

Abstract
Thalassemia major and sickle cell disease are the two most widely disseminated hereditary hemoglobinopathies in the world. The outlook for affected individuals has improved in recent years due to advances in medical management in the prevention and treatment of complications. However, hematopoietic stem cell transplantation is still the only available curative option. The use of hematopoietic stem cell transplantation has been increasing, and outcomes today have substantially improved compared with the past three decades. Current experience world-wide is that more than 90% of patients now survive hematopoietic stem cell transplantation and disease-free survival is around 80%. However, only a few controlled trials have been reported, and decisions on patient selection for hematopoietic stem cell transplantation and transplant management remain principally dependent on data from retrospective analyses and on the clinical experience of the transplant centers. This consensus document from the European Blood and Marrow Transplantation Inborn Error Working Party and the Paediatric Diseases Working Party aims to report new data and provide consensus-based recommendations on indications for hematopoietic stem cell transplantation and transplant management.
Introduction
Thalassemia major (TM) originated in Mediterranean, Middle Eastern, and Asian regions, and sickle cell disease (SCD) originated from throughout central Africa. However, because of migration, both diseases now occur globally and represent a growing health problem in many countries.1 Despite the remarkable improvements in medical therapy for hemoglobinopathies,2,3 hematopoietic stem cell transplantation (HSCT) still remains the only available curative approach. Although both TM and SCD are hemoglobinopathies, they are two distinct diseases requiring different approaches to HSCT, based on their different clinical features and course of disease. While transfusion dependency for TM is a priori an indication for HSCT, the indications for HSCT in SCD are less clearly defined because of the variability of the disease course. The literature shows there is a wide experience in transplantation for TM, whereas only a few hundred transplants have been performed for SCD.4,5 There are a number of possible reasons for this difference, including a lack of consensus about the indications and time point for HSCT in SCD, and the low chance of identifying an unrelated donor for SCD patients.6,7 Table 1 shows the principal differences between TM and SCD from the transplant perspective.
Table 1.
Major differences between thalassemia major (TM) and sickle cell disease (SCD) on HSCT perspective.

So far, only a few prospective clinical trials have been reported in these diseases, and the decision to perform HSCT and the details of transplant management remain principally dependent on data derived from predominantly retrospective investigations and on the clinical expertise of the different transplant centers.8 For these reasons, the EBMT Paediatric Diseases Working Party and Inborn Error Working Party recognized the importance of establishing common guidelines. In October 2011, a Consensus Committee was convened with the aim of reviewing the literature and creating a consensus document addressing the current treatment strategies for TM and SCD. This paper summarizes the results of this review and the panel’s recommendations on indications and the approach to HSCT in these disorders.
Methodology
The committee panel was made up of experts jointly selected by the European Blood and Marrow Transplantation (EBMT) Inborn Error Working Party and the Paediatric Diseases Working Party. It was composed of 20 members, with recognized clinical and scientific experience in HSCT and/or medical management of TM and SCD. Members came from 18 institutions in 8 countries.
Pertinent published literature was identified from a search using the terms “TRANSPLANTATION AND THALASSAEMIA” or “SICKLE CELL DISEASE” using the National Library of Medicine PubMed database. In addition, abstracts from recent international hematology and annual stem cell transplant meetings (ASH, EBMT, and CIBMTR), meetings of the EBMT Inborn Errors and Paediatric Disease working parties and other educational meetings were also reviewed for relevance, supplemented by unpublished data from HSCT centers in Europe. The consensus meeting was prepared in the six months preceding the meeting by two working groups for TM (chaired by EA) and for SCD (chaired by SM). The two working groups assessed and weighted evidence using the GRADe approach for the two diseases separately from the abovementioned sources.9 Each member of the respective working group received a draft statement on the different topics, and checked and revised the suggested recommendations. During the final joint working group meeting, the recommendation statements were discussed and approved. Subsequently, a draft manuscript was sent to all committee members and was finally accepted by all of them. Any industrial influence on the process of consensus development was avoided.
Thalassemia major
HSCT in children and adolescents
Thirty years have elapsed since the first HSCT was performed for patients with TM, and allogeneic transplantation in TM is now accepted as standard clinical practice.10 In the 1980s and early 1990s, more than 1000 TM patients were transplanted at the transplant center in Pesaro, Italy. They reported a 20-year probability of thalassemia-free survival of 73% in 900 consecutive unselected patients transplanted from an HLA-identical sibling donor.11 Subsequently, several centers worldwide have started their own transplant programs.12–29 Recent results show that, with modern transplantation approaches, and careful patient selection, even better results could be obtained (Table 2). At the same time, however, survival without transplantation of TM patients has improved as the result both of a better understanding of the pathophysiology of iron overload and improvements in the medical therapy of TM; survival into the fourth or fifth decade of life is now possible for well-treated patients.30 Moreover, the first successful use of the long-awaited gene therapy approach has recently been reported, and the possibility that in a not so remote future gene therapy will become available for TM patients must also be included in the evaluation.31 However, the availability of optimal conventional medical therapy and the prospect of gene therapy are both limited to industrialized countries with longstanding experience where only a minority of TM patients live. Thus, at present, transplantation remains the only available curative approach for TM, but its role needs to be carefully evaluated, particularly in a global setting.
Table 2.
Recent reports on matched sibling donor HSCT in children and adults with thalassaemia major.
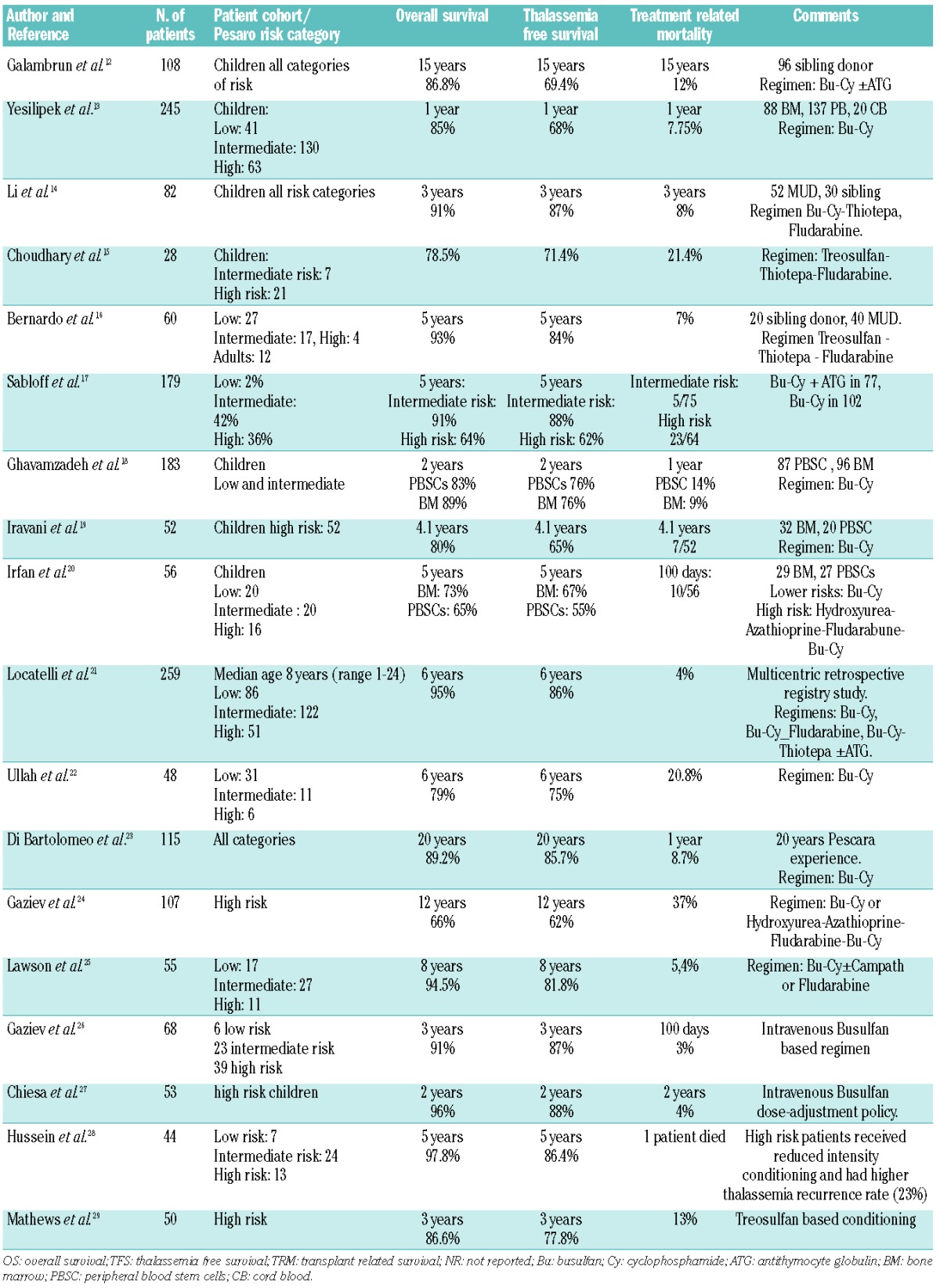
The Pesaro experience has clearly indicated that patient status at time of transplantation is the critical element predicting outcome. This was formalized with the identification and the adoption in the clinical practice of three risk factors and three risk classes.32 In the last decade, almost all transplant centers have followed this simple classification to predict the risks and benefits of HSCT in TM patients and perform HSCT in the first years of life before iron-related complications have developed.
In addition to this, transplantation techniques have improved and transplantation-related mortality (TRM) has fallen to 5% or even lower in young low-risk children transplanted from an HLA-matched sibling donor (MSD) (Table 2). In a large EBMT survey of 1061 cases of MSD-transplantation performed in the last decade, overall survival (OS) and disease-free survival (DFS) were 91±0.01 and 83±0.01 months, respectively. Moreover, in this report there was a significant age threshold of 14 years for optimal results (96% vs. 82% and 86% vs. 74% for OS and DFS, respectively).33
The use of peripheral blood stem cell transplantation (PBSCT) from MSD instead of bone marrow (BM) has been proposed to prevent graft failure in TM. Four studies report this type of transplant in TM. Overall, 886 patients receiving PBSCT have been described with substantially the same conclusions: the procedure is feasible in high-risk patients, with one study showing some advantages of PBSCT, but three studies showed an increased risk of chronic graft-versus-host disease (GvHD).18–20 Recently, the fourth study reported from a Chinese group demonstrated no difference in terms of rate of acute and chronic GvHD using a novel conditioning regimen.14
In 2003, Locatelli et al. first reported the feasibility of using HLA-identical sibling cord blood (CB) for HSCT in TM.34 This kind of allograft was associated with a decreased risk of both acute GvHD (aGvHD) and chronic GvHD (cGvHD) and of TRM, provided the CB unit had an adequate number of nucleated cells (i.e. > 3.5×107/kg). A large, international study retrospectively comparing CB to BM recipients has recently been completed. Analyses showed that OS, DFS, acute and chronic GvHD were 95%, 88%, 20%, and 12%, respectively for BM recipients (n=389), and 96%, 81%, 10%, and 5%, respectively for CB recipients (n=70). In this cohort, the cell dose (median 3.9×107/kg) did not influence outcome of patients given cord blood.21
Recommendations
Young TM patients with an available HLA identical sibling should be offered HSCT as soon as possible before development of iron overload and iron-related tissue damage.
Transplant-related risk factors should be evaluated according to the Pesaro risk score.
HLA genoidentical CB and BM are equally effective stem cell sources.
PBSCT should be avoided because of the increased risk of cGVHD.
HSCT in adult patients
Experience of HSCT in adult patients remains very limited, with very few centers performing HSCT in patients over the age of 18 years, and with TRM being persistently around 25%.24
The Pesaro experience demonstrated that constant control of iron overload is the main factor determining transplant outcome.35 As medical therapy of TM has improved substantially over the last years, and, therefore, nowadays adult patients are in a much better condition compared to those who underwent HSCT in the past, outcome after HSCT should also improve.36,37
Recommendations
HSCT in adults who have been well-chelated since infancy should be offered within controlled trials.
Assessment of clinical condition according to the Pesaro risk score and adequate transfusions/chelation regimen are the major issues to be evaluated before deciding to perform HSCT.
Use of donors other than an HLA matched sibling
On average, 25–30% of patients have an available MSD. However, in China, because of the ‘one-child’ policy, the possibility of having an MSD will remain unchanged. In contrast, in countries with large families, the likelihood of finding an MSD could be as high as 60–70%.38
So far, the experience of HSCT from HLA-disparate relatives is limited and the results are much inferior to those obtained with an HLA-identical sibling as donor.39 Recently, important results have been published from a small series using related donors who were not MSD (11 pheno-identical and 5 1-antigen mismatched related donors) using a pre-conditioning phase with hydroxyurea, azathioprine and fludarabine and a conditioning regimen including busulfan, thiotepa, cyclophosphamide and rabbit ATG. In this series, TM-free survival was 94%.40 The option of a haploidentical related donor has been explored in a limited series (n=22) of heterogeneous TM patients using a haploidentical related donor and a “megadose” of positively-selected CD34+ cells.41
Recommendations
HSCT from an HLA-mismatched family member in TM should still be considered an experimental approach and has to be conducted in the context of well-designed controlled trials.
HLA typing of of the entire family is advisable. HSCT from an HLA-phenotypically-identical donor is an option to be performed in expert centers.
Matched unrelated donors
A number of studies have shown that unrelated-donor (UD)-HSCT can cure a large proportion of patients with TM, provided that the UD is selected using high-resolution molecular typing for both HLA class I and II molecules and according to stringent criteria of compatibility with the recipient (i.e. identity or single allelic disparity for the loci for HLA-A, B, C, DRB1, and DQB1 loci).42 Using this approach, a suitable donor can be found in approximately one-third of Caucasian patients with TM; a higher probability could be expected in China because of the relatively genetic homogeneity of the Chinese population.14 In addition, the risk of rejection can be reduced by selecting UD who do not have non-permissive mismatches at HLA-DPB1 locus in the host-versus-graft direction.43
Recommendations
If a well-matched UD is available, allogeneic HSCT is a suitable option for a child with life-long control of iron overload and absence of iron-related tissue complications.
The UD must be selected using high-resolution molecular typing for both HLA class I and II loci, and according to stringent criteria of compatibility with the recipient.
Unrelated cord blood transplantation
The use of unrelated cord blood transplantation (UCB) as source of stem cells in TM has not been explored in well-designed clinical trials. Evaluation of reported results is hampered by differences in single center experience, conditioning regimens and accepted degree of HLA disparity. Recently, Jaing et al. reported results of UCBT in 35 TM patients. OS was found to be 88%, while DFS was 74%. The cumulative incidence of TRM was 11%. These remarkably good results are likely attributable to the high number of total nucleated cells/kg infused (7.8×107/kg).44 Combining data from 3 different registries, Ruggeri et al. found the outcome of UCBT in TM to be much less favorable; in 35 TM patients, an OS of 62% with a DFS of only 21% was reported. The cumulative incidence of graft failure was 52%.45
Recommendations
Unrelated cord blood transplantation must be performed in the context of well-controlled clinical trials in centers with specific UCBT programs.
Transplant management
Conditioning regimen
The biological aspects of allogeneic HSCT in TM are different from those for hematologic malignancies. There is no necessity to eradicate a malignant clone and the graft-versus-tumor effect is not required.11 Moreover, TM patients have not received previous chemotherapy and their immunological system is not impaired. TM patients have a hypercellular and expanded marrow.46 Additionally, in adolescent or adults undergoing HSCT, sensitization to red blood cell antigens may have occurred, possibly together with development of anti-HLA antibodies.47 In these circumstances, the ideal conditioning regimen should be capable of eradicating the diseased marrow and be sufficiently immunosuppressive to permit a sustained engraftment. For many years, the preferred regimen included oral busulfan (Bu) at 14 mg/kg and cyclophosphamide (Cy) at 120–200 mg/kg. The addition of azathioprine, hydroxyurea and fludarabine to the BuCy regimen made an important contribution to improving the results in high-risk patients.48 In the last decade, new conditioning regimens for TM patients have been introduced with improved results, such as intravenous Bu, or treosulfan associated with thiotepa and fludarabine.14,16 Particularly fludarabine has been included in the conditioning regimen by several groups in the last decade with low TRM and reduced rejection risk (Table 2).
To overcome the non-negligible TRM and morbidity, especially in high-risk or adult patients, reduced intensity conditioning (RIC) regimens have been tested.49,50 Although transplant-related toxicity was minimal, many patients showed only transient and incomplete engraftment, and most ultimately developed graft rejection, thus indicating that TM patients need a more intensive, myeloablative conditioning regimen (MAC).29,51 However, to avoid the well-known late effects of radiotherapy on the growing organism, and in particular the risk of secondary malignancies, irradiation should not be an option for nonmalignant disorders.52
A newer experience has shown that approximately 11% of transplanted patients develop long-term, stable mixed chimerism (MC) after HSCT.53 This percentage is higher in patients given CB transplantation from an HLA-identical relative.54 MC patients, despite the limited engraftment (even if no higher than 20%), may achieve a functioning graft status characterized by a normal hemoglobin level, no red blood cell transfusion requirement, no increment in iron stores, and a limited degree of erythroid hyperplasia which is not of any clinical relevance.55 Thus the disease can be adequately controlled without complete eradication of the pathological hematopoiesis. Another tool to prevent graft failure could be the systematic use of antithymocyte globulin (ATG) in addition to an MAC.12
Recommendations
MAC without irradiation should always be used for standard transplantation. In case of BU-containing regimen, intravenous formulation should be used.
Reduced toxicity regimens are under investigation and are to be used in the context of clinical trials.
Any prospective attempt to induce stable MC should be considered experimental.
Graft-versus-host disease prophylaxis
The preferred GvHD prophylaxis in the majority of published studies of HSCT from matched sibling donors consisted of cyclosporine and methotrexate (4 doses intravenous (IV) on Days +1, +3, +6 and +11 post transplantation).48 The addition of ATG to this regimen was successfully used both in HLA-identical sibling HSCT recipients and, especially, in those transplanted from an unrelated volunteer or an HLA-partially matched relative.56,57
Recommendations
The combination of cyclosporine and methotrexate represents the gold standard for GvHD prophylaxis for HSCT from MSD.
Mono- or polyclonal antibodies like ATG or alemtuzumab could contribute to better prevent rejection and GVHD in the context of MSD HSCT and should be explored in prospective trials.
These antibodies should be routinely used for GVHD prevention in non-sibling HSCT.
Sickle cell disease
Sickle cell disease (SCD) is associated with substantial morbidity, leading to both reduced quality of life and shortened life expectancy.58–63 Survival has improved significantly in the last two decades and 94% of children with SCD now survive until the age of 18 years thanks to better surveillance, pneumoccocus vaccination, penicillin prophylaxis and treatment with hydroyurea.64,65 However, mortality is still significant once patients reach adulthood.61,66
SCD-associated morbidity and mortality in young adults is largely due to as yet unpreventable complications such as priapism, avascular necrosis, chronic pulmonary impairment, hypertension, stroke and recurrent venoocclusive crises.58,67–70
The only curative approach for SCD is HSCT. Historically, the indication for HSCT in SCD was mainly based on SCD-associated morbidity: the sicker the child, the stronger the indication.71 With the reduction in TRM in recent years, and with the increasing knowledge of the severity of complications in untreated patients, the accepted indications for HSCT have become less restrictive.72,73
Matched sibling transplantation
In the last ten years, the outcome data of more than 200 patients who underwent HSCT from an HLA-matched sibling donor have been analyzed and published. OS was found to be around 95%.72–75 Bernaudin et al. reported 121 patients transplanted in France after 2000 with a DFS of 95% at three years.76 Taking into account that: 1) overall survival has become equal or even better in patients who undergo HSCT compared to those on supportive treatment, and disease-free survival is over 95%; 2) disease-related morbidity and mortality are increasing with age, and event-free survival following HSCT is significantly better in patients transplanted before developing SCD-related organ damage; and 3) in children, TRM increases with age, the use of early HSCT is justified in children with any SCD-associated symptoms or event.5,64,77 Young adults who experience more severe disease as they get older also benefit from HSCT.78 However, severe chronic GvHD has been described following HSCT in heavily-transfused end-stage adult SCD patients.79
The main obstacle for HSCT in patients with SCD is the restricted donor availability. Overall, only 18% of patients with SCD have a healthy matched sibling donor, and the probability of finding a matched unrelated donor is extremely low.80
It has been shown that the incidence of chronic GvHD following matched sibling HSCT for non-malignant diseases is significantly higher in patients receiving PBSC as compared to BM.81,82 This justifies the recommendation that BM be employed for transplanting SCD patients. Very promising results with a low rejection rate and a low risk of GvHD in patients with SCD transplanted with related UCB have been published by Locatelli et al.34
Recommendations
Young patients with symptomatic SCD who have an HLA-matched sibling donor should be transplanted as early as possible, preferably at pre-school age.
Unmanipulated BM or UCB (whenever available) from matched sibling donors are the recommended stem cell source.
Alternative donor transplantation
Even if results of matched unrelated HSCT can be expected to be comparable to those obtained in TM, due to the lack of matched donors in the registries there are no firm data on outcome of HSCT from UD for SCD, and, therefore, the advantages and disadvantages of this option cannot be adequately addressed.
For many patients, however, a 4/6 or higher HLA-matched unrelated CB unit can be found. In France, a large effort is being undertaken to set up a programme of ‘tailored’ CB banking in SCD-affected families. The CB units not HLA compatible with the affected sibling are cryopreserved so that to establish representation of the ethnic minority (patients of African descent) in the French CB banks (M Cavazzana, personal communication, 2014). So far, a total of 32 patients with SCD transplanted with an UCB unit have been published, 18 of them following an RIC regimen, with an overall survival of 91%, but a disease-free survival of only 50% because of rejection.7,45,83,84 The Sickle Cell Unrelated Donor Transplant Trial (SCURT trial) of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN) was a phase II study on the toxicity and efficacy of UCB or BMT in children with severe SCD using RIC. Eight children with severe SCD underwent unrelated donor CB-transplantation following alemtuzumab, fludarabine, and melphalan. Three patients who engrafted had 100% donor cells by Day 100 which was sustained, while 5 patients had autologous hematopoietic recovery. Based upon this high incidence of graft rejection, enrolment into the CB arm of the SCURT trial was suspended. However, because this RIC has demonstrated a favorable safety profile, the trial remains open to enrolment for unrelated BMT.7 Another concern to be considered is the high incidence of chronic GvHD in the context of unrelated CBT.85
Recently, promising results from haploidentical HSCT following an RIC regimen have been published.86 However, for both unrelated CB and haploidentical HSCT, rejection remains a major obstacle in the context of RIC.
Recommendations
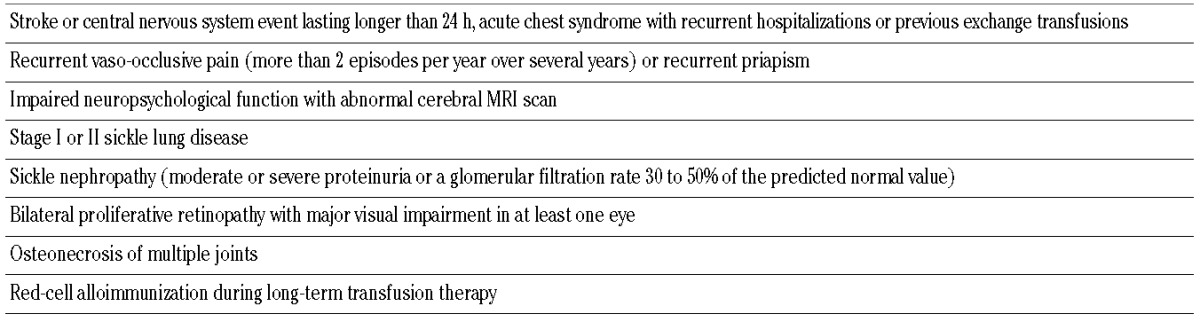
SCT from unrelated BM or CB donors should only be considered in the presence of at least one of the indications suggested by Walters et al.,71 and should be performed only in the context of controlled trials in experienced centers (Table 3).
Table 3.
Indication for allogeneic HSCT suggested by Walters et al.
Transplant management
Conditioning regimen
The most widely used myeloablative conditioning regimen for patients with SCD consists of busulfan 14–16 mg/kg and cyclophosphamide 200 mg/kg +/− ATG.34,73–75 Growth does not seem to be impaired following myeloablative conditioning as long as it is not performed near or during the adolescent growth spurt. However, the risks of infertility and secondary neoplasm are a major concern.34,74,87 Cryopreservation of sperm and ovarian tissue, respectively, has been proposed.88,89 HSCT following an RIC regimen is currently being evaluated in several centers. Small patient series using mainly fludarabine-based regimens have shown promising results with an OS of 100% and a DFS of 95%.78,90,91 The use of either alemtuzumab (Campath-1H) or ATG in the context of RIC seems to be indispensable for stable engraftment.92 In Austria, a cohort of 8 patients underwent matched sibling HSCT following conditioning with fludarabine, melphalan, thiotepa and ATG or Campath-1H with a disease-free survival of 100% and stable engraftment.93
Recommendations
The gold standard for conditioning in patients with SCD is busulfan, cyclophosphamide and ATG.
RIC regimen should be explored in and confirmed by prospective trials.
Graft-versus-host disease prophylaxis
A survey on patient perception of HSCT with RIC performed by Chakrabarti et al. showed that chronic GvHD was considered unacceptable for 80% of patients with SCD.94 With the use of ATG, cyclosporine A and MTX an incidence of over grade II aGvHD between 11% and 44% and an incidence of cGvHD between 6% and 44% has been reported.34,72,74 Two small series have been published on the combination of RIC and alemtuzumab and no aGvHD or cGVHD was reported.78,90 The use of alemtuzumab in the context of RIC seems, in effect, to be associated with a lower incidence of acute and chronic GvHD, but at the price of an increased incidence of viral complications.95
Recommendations
The use of ATG and post-transplantation cyclosporine A plus MTX is the gold standard for patients with SCD following myeloablative conditioning.
Follow-up evaluation and chimerism testing for TM and SCD
A standardized pre-transplantation assessment based on existing standards for TM and SCD care, and individualized re-assessment post transplantation, are a prerequisite for objective evaluation of the impact of HSCT in terms of improvement or reversal of pre-transplantation morbidity.
All TM and SCD-specific conditions must be addressed and treated if necessary (e.g. iron overload in TM and vascular impairment in SCD). As demonstrated, persistent MC does not impact DFS and OS either in TM or in SCD; however, chimerism should be regularly checked in both diseases after HSCT.34,75,78,90
Table 4.
Recommendation summary for HSCT in TM.
Table 5.
Recommendation summary for HSCT in SCD.

Recommendations
Post-transplantation evaluation and care should be undertaken in cooperation with hematologists experienced in TM or SCD.
Cost and cost effectiveness for TM and SCD patients
The care of TM, both by transplantation and medical therapy, has dramatically improved during recent decades. However, this improvement has been limited to patients living in industrialized countries, while the large majority of patients are born and live in other, non-industrialized countries.96 TM care is a complex, multidisciplinary and expensive tool requiring dedicated and experienced units. From a global health perspective, in some countries, TM represents an enormous burden of care. For example, in Cyprus it was calculated in the 1970s that in 40 years 80,000 units of blood would be needed annually to transfuse the country’s TM patients and the overall cost would exceed the total national health budget.97 An Italian study investigated the cost/benefit estimation from a societal perspective and quantified tariffs, expenses and net income in 2006 for TM patients. The mean costs were € 1242/patient/month, 55.5% attributed to iron chelation therapy and 33.2% attributable to transfusions.98 Moreover, proper medical therapy for TM requires advanced technologies such as cardiac or hepatic MRI.30,99 With the combined cost of blood transfusions, chelation and management of complications, the requirements for optimal thalassemia care clearly exceed the health resources available in most non-industrialized countries.
For SCD, it has been calculated that 50,000 patients in the USA need more than 100,000 hospital admissions per year with associated costs of approximately US$ 10,000 US/SCD patient/year.100 Kauf et al. calculated that the average lifetime cost of care was US$460,151/SCD patient. These data compare to the costs of HSCT from sibling donors for non-malignant diseases of US$ 112,000–150,000 US, which would translate into US$ 1900 US/expected life year in case of HSCT in infancy.101 In conclusion, for both TM and SCD, HSCT is cost-effective in comparison to life-long supportive therapy.
Conclusion
Hematopoietic stem cell transplantation currently remains the only available curative treatment for hemoglobinopathies. In contrast with some recommendations, our group strongly suggests early transplantation for TM and SCD if a suitable donor is available and if the patient can be treated in an experienced transplantation center. The development of improved supportive care, including transfusion services, chelation therapy and prevention of infectious complications, does not modify this position. However, much more uncertainty applies to the complex challenge of where to place the curative, but potentially lethal, HSCT treatment as an alternative to a medical, non-curative therapy in adults and patients with advanced disease. Transplantation outcomes today are much improved compared with the 1980s and 1990s, with more than 90% of patients surviving transplantation and more than 80% of them being disease-free after having been treated in a number of different centers worldwide, including centers outside industrialized countries. If in the future gene therapy could provide a cure, it needs to demonstrate at least equivalent results in terms of cost/benefit ratio with HSCT, which is today a widely applied, standard practice for the cure of hemoglobinopathies.
Acknowledgments
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals Corporation. The authors would like to thank Nene Anadu, PhD, for medical editorial assistance with this manuscript.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Weatherall DJ, Williams TN, Allen SJ, O’Donnell A. The population genetics and dynamics of the thalassemias. Hematol Oncol Clin North Am. 2010;24(6):1021–31 [DOI] [PubMed] [Google Scholar]
- 2.Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood. 2011;118(13):3479–88 [DOI] [PubMed] [Google Scholar]
- 3.Sharef SW, Al-Hajri M, Beshlawi I, Al-Shahrabally A, Elshinawy M, Zachariah M, et al. Optimizing Hydroxyurea use in children with sickle cell disease: low dose regimen is effective. Eur J Haematol. 2013;90(6):519–24 [DOI] [PubMed] [Google Scholar]
- 4.Angelucci E. Hematopoietic stem cell transplantation in thalassemia. Hematology Am Soc Hematol Educ Program. 2010;2010:456–62 [DOI] [PubMed] [Google Scholar]
- 5.Bernaudin F, Socie G, Kuentz M, Chevret S, Duval M, Bertrand Y, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110(7):2749–56 [DOI] [PubMed] [Google Scholar]
- 6.Omondi NA, Ferguson SE, Majhail NS, Denzen EM, Buchanan GR, Haight AE, et al. Barriers to Hematopoietic Cell Transplantation Clinical Trial Participation of African American and Black Youth With Sickle Cell Disease and Their Parents. J Pediatr Hematol Oncol. 2013;35(4):289–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamani NR, Walters MC, Carter S, Aquino V, Brochstein JA, Chaudhury S, et al. Unrelated donor cord blood transplantation for children with severe sickle cell disease: results of one cohort from the phase II study from the Blood and Marrow Transplant Clinical Trials Network (BMT CTN). Biol Blood Marrow Transplant. 2012;18(8):1265–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jagannath VA, Fedorowicz Z, Al Hajeri A, Hu N, Sharma A. Hematopoietic stem cell transplantation for people with ss-thalassaemia major. Cochrane Database Syst Rev. 2011;(10):CD008708. [DOI] [PubMed] [Google Scholar]
- 9.Guyatt GH, Oxman AD, Schunemann HJ, Tugwell P, Knottnerus A. GRADE guidelines: a new series of articles in the Journal of Clinical Epidemiology. J Clin Epidemiol. 2011;64(4):380–2 [DOI] [PubMed] [Google Scholar]
- 10.Thomas ED, Buckner CD, Sanders JE, Papayannopoulou T, Borgna-Pignatti C, De Stefano P, et al. Marrow transplantation for thalassaemia. Lancet. 1982;2(8292):227–9 [DOI] [PubMed] [Google Scholar]
- 11.Angelucci E, Baronciani D. Allogeneic stem cell transplantation for thalassemia major. Haematologica. 2008;93(12):1780–4 [DOI] [PubMed] [Google Scholar]
- 12.Galambrun C, Pondarre C, Bertrand Y, Loundou A, Bordigoni P, Frange P, et al. French multicenter 22-year experience in stem cell transplantation for beta-thalassemia major: lessons and future direc tions. Biol Blood Marrow Transplant. 2013;19(1):62–8 [DOI] [PubMed] [Google Scholar]
- 13.Yesilipek MA, Ertem M, Cetin M, Oniz H, Kansoy S, Tanyeli A, et al. HLA-matched family hematopoetic stem cell transplantation in children with beta thalassemia major: the experience of the Turkish Pediatric Bone Marrow Transplantation Group. Pediatr Transplant. 2012;16(8):846–51 [DOI] [PubMed] [Google Scholar]
- 14.Li C, Wu X, Feng X, He Y, Liu H, Pei F, et al. A novel conditioning regimen improves outcomes in beta-thalassemia major patients using unrelated donor peripheral blood stem cell transplantation. Blood. 2012;120(19):3875–81 [DOI] [PubMed] [Google Scholar]
- 15.Choudhary D, Sharma SK, Gupta N, Kharya G, Pavecha P, Handoo A, et al. Treosulfan-thiotepa-fludarabine-based conditioning regimen for allogeneic transplantation in patients with thalassemia major: a single-center experience from north India. Biol Blood Marrow Transplant. 2013;19(3):492–5 [DOI] [PubMed] [Google Scholar]
- 16.Bernardo ME, Piras E, Vacca A, Giorgiani G, Zecca M, Bertaina A, et al. Allogeneic hematopoietic stem cell transplantation in thalassemia major: results of a reduced-toxicity conditioning regimen based on the use of treosulfan. Blood. 2012;120(2):473–6 [DOI] [PubMed] [Google Scholar]
- 17.Sabloff M, Chandy M, Wang Z, Logan BR, Ghavamzadeh A, Li CK, et al. HLA-matched sibling bone marrow transplantation for beta-thalassemia major. Blood. 2011;117(5):1745–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghavamzadeh A, Iravani M, Ashouri A, Mousavi SA, Mahdavi N, Shamshiri A, et al. Peripheral blood versus bone marrow as a source of hematopoietic stem cells for allogeneic transplantation in children with class I and II beta thalassemia major. Biol Blood Marrow Transplant. 2008;14(3):301–8 [DOI] [PubMed] [Google Scholar]
- 19.Iravani M, Tavakoli E, Babaie MH, Ashouri A, Khatami F, Ghavamzadeh A. Comparison of peripheral blood stem cell transplant with bone marrow transplant in class 3 thalassemic patients. Exp Clin Transplant. 2010;8(1):66–73 [PubMed] [Google Scholar]
- 20.Irfan M, Hashmi K, Adil S, Shamsi T, Farzana T, Ansari S, et al. Beta-thalassaemia major: bone marrow versus peripheral blood stem cell transplantation. J Pak Med Assoc. 2008;58(3):107–10 [PubMed] [Google Scholar]
- 21.Locatelli F, Kabbara N, Ruggeri A, Ghavamzadeh A, Roberts I, Li CK, et al. Outcome of patients with hemoglobinopathies given either cord blood or bone marrow transplantation from an HLA-identical sibling. Blood. 2013;122(6):1072–8 [DOI] [PubMed] [Google Scholar]
- 22.Ullah K, Khan B, Raza S, Ahmed P, Satti TM, Butt T, et al. Bone marrow transplant cure for beta-thalassaemia major: initial experience from a developing country. Ann Hematol. 2008;87(8):655–61 [DOI] [PubMed] [Google Scholar]
- 23.Di Bartolomeo P, Santarone S, Di Bartolomeo E, Olioso P, Bavaro P, Papalinetti G, et al. Long-term results of survival in patients with thalassemia major treated with bone marrow transplantation. Am J Hematol. 2008;83(7):528–30 [DOI] [PubMed] [Google Scholar]
- 24.Gaziev J, Sodani P, Polchi P, Andreani M, Lucarelli G. Bone marrow transplantation in adults with thalassemia: Treatment and long-term follow-up. Ann NY Acad Sci. 2005;1054:196–205 [DOI] [PubMed] [Google Scholar]
- 25.Lawson SE, Roberts IA, Amrolia P, Dokal I, Szydlo R, Darbyshire PJ. Bone marrow transplantation for beta-thalassaemia major: the UK experience in two paediatric centres. Br J Haematol. 2003;120(2):289–95 [DOI] [PubMed] [Google Scholar]
- 26.Gaziev J, Nguyen L, Puozzo C, Mozzi AF, Casella M, Perrone Donnorso M, et al. Novel pharmacokinetic behavior of intravenous busulfan in children with thalassemia undergoing hematopoietic stem cell transplantation: a prospective evaluation of pharmacokinetic and pharmacodynamic profile with therapeutic drug monitoring. Blood. 2010;115(22):4597–604 [DOI] [PubMed] [Google Scholar]
- 27.Hussein AA, Al-Zaben A, Ghatasheh L, Natsheh A, Hammada T, Abdel-Rahman F, et al. Risk adopted allogeneic hematopoietic stem cell transplantation using a reduced intensity regimen for children with thalassemia major. Pediatr Blood Cancer. 2013;60(8):1345–9 [DOI] [PubMed] [Google Scholar]
- 28.Chiesa R, Cappelli B, Crocchiolo R, Frugnoli I, Biral E, Noe A, et al. Unpredictability of intravenous busulfan pharmacokinetics in children undergoing hematopoietic stem cell transplantation for advanced beta thalassemia: limited toxicity with a dose-adjustment policy. Biol Blood Marrow Transplant. 2010;16(5):622–8 [DOI] [PubMed] [Google Scholar]
- 29.Mathews V, George B, Viswabandya A, Abraham A, Ahmed R, Ganapule A, et al. Improved Clinical Outcomes of High Risk beta Thalassemia Major Patients Undergoing a HLA Matched Related Allogeneic Stem Cell Transplant with a Treosulfan Based Conditioning Regimen and Peripheral Blood Stem Cell Grafts. PLoS One. 2013;8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Angelucci E, Barosi G, Camaschella C, Cappellini MD, Cazzola M, Galanello R, et al. Italian Society of Hematology practice guidelines for the management of iron overload in thalassemia major and related disorders. Haematologica. 2008;93(5):741–52 [DOI] [PubMed] [Google Scholar]
- 31.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467(7313):318–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lucarelli G, Galimberti M, Polchi P, Angelucci E, Baronciani D, Giardini C, et al. Marrow transplantation in patients with thalassemia responsive to iron chelation therapy. N Engl J Med. 1993;329(12):840–4 [DOI] [PubMed] [Google Scholar]
- 33.Baronciani D, Pilo F, Lyon-Caen S, Poetschger U, Dini G, Peters C. Hematopoietic Stem Cell Transplantation in Thalassemia Major. Report from the EBMT Hemoglobinopathy Registry. ASH Annual Meeting; 2011 [Google Scholar]
- 34.Locatelli F, Rocha V, Reed W, Bernaudin F, Ertem M, Grafakos S, et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood. 2003;101(6):2137–43 [DOI] [PubMed] [Google Scholar]
- 35.Angelucci E. Hematopoietic stem cell transplantation in thalassemia. Hematology Am Soc Hematol Educ Program. 2010:456–62 [DOI] [PubMed] [Google Scholar]
- 36.Muretto P, Angelucci E, Lucarelli G. Reversibility of cirrhosis in patients cured of thalassemia by bone marrow transplantation. Ann Intern Med. 2002;136(9):667–72 [DOI] [PubMed] [Google Scholar]
- 37.Taher AT, Porter J, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, et al. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood. 2012;120(5):970–7 [DOI] [PubMed] [Google Scholar]
- 38.Jawdat DM, Al Saleh S, Sutton P, Al Anazi H, Shubaili A, Tamim H, et al. Chances of finding an HLA-matched sibling: The Saudi experience. Biol Blood Marrow Transplant. 2009;15(10):1342–4 [DOI] [PubMed] [Google Scholar]
- 39.Gaziev D, Galimberti M, Lucarelli G, Polchi P, Giardini C, Angelucci E, et al. Bone marrow transplantation from alternative donors for thalassemia: HLA-phenotypically identical relative and HLA-nonidentical sibling or parent transplants. Bone Marrow Transplant. 2000;25(8):815–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaziev J, Marziali M, Isgro A, Sodani P, Paciaroni K, Gallucci C, et al. Bone marrow transplantation for thalassemia from alternative related donors: improved outcomes with a new approach. Blood. 2013;122(15):2751–6 [DOI] [PubMed] [Google Scholar]
- 41.Sodani P, Isgro A, Gaziev J, Polchi P, Paciaroni K, Marziali M, et al. Purified T-depleted, CD34+ peripheral blood and bone marrow cell transplantation from haploidentical mother to child with thalassemia. Blood. 2010;115(6):1296–302 [DOI] [PubMed] [Google Scholar]
- 42.Flomenberg N, Baxter-Lowe LA, Confer D, Fernandez-Vina M, Filipovich A, Horowitz M, et al. Impact of HLA class I and class II high-resolution matching on outcomes of unrelated donor bone marrow transplantation: HLA-C mismatching is associated with a strong adverse effect on transplantation outcome. Blood. 2004;104(7):1923–30 [DOI] [PubMed] [Google Scholar]
- 43.Fleischhauer K, Locatelli F, Zecca M, Orofino MG, Giardini C, De Stefano P, et al. Graft rejection after unrelated donor hematopoietic stem cell transplantation for thalassemia is associated with nonpermissive HLA-DPB1 disparity in host-versus-graft direction. Blood. 2006;107(7):2984–92 [DOI] [PubMed] [Google Scholar]
- 44.Jaing TH, Hung IJ, Yang CP, Chen SH, Chung HT, Tsay PK, et al. Unrelated cord blood transplantation for thalassaemia: a single-institution experience of 35 patients. Bone Marrow Transplant. 2012;47(1):33–9 [DOI] [PubMed] [Google Scholar]
- 45.Ruggeri A, Eapen M, Scaravadou A, Cairo MS, Bhatia M, Kurtzberg J, et al. Umbilical cord blood transplantation for children with thalassemia and sickle cell disease. Biol Blood Marrow Transplant. 2011;17(9):1375–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Centis F, Tabellini L, Lucarelli G, Buffi O, Tonucci P, Persini B, et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with beta-thalassemia major. Blood. 2000;96(10):3624–9 [PubMed] [Google Scholar]
- 47.Thompson AA, Cunningham MJ, Singer ST, Neufeld EJ, Vichinsky E, Yamashita R, et al. Red cell alloimmunization in a diverse population of transfused patients with thalassaemia. Br J Haematol. 2011;153(1):121–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sodani P, Gaziev D, Polchi P, Erer B, Giardini C, Angelucci E, et al. New approach for bone marrow transplantation in patients with class 3 thalassemia aged younger than 17 years. Blood. 2004;104(4):1201–3 [DOI] [PubMed] [Google Scholar]
- 49.Locatelli F, De Stefano P. Innovative approaches to hematopoietic stem cell transplantation for patients with thalassemia. Haematologica. 2005;90(12):1592–4 [PubMed] [Google Scholar]
- 50.Lucarelli G, Gaziev J. Advances in the allogeneic transplantation for thalassemia. Blood Rev. 2008;22(2):53–63 [DOI] [PubMed] [Google Scholar]
- 51.Hussein AA, Al-Zaben A, Ghatasheh L, Natsheh A, Hammada T, Abdel-Rahman F, et al. Risk Adopted Allogeneic Hematopoietic Stem Cell Transplantation Using a Reduced Intensity Regimen for Children with Thalassemia Major. Pediatr Blood Cancer. 2013;19(10):24493. [DOI] [PubMed] [Google Scholar]
- 52.Baker KS, DeFor TE, Burns LJ, Ramsay NK, Neglia JP, Robison LL. New malignancies after blood or marrow stem-cell transplantation in children and adults: incidence and risk factors. J Clin Oncol. 2003;21(7):1352–8 [DOI] [PubMed] [Google Scholar]
- 53.Andreani M, Nesci S, Lucarelli G, Tonucci P, Rapa S, Angelucci E, et al. Long-term survival of ex-thalassemic patients with persistent mixed chimerism after bone marrow transplantation. Bone Marrow Transplant. 2000;25(4):401–4 [DOI] [PubMed] [Google Scholar]
- 54.Lisini D, Zecca M, Giorgiani G, Montagna D, Cristantielli R, Labirio M, et al. Donor/recipient mixed chimerism does not predict graft failure in children with beta-thalassemia given an allogeneic cord blood transplant from an HLA-identical sibling. Haematologica. 2008;93(12):1859–67 [DOI] [PubMed] [Google Scholar]
- 55.Andreani M, Testi M, Gaziev J, Condello R, Bontadini A, Tazzari PL, et al. Quantitatively different red cell/nucleated cell chimerism in patients with long-term, persistent hematopoietic mixed chimerism after bone marrow transplantation for thalassemia major or sickle cell disease. Haematologica. 2011;96(1):128–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sauer M, Bettoni C, Lauten M, Ghosh A, Rehe K, Grigull L, et al. Complete substitution of cyclophosphamide by fludarabine and ATG in a busulfan-based preparative regimen for children and adolescents with beta-thalassemia. Bone Marrow Transplant. 2005;36(5):383–7 [DOI] [PubMed] [Google Scholar]
- 57.Goussetis E, Peristeri I, Kitra V, Vessalas G, Paisiou A, Theodosaki M, et al. HLA-matched sibling stem cell transplantation in children with beta-thalassemia with anti-thymocyte globulin as part of the preparative regimen: the Greek experience. Bone Marrow Transplant. 2012;47(8):1061–6 [DOI] [PubMed] [Google Scholar]
- 58.Klings ES, Wyszynski DF, Nolan VG, Steinberg MH. Abnormal pulmonary function in adults with sickle cell anemia. Am J Respir Crit Care Med. 2006;173(11):1264–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McClish DK, Penberthy LT, Bovbjerg VE, Roberts JD, Aisiku IP, Levenson JL, et al. Health related quality of life in sickle cell patients: the PiSCES project. Health Qual Life Outcomes. 2005;3:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ngo C, Kayem G, Habibi A, Benachi A, Goffinet F, Galacteros F, et al. Pregnancy in sickle cell disease: maternal and fetal outcomes in a population receiving prophylactic partial exchange transfusions. Eur J Obstet Gynecol Reprod Biol. 2010;152(2):138–42 [DOI] [PubMed] [Google Scholar]
- 61.van der Plas EM, van den Tweel XW, Geskus RB, Heijboer H, Biemond BJ, Peters M, et al. Mortality and causes of death in children with sickle cell disease in the Netherlands, before the introduction of neonatal screening. Br J Haematol. 2011;155(1):106–10 [DOI] [PubMed] [Google Scholar]
- 62.Wang WC. Sickle-cell disease and compromised cognition. Pediatr Blood Cancer. 2011;56(5):705–6 [DOI] [PubMed] [Google Scholar]
- 63.McCavit TL, Xuan L, Zhang S, Flores G, Quinn CT. Hospitalization for invasive pneumococcal disease in a national sample of children with sickle cell disease before and after PCV7 licensure. Pediatr Blood Cancer. 2012;58(6):945–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115(17):3447–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dowling MM, Lee N, Quinn CT, Rogers ZR, Boger D, Ahmad N, et al. Prevalence of intracardiac shunting in children with sickle cell disease and stroke. J Pediatr. 2010;156(4):645–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nouraie M, Lee JS, Zhang Y, Kanias T, Zhao X, Xiong Z, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica. 2013;98(3):464–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377(9778):1663–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–44 [DOI] [PubMed] [Google Scholar]
- 69.Switzer JA, Hess DC, Nichols FT, Adams RJ. Pathophysiology and treatment of stroke in sickle-cell disease: present and future. Lancet Neurol. 2006;5(6):501–12 [DOI] [PubMed] [Google Scholar]
- 70.Mukisi-Mukaza M, Saint Martin C, Etienne-Julan M, Donkerwolcke M, Burny ME, Burny F. Risk factors and impact of orthopaedic monitoring on the outcome of avascular necrosis of the femoral head in adults with sickle cell disease: 215 patients case study with control group. Orthop Traumatol Surg Res. 2011;97(8):814–20 [DOI] [PubMed] [Google Scholar]
- 71.Walters MC, Patience M, Leisenring W, Eckman JR, Scott JP, Mentzer WC, et al. Bone marrow transplantation for sickle cell disease. N Engl J Med. 1996;335(6):369–76 [DOI] [PubMed] [Google Scholar]
- 72.Majumdar S, Robertson Z, Robinson A, Starnes S, Iyer R, Megason G. Outcome of hematopoietic cell transplantation in children with sickle cell disease, a single center’s experience. Bone Marrow Transplant. 2010;45(5):895–900 [DOI] [PubMed] [Google Scholar]
- 73.Panepinto JA, Walters MC, Carreras J, Marsh J, Bredeson CN, Gale RP, et al. Matched-related donor transplantation for sickle cell disease: report from the Center for International Blood and Transplant Research. Br J Haematol. 2007;137(5):479–85 [DOI] [PubMed] [Google Scholar]
- 74.Brachet C, Heinrichs C, Tenoutasse S, Devalck C, Azzi N, Ferster A. Children with sickle cell disease: growth and gonadal function after hematopoietic stem cell transplantation. J Pediatr Hematol Oncol. 2007;29(7):445–50 [DOI] [PubMed] [Google Scholar]
- 75.McPherson ME, Hutcherson D, Olson E, Haight AE, Horan J, Chiang KY. Safety and efficacy of targeted busulfan therapy in children undergoing myeloablative matched sibling donor BMT for sickle cell disease. Bone Marrow Transplant. 2011;46(1):27–33 [DOI] [PubMed] [Google Scholar]
- 76.Bernaudin F, Robin M, Ferry C, Yacouben K, Dalle JH, Peffault de Latour R, et al. Related mmyeloablative stem cell transplantation (SCT) to cure sickle cell anemia (SCA): update of French results. Blood. 2010;116(3518). [Google Scholar]
- 77.Matthes-Martin S, Potschger U, Bergmann K, Frommlet F, Brannath W, Bauer P, et al. Risk-adjusted outcome measurement in pediatric allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2008;14(3):335–43 [DOI] [PubMed] [Google Scholar]
- 78.Hsieh MM, Kang EM, Fitzhugh CD, Link MB, Bolan CD, Kurlander R, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med. 2009;361(24):2309–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van Besien K, Bartholomew A, Stock W, Peace D, Devine S, Sher D, et al. Fludarabine-based conditioning for allogeneic transplantation in adults with sickle cell disease. Bone Marrow Transplant. 2000; 26(4):445–9 [DOI] [PubMed] [Google Scholar]
- 80.Krishnamurti L, Abel S, Maiers M, Flesch S. Availability of unrelated donors for hematopoietic stem cell transplantation for hemoglobinopathies. Bone Marrow Transplant. 2003;31(7):547–50 [DOI] [PubMed] [Google Scholar]
- 81.Eapen M, Horowitz MM, Klein JP, Champlin RE, Loberiza FR, Jr, Ringden O, et al. Higher mortality after allogeneic peripheral-blood transplantation compared with bone marrow in children and adolescents: the Histocompatibility and Alternate Stem Cell Source Working Committee of the International Bone Marrow Transplant Registry. J Clin Oncol. 2004;22(24):4872–80 [DOI] [PubMed] [Google Scholar]
- 82.Levine JE, Wiley J, Kletzel M, Yanik G, Hutchinson RJ, Koehler M, et al. Cytokine-mobilized allogeneic peripheral blood stem cell transplants in children result in rapid engraftment and a high incidence of chronic GVHD. Bone Marrow Transplant. 2000;25(1):13–8 [DOI] [PubMed] [Google Scholar]
- 83.Adamkiewicz TV, Szabolcs P, Haight A, Baker KS, Staba S, Kedar A, et al. Unrelated cord blood transplantation in children with sickle cell disease: review of four-center experience. Pediatr Transplant. 2007;11(6):641–4 [DOI] [PubMed] [Google Scholar]
- 84.Mazur M, Kurtzberg J, Halperin E, Ciocci G, Szabolcs P. Transplantation of a child with sickle cell anemia with an unrelated cord blood unit after reduced intensity conditioning. J Pediatr Hematol Oncol. 2006;28(12):840–4 [DOI] [PubMed] [Google Scholar]
- 85.Fernandes JF, Rocha V, Labopin M, Neven B, Moshous D, Gennery AR, et al. Transplantation in patients with SCID: mismatched related stem cells or unrelated cord blood¿ Blood. 2012;119(12):2949–55 [DOI] [PubMed] [Google Scholar]
- 86.Bolanos-Meade J, Fuchs EJ, Luznik L, Lanzkron SM, Gamper CJ, Jones RJ, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120(22):4285–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eggleston B, Patience M, Edwards S, Adamkiewicz T, Buchanan GR, Davies SC, et al. Effect of myeloablative bone marrow transplantation on growth in children with sickle cell anaemia: results of the multicenter study of haematopoietic cell transplantation for sickle cell anaemia. Br J Haematol. 2007;136(4):673–6 [DOI] [PubMed] [Google Scholar]
- 88.Jadoul P, Donnez J. How does bone marrow transplantation affect ovarian function and fertility¿ Curr Opin Obstet Gynecol. 2012;24(3):164–71 [DOI] [PubMed] [Google Scholar]
- 89.Kuentz M, Robin M, Dhedin N, Hicheri Y, de Latour RP, Rohrlich P, et al. Is there still a place for myeloablative regimen to transplant young adults with sickle cell disease¿ Blood. 2011;118(16):4491–2 [DOI] [PubMed] [Google Scholar]
- 90.Horwitz ME, Spasojevic I, Morris A, Telen M, Essell J, Gasparetto C, et al. Fludarabine-based nonmyeloablative stem cell transplantation for sickle cell disease with and without renal failure: clinical outcome and pharmacokinetics. Biol Blood Marrow Transplant. 2007;13(12):1422–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krishnamurti L, Kharbanda S, Biernacki MA, Zhang W, Baker KS, Wagner JE, et al. Stable long-term donor engraftment following reduced-intensity hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2008;14(11):1270–8 [DOI] [PubMed] [Google Scholar]
- 92.Iannone R, Casella JF, Fuchs EJ, Chen AR, Jones RJ, Woolfrey A, et al. Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant. 2003;9(8):519–28 [DOI] [PubMed] [Google Scholar]
- 93.Matthes-Martin S, Lawitschka A, Fritsch G, Lion T, Grimm B, Breuer S, et al. Stem cell transplantation after reduced-intensity conditioning for sickle cell disease. Eur J Haematol. 2013;90(4):308–12 [DOI] [PubMed] [Google Scholar]
- 94.Chakrabarti S, Bareford D. A survey on patient perception of reduced-intensity transplantation in adults with sickle cell disease. Bone Marrow Transplant. 2007;39(8):447–51 [DOI] [PubMed] [Google Scholar]
- 95.Marsh JC, Gupta V, Lim Z, Ho AY, Ireland RM, Hayden J, et al. Alemtuzumab with fludarabine and cyclophosphamide reduces chronic graft-versus-host disease after allogeneic stem cell transplantation for acquired aplastic anemia. Blood. 2011;118(8):2351–7 [DOI] [PubMed] [Google Scholar]
- 96.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weatherall DJ. Thalassemia as a global health problem: recent progress toward its control in the developing countries. Ann N Y Acad Sci. 2010;1202:17–23 [DOI] [PubMed] [Google Scholar]
- 98.Scalone L, Mantovani LG, Krol M, Rofail D, Ravera S, Bisconte MG, et al. Costs, quality of life, treatment satisfaction and compliance in patients with beta-thalassemia major undergoing iron chelation therapy: the ITHACA study. Curr Med Res Opin. 2008;24(7):1905–17 [DOI] [PubMed] [Google Scholar]
- 99.Modell B, Khan M, Darlison M, Westwood MA, Ingram D, Pennell DJ. Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2008;10(42):10–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Amendah DD, Mvundura M, Kavanagh PL, Sprinz PG, Grosse SD. Sickle cell disease-related pediatric medical expenditures in the US. Am J Prev Med. 2010;38(4 Suppl):S550–6 [DOI] [PubMed] [Google Scholar]
- 101.Matthes-Martin S, Potschger U, Barr R, Martin M, Boztug H, Klingebiel T, et al. Costs and cost-effectiveness of allogeneic stem cell transplantation in children are predictable. Biol Blood Marrow Transplant. 2012;18(10):1533–9 [DOI] [PubMed] [Google Scholar]