abdominal pain and chest pain have been associated with a greater risk of thrombosis and mortality.27
PNH has wide-ranging effects on patients’ lives. The symptoms associated with the disease have serious consequences and are frequently devastating; one-quarter of PNH patients in the registry had been hospitalized and one-third had missed work due to PNH. Approximately one in 7 patients were not working or worked less due to PNH.
This large, global PNH registry of patients observed in clinical practice will continue to prospectively evaluate disease burden and the long-term natural history of PNH and treatment outcomes. The breadth and diversity of the registry provides an excellent basis for investigating research questions that may not be answerable from a single institution or country, and the registry has the additional advantages of collecting both clinical and patient-reported data and collecting these data both at enrollment and prospectively. The International PNH Registry will provide a valuable ongoing resource to further the clinical understanding of this rare, life-threatening disease.
Acknowledgments
The authors would like to thank Eric Elkin of ICON for statistical analysis and Mark Hughes, PhD, and Joshua Safran of Infusion Communications for writing and editorial support, which was funded by Alexion Pharmaceuticals.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hillmen P, Elebute M, Kelly R, Urbano-Ispizua A, Hill A, Rother RP, et al. Long-term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2010;85(8):553–9 [DOI] [PubMed] [Google Scholar]
- 3.Kelly RJ, Hill A, Arnold LM, Brooksbank GL, Richards SJ, Cullen M, et al. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011; 117(25):6786–92 [DOI] [PubMed] [Google Scholar]
- 4.Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253–8 [DOI] [PubMed] [Google Scholar]
- 5.Socié G, Mary JY, de Gramont A, Rio B, Leporrier M, Rose C, et al. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology. Lancet. 1996;348(9027):573–7 [DOI] [PubMed] [Google Scholar]
- 6.de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008;112(8):3099–106 [DOI] [PubMed] [Google Scholar]
- 7.Hillmen P, Muus P, Dührsen U, Risitano AM, Schubert J, Luzzatto L, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(12):4123–8 [DOI] [PubMed] [Google Scholar]
- 8.Young NS, Meyers G, Schrezenmeier H, Hillmen P, Hill A. The management of paroxysmal nocturnal hemoglobinuria: recent advances in diagnosis and treatment and new hope for patients. Semin Hematol. 2009;46(1 Suppl 1):S1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegenbart U, Niederwieser D, Forman S, Holler E, Leiblein S, Johnston L, et al. Hematopoietic cell transplantation from related and unrelated donors after minimal conditioning as a curative treatment modality for severe paroxysmal nocturnal hemoglobinuria. Biol Blood Marrow Transplant. 2003;9(11):689–97 [DOI] [PubMed] [Google Scholar]
- 10.Santarone S, Bacigalupo A, Risitano AM, Tagliaferri E, Di Bartolomeo E, Paola Iori A, et al. Hematopoietic stem cell transplantation for paroxysmal nocturnal hemoglobinuria: long-term results of a retrospective study on behalf of the Gruppo Italiano Trapianto Midollo Osseo (GITMO). Haematologica. 2010;95(6):983–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peffault de Latour R, Schrezenmeier H, Bacigalupo A, Blaise D, De Souza CA, Vigouroux S, et al. Allogeneic stem cell transplantation in paroxysmal nocturnal hemoglobinuria. Haematologica. 2012;97(11):1666–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill A, Reid SA, Rother RP, Gladwin MT, Collinson MO, Gaze DC, et al. High definition contrast-enhanced MR imaging in paroxysmal nocturnal hemoglobinuria (PNH) suggests a high frequency of subclinical thrombosis. Haematologica. 2013; 92(1):24–5 [Google Scholar]
- 13.Palareti G, Leali N, Coccheri S, Poggi M, Manotti C, D'Angelo A, et al. Bleeding complications of oral anticoagulant treatment: an inception-cohort, prospective collaborative study (ISCOAT). Italian Study on Complications of Oral Anticoagulant Therapy. Lancet. 1996;348(9025):423–8 [DOI] [PubMed] [Google Scholar]
- 14.Hillmen P, Hall C, Marsh JC, Elebute M, Bombara MP, Petro BE, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350(6):552–9 [DOI] [PubMed] [Google Scholar]
- 15.Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–43 [DOI] [PubMed] [Google Scholar]
- 16.Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111(4):1840–7 [DOI] [PubMed] [Google Scholar]
- 17.Hill A, Rother RP, Wang X, Morris SM, Jr, Quinn-Senger K, Kelly R, et al. Effect of eculizumab on haemolysis-associated nitric oxide depletion, dyspnoea, and measures of pulmonary hypertension in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2010;149(3):414–25 [DOI] [PubMed] [Google Scholar]
- 18.Moyo VM, Mukhina GL, Garrett ES, Brodsky RA. Natural history of paroxysmal nocturnal haemoglobinuria using modern diagnostic assays. Br J Haematol. 2004; 126(1):133–8 [DOI] [PubMed] [Google Scholar]
- 19.Pu JJ, Mukhina G, Wang H, Savage WJ, Brodsky RA. Natural history of paroxysmal nocturnal hemoglobinuria clones in patients presenting as aplastic anemia. Eur J Haematol. 2011;87(1):37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cella D. The effects of anemia and anemia treatment on the quality of life of people with cancer. Oncology. 2002;16(9 Suppl 10):125–32 [PubMed] [Google Scholar]
- 21.Aaronson NK, Ahmedzai S, Bergman B, Bullinger M, Cull A, Duez NJ, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365–76 [DOI] [PubMed] [Google Scholar]
- 22.Lee JW, Jang JH, Kim JS, Yoon SS, Lee JH, Kim YK, et al. Uncontrolled complement activation and the resulting chronic hemolysis as measured by LDH serum level at diagnosis as predictor of thrombotic complications and mortality in a large cohort of patients with paroxysmal nocturnal hemoglobinuria (PNH). Blood. 2011;118(21):(Abstract 3166) [Google Scholar]
- 23.Lee JW, Jang JH, Kim JS, Yoon S-S, Lee JH, Kim Y-K, et al. Clinical signs and symptoms associated with increased risk for thrombosis in patients with paroxysmal nocturnal hemoglobinuria from a Korean Registry. Int J Hematol. 2013;97(6):749–57 [DOI] [PubMed] [Google Scholar]
- 24.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139–44 [DOI] [PubMed] [Google Scholar]
- 25.Cella D, Eton DT, Lai JS, Peterman AH, Merkel DE. Combining anchor and distribution-based methods to derive minimal clinically important differences on the Functional Assessment of Cancer Therapy (FACT) anemia and fatigue scales. J Pain Symptom Manage. 2002;24(6):547–61 [DOI] [PubMed] [Google Scholar]
- 26.Nishimura J, Kanakura Y, Ware RE, Shichishima T, Nakakuma H, Ninomiya H, et al. Clinical course and flow cytometric analysis of paroxysmal nocturnal hemoglobinuria in the United States and Japan. Medicine (Baltimore). 2004;83(3):193–207 [DOI] [PubMed] [Google Scholar]
- 27.Lee J, Jun Ho J, Sung Soo Y, Jin Seok K, Yeo Kyung K, Deog Yeon C, et al. High prevalence and mortality associated with thromboembolism in Asian patients with paroxysmal nocturnal hemglobinuria (PNH) [abstract]. Haematologica. 2010;95(Suppl 2):205–6 Abstract 0505 [Google Scholar]
- 28.Cella D, Lai JS, Chang CH, Peterman A, Slavin M. Fatigue in cancer patients compared with fatigue in the general United States population. Cancer. 2002;94(2):528–38 [DOI] [PubMed] [Google Scholar]
- 29.Cella D, Webster K, Beaumont J. The FACIT-fatigue scale: description, reliability and validity. Evanston, IL: Center on Outcomes, Research and Education; 2003 [Google Scholar]
- 30.Scott NW, Fayers PM, Aaronson NK, Bottomley A, de Graeff A, Groenvold M, et al. EORTC Reference Values. Brussels, Belgium: EORTC Quality Of Life Group; 2008 [Google Scholar]