

Abstract
Neurodegeneration, the progressive dysfunction and loss of neurons in the central nervous system (CNS), is the major cause of cognitive and motor dysfunction. While neuronal degeneration is well-known in Alzheimer's and Parkinson's diseases, it is also observed in neurotrophic infections, traumatic brain and spinal cord injury, stroke, neoplastic disorders, prion diseases, multiple sclerosis and amyotrophic lateral sclerosis, as well as neuropsychiatric disorders and genetic disorders. A common link between these diseases is chronic activation of innate immune responses including those mediated by microglia, the resident CNS macrophages. Such activation can trigger neurotoxic pathways leading to progressive degeneration. Yet, microglia are also crucial for controlling inflammatory processes, and repair and regeneration. The adaptive immune response is implicated in neurodegenerative diseases contributing to tissue damage, but also plays important roles in resolving inflammation and mediating neuroprotection and repair. The growing awareness that the immune system is inextricably involved in mediating damage as well as regeneration and repair in neurodegenerative disorders, has prompted novel approaches to modulate the immune system, although it remains whether these approaches can be used in humans. Additional factors in humans include ageing and exposure to environmental factors such as systemic infections that provide additional clues that may be human specific and therefore difficult to translate from animal models. Nevertheless, a better understanding of how immune responses are involved in neuronal damage and regeneration, as reviewed here, will be essential to develop effective therapies to improve quality of life, and mitigate the personal, economic and social impact of these diseases.
Keywords: central nervous system, inflammation, innate, neurodegeneration, neuroprotection, repair
Introduction
The demographics of dementia published by the World Health Organization (WHO) predict that neurodegenerative diseases will reach over 70 million in 2030 and 106 million in 2050.1 As in dementia, neurodegeneration associated with immune activation is observed in many other disorders including stroke, spinal cord injury, multiple sclerosis (MS), brain injury (Table 1), and the animal models of these diseases (Table 2). Hence, while the central nervous system (CNS) is an immune-privileged site, it is clear that innate and adaptive immune responses do take place in the CNS to limit neurotrophic viral and bacterial infections, limit tumour growth, remove necrotic cells following ischaemia, shape the brain during development, and promote regeneration and repair following damage. For example, both macrophages and microglia clear debris as a result of damage in the CNS and when such phagocytosis is impeded, regeneration is delayed. Likewise, inhibiting migration of adaptive immune cells into the brain renders the CNS susceptible to devastating infections.
Table 1.
Immune responses in neurodegenerative diseases
| Disease | Clinical and neuropathology | Innate immunity | Adaptive immunity | References |
|---|---|---|---|---|
| AD | Cognitive decline Atrophy of hippocampus and neocortex Neuronal loss with amyloid plaques and neurofibrillary tangles | Activated microglia ↑ Cytokines, chemokines and complement ↑ TLRs and PRRs | ↑ Antibody and T-cell responses to Aβ peptide | 3 |
| FTD | Also called Pick's disease Changes in personality behaviour, and aphasia Microvacuolation, neuronal loss, demyelination, astrocytic gliosis Mutations in the progranulin gene present in familial FTD | Activated microglia ↑ Macrophages ↑ IL-1β and COX-2 | Not reported | 4 |
| PD and LBD | Resting tremor, bradykinesia, rigidity, gait dysfunction Degeneration of dopaminergic neurons due to aggregates of α-synuclein and ubiquitin | Microglia activation ↑ NK cells ↑ IL-1β, IL-6, TNF-α in CSF ↑ TLR2, TLR5, CD14 in CNS ↑ CD14 and TLR4 in substantia nigra of MPTP animal model | Antibodies to neuronal antigens In LBD autoantibodies serum directed to Aβ and S100B, α-synuclein ↑ T cells in periphery and CNS | 5,6 |
| HD | Chorea, dystonia, atrophy of caudate and putamen, cognitive changes Diffuse atrophy of neostriatum Protein aggregates in cytoplasm and nucleus of neurons | ↑ Microglia proliferation ↑ Complement factor 3 and 9 ↑ Inflammatory factors ↑ Kynurenine pathway activity | Not reported | 7 |
| SMA | Autosomal-recessive disorder Hypotonia and weakness in newborn Degeneration of anterior horn cells in spinal cord and lower brain stem | ↑ IL-6, IL-1β | Not reported | 8 |
| SCA | Polyglutamine disease Progressive ataxia and neurodegeneration Mutant protein expressed in glia | ↑ Microglia proliferation ↑ C3 and C9 expression | Not reported | 9 |
| PNND | Heterogeneous clinical symptoms Immune response to antigens expressed on tumours | ↑ Microglia activation ↑ Immune regulatory molecules | Antibodies to neuronal antigens ↑ Regulatory, memory T cells, B cells | 10,11 |
| Lysoso- mal storage disorders | Congenital metabolic disorders, due to defects in enzymes required for lysosomal acid hydrolysis, resulting in heterogeneous clinical symptoms | ↑ Activation of glia before neuronal damage ↑ Inflammatory genes and factors | ↑ Genes encoding CD3, CD8, CD28, CTLA-4, and CD22 | 12 |
| ALS | Progressive paralysis Progressive severe motor neuron loss in anterior horn, ubiquitin aggregation and axonopathy | ↑ Complement, TLRs, CD14 RAGE, HMGB1 ↑ Genes for TRAF6, CNTF, HGF and GDNF | Antibodies to neurons in blood ↑ CD4+, CD8+ T cells | 13,14 |
| MS | Heterogeneous neurological symptoms, depending on lesion location Chronic inflammation Demyelination and progressive neurodegeneration Different lesion staging | ↑ Microglial activation ↑ NK cells ↑ Innate receptors Phagocytosis of neuronal debris ↑ Cytokines, chemokines, complement, growth factors | Antibodies and T-cells to neurons, myelin and myelin associated antigens ↑ Heat-shock proteins Neurotrophins secreted by T cells | 15–18 |
| NMO | Autoimmune disease with demyelination of spinal cord and optic nerve in which astrocytes are targeted | ↑ Microglia activation ↑ Chemokines, cytokines | Pathogenic antibodies to AQP4 | 19,20 |
| Prion diseases | Progressive dementia, ataxia, monoclonus, seizures | ↑ Microglia activation ↑ IL-1B, IL-6, ROS ↑ Complement Mast cell express PrP | B cells aid transport of PrP | 6,21 |
| Infection | Neurotrophic and peripheral pathology varies with pathogen | ↑ Microglia activation astrocytosis ↑ Innate receptors, cytokines, complement | ↑ (Auto) antibodies ↑ T cells ↑ B cells | 2 |
| Toxins and metals | For example, manganese induces neurodegeneration Diesel exhaust particles Heavy metals | ↑ Microglia activation ↑ MMP production ↑ TNF-α, IL-6 | ↑ Autoantibodies ↑ T cells ↑ B cells | 22 |
| CVD | Brain ischaemia due to several pathological processes involving the blood vessels | ↑ Activated microglia, astrocytes Phagocytic macrophages | T cells contribute to early post-stroke neuronal injury | 23,24 |
| Epilepsy and Epileptic syndro-mes | Seizures associated with cognitive and psychological sequelae. Occurs in about 1% of the population | Microglia, astrocytes activation ↑ CCL3, CCL4 genes in CNS ↑ IL-1β, IL-6 in the CSF ↑ TLR2 and TLR4, RAGE, HMGB1 ↑ COX-2 in neurons, astrocytes | ↑ Antibodies ↑ CD8+ granzyme B, T cells in parenchyma | 25 |
| TBI | Mechanical injuries of brain or spinal cord resulting in inflammatory responses enhancing tissue damage | ↑ C1q, C3b, d, C4 and C5b-9 ↑ Proinflammatory cytokines ↑ TLRs | ↑ T cells | 26 |
Aβ, amyloid-β; AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; AQP4, Aquaporin 4; COX-2, cyclooxygenase-2; CTLA-4, cytotoxic T-lymphocyte antigen 4; CVD, cerebrovascular diseases; FTD, frontotemporal dementia; GluR3, glutamate receptor 3; HD, Huntington's disease; IL, interleukin; LBD, Lewy body disease; MHC, major histocompatibility complex; MMP, matrix metalloproteinase; MS, multiple sclerosis; NK, natural killer; NMO, neuromyelitis optica; PD, Parkinson's disease; PNND, paraneoplastic neurological disorders; PrP, prion protein; PRR, pattern recognition receptor; ROS, reactive oxygen species; SCA, spinocerebellar ataxia; SMA, spinal muscle atrophy; TBI, traumatic brain injury; TNF, tumour necrosis factor; TSI, traumatic spinal cord injury.
Table 2.
Animal models of neurodegenerative diseases and involvement of immune responses
| Disease | Model | Clinical features | References |
|---|---|---|---|
| AD | APP transgenic mice: Tg2576, PDAPP, TgAPP23 Tg-APPswe/PS1dE9, 5XFAD, 3xTg-AD mice, which carry APP and PS1 Tau transgenic mice | Behavioural and cognitive changes, amyloid pathology, accumulation of Aβ, loss of neuronal subpopulations, mitochondrial oxidative damage, synaptic loss, proliferation and activation of astrocytes and microglia | 42 |
| Ageing | Senescence accelerated mice | Age-related changes in the brain. Brain atrophy, loss of cortical neurons, and dendritic spines, loss of synapses, impaired learning and memory | 43 |
| ALS | SOD-1 mutation Wobbler mouse (point mutation in the Vps54 gene) Immunization with motor neurons ALS in knockout mouse hTDP-43-associated familial ALS (transgenic) | ER stress-related toxicity, neurofilament aggregation, neuronal hyper-excitability, neuroinflammation, antibodies to neurons, reactive astrocytes, microglia activation and proliferation, increased TDP-43 expression | 44 |
| HD and other polyglutamine disorders | Many transgenic mice in which each disease possesses unique features as a result of the polyQ mutation | Dependent on model. Cognitive deficits, ataxia seizures, neuronal loss, microglia activation and reactive astrocytes | 45 |
| PD | Over-expression of α-synuclein Neurotoxic models, e.g. MPTP; 6-OHDA, rotenone | Microglial activation, adaptive immunity directed to neurons expressing α-synuclein, CD4+ T-cell-mediated damage | 46 |
| Prion | Tg(Hu-PrP)110, Tg(HuPrP)152 transgenic mice Tg(MHuPrP)-segment of mouse PrP gene replaced with human PrP | Expression of PrP plaques, neuronal loss, and spongiosis depends on transgenic mouse | 47 |
| Stroke | Middle cerebral artery occlusion model, photothrombotic model (non-invasive); thrombin injections, vessel occlusion | Leucocytes, macrophages, activated microglia and astrocytes are considered to contribute to secondary damage. | 48 |
| MS | Viral models: SFV, TMEV, MHV infection Autoimmune models: acute, chronic and secondary progressive EAE immunized with myelin antigens. Spasticity in mice immunized with NF-L. Toxin models: cuprizone, lysolecithin, ethidium bromide | SFV induces T-cell-mediated inflammatory demyelination, TMEV induces neurological symptoms depending route and strain, autoimmune models immunized with spinal cord homogenate, myelin protein or neuronal antigens in strong adjuvants | 49,50 |
AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; APP, amyloid precursor protein; ER, endoplasmic reticulum; EAE, experimental autoimmune encephalomyelitis; HD, Huntington's disease; MHV, mouse hepatitis virus; MTPT, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MS, multiple sclerosis; NF-L, neurofilaments light; PD, Parkinson's disease; PrP, prion protein; TDP-43, TAR DNA-binding protein 43 kd; TMEV, Theiler's murine encephalomyelitis virus; SFV, Semliki Forest virus; SOD, superoxide dismutase; 6-OHDA, 6-hydroxydopamine.
The field of neuroimmunology has uncovered many examples of cross-talk between the nervous and immune systems, some of which have been exploited to design therapeutic strategies. Given the major steps this field has taken in recent years we provide an update of our original paper on immune responses in the CNS,2 incorporating recent knowledge of the delicate balance between pathogenic and reparative processes. We also review emerging evidence showing that immune responses differ in the ageing brain, or following peripheral infections and other previous insults, indicating that a so-called ‘primed’ environment may make the CNS more susceptible to damage. These factors may be key in explaining why neurodegenerative diseases are more prevalent in the elderly, and why even peripheral infections are linked to chronic CNS diseases such as MS. A better understanding of the cross-talk between the innate and adaptive immune response and the CNS will be crucial to harness natural beneficial responses for therapeutic strategies.
Immune privilege in the CNS
That the CNS enjoys a state of ‘immune privilege’ was recognized by Sir Peter Medawar who, with Sir Frank Macfarlane Burnet was awarded the Nobel Prize in 1960. Medawar's idea of ‘immune privilege’ led to the notion that immune responses are tightly regulated in the brain, such that antigens within the brain do not elicit immune responses but can only be targets of an immune response initiated in the periphery. The CNS lacks recognizable parenchymal dendritic cells and is devoid of a classical lymphatic system. While several studies have revealed that CNS antigens can be drained to cervical lymph nodes, it is generally believed that this leads to immune tolerance rather than priming.27 The ‘immune privilege’ status of the CNS is dependent on several elements, notably including the physical blood–brain barrier and blood–spinal cord barrier. The unusually tight vessel walls within the CNS are designed to limit the entry of solutes and ions into the CNS. These barriers, together with neurons, glia and extracellular matrix, form the neurovascular unit that regulates immune responses in the CNS, in concert with local production of neuroimmunoregulatory molecules.28 The majority of these local cues come from neurons, although microglia,29 astrocytes and oligodendrocytes30 also play important roles in maintaining this integrity. In concert, these ‘don't eat me’ cell-contact-dependent signals and soluble mediators maintain a muted immune response that inhibits microglia activation and maturation of antigen-presenting cells, and restricts survival, especially of activated lymphocytes. Such regulation is mediated by several factors, including CX3CL1 (fractalkine), CD22, CD27, CD47, CD200 and neuronal cell adhesion molecule, known to be expressed by neurons and glia (Table 3). Neurons also produce a range of soluble mediators including chemokines, neuropeptides, neurotransmitters and neurotrophins, all of which contribute to the control and tight regulation of local immune responses. For example, TREM2 ligands on neurons help preserve the ‘immune privilege’ environment,31 and neuronal semaphorins additionally assist in immune regulation and repair by mediating oligodendrocyte precursor cell migration (Table 3). Glial cells, and especially astrocytes, can additionally produce an array of molecules to inhibit or kill activated lymphocytes, for example by Fas–Fas ligand (FasL) interactions. Defects in such regulatory circuits are known to contribute to inflammatory neurodegeneration. Some semaphorins are aberrantly expressed in neurons during Alzheimer's disease (AD), at the neuromuscular junction in amyotrophic lateral sclerosis (ALS) and in MS lesions where they control oligodendrocyte precursor cell migration whereas others regulate inflammation suppressing disease.32,33 When neurons are lost altogether, immunoregulatory signals otherwise delivered by these cells are impaired, rendering the CNS less able to maintain its protective shield.
Table 3.
Molecules expressed or secreted imparting immune regulation in the central nervous system
| Microglia | Neurons | Astrocytes | Oligodendrocytes | References |
|---|---|---|---|---|
| CD200R | CD200 | CD200 | CD200 | 28,29 |
| CX3CR1 | CX3CL1 | CX3CL1 | Unknown | 28,29 |
| CD172a/Sirp1α thrombospondin | CD47 | CD47 | CD47 | 28,29 |
| TREM2 | HSP60 TREM2L | HSP60 | HSP60 | 28,30,31 |
| Plexins | Semaphorins | Unknown | Plexin4 assists in OPC migration but semaphorins likely to be pathogenic | 32,33 |
| ICAM | sICAM5 secreted | Unknown | Unknown | 34 |
| Csf1r | IL-34 | IL-34 | Unknown | 35 |
| CD45 | CD22 | Not reported | Unknown | 36 |
| NCAM/CD56 | NCAM/CD56 | NCAM/CD56 | Unknown | 37 |
| Complement, MAC, C3, C5, | Regulators of complement activation, CD46, factor H | CD46, factor H | Unknown | 28 |
| Soluble factors: IL-4, IL-1β, IFN-γ, TGF-β, BDNF, GDNF, TIMPS, IDO | CX3CL1, TGF- β, C22, NT3 neuropeptides, sCD95L, IL-10, Sema-3A dopamine, GABA | IL-4, IL-10, IFN-γ, TGF-β, Proteoglycans, factor H | TGF-β isoforms expressed in vitro | 28,29,37 |
BDNF, brain-derived neurotrophic factor; Csf1R, colony-stimulating factor 1; GABA, γ-aminobutyric acid; GDNF, glial cell-derived neurotrophic factor; HSP, heat-shock protein; ICAM, intercellular adhesion molecule 1; IDO, indoleamine-pyrrole 2,3-dioxygenase; IFN-γ, interferon- γ; IL, interleukin; MAC, membrane attack complex; NCAM, neural cell adhesion molecule; NT, neurotrophin; Sirp1α, signal regulatory protein α; TGF, transforming growth factor; TIMPS, tissue inhibitors of metalloproteinases; TREM, triggering receptor expressed on myeloid cells 2.
Innate and adaptive immune responses in the CNS
Local innate immune responses play a key role in the first line of defence against bacterial, viral, fungal and parasitic infections in the CNS, and they are also essential to clear apoptotic cells, misfolded or aggregated proteins. In this way, innate responses pave the way for tissue repair and full restoration of homeostasis. Yet, exaggerated or prolonged innate responses may cause damage. Such damaging responses may result not only from activation of microglia, astrocytes or oligodendrocytes,29,30 but also of peripheral innate immune cells including natural killer cells, natural killer T cells, mast cells,38 granulocytes and γδ T cells. Such cells can contribute significantly to local inflammatory processes (Table 1, Fig. 1).
Figure 1.

Immune responses in human and experimental inflammatory neurodegenerative disorders. In an ischaemic area in stroke, HLA class II+ cells (blue, arrow) can be seen phagocytosing myelin basic protein (red) (a). In Alzheimer's disease, activated microglia (HLA class II+; blue) cluster around neurons and amyloid deposits (red) (b). A meningeal infiltrate in acute bacterial meningitis contains a single CD20+ B cell (brown; c), yet large numbers of activated microglia (HLA class II+; brown) in the meninges as well as in the brain parenchyma (d). In multiple sclerosis (MS), macrophages and microglia phagocytose myelin, and turn into so-called foam cells that are found in the perivascular space (e) as well as in the cerebrospinal fluid (f). Large numbers of inflammatory cells around a blood vessel are found in the centre of MS lesions that show extensive loss of myelin (g), as well as at the rim of the demyelinating zone (h). In the autoimmune mouse model of MS, perivascular infiltrates of HLA-class II positive cells can be observed in the white and grey matter of the spinal cord (i). In the meninges of mice that have been immunized with the neuronal antigen NF-L, the presence of B220+ B cells (j) and CD4+ T cells (k) is closely associated with neuronal damage (l).
Innate immune responses are generally initiated following recognition of pathogen-associated molecular patterns (PAMPS), conserved structures expressed by infectious agents. Equally important are endogenous signals for innate responses, known as danger-associated molecular patterns (DAMPS). DAMPS include a plethora of different molecules such as nucleic acids, heat-shock proteins, ATP, high mobility group box chromosomal protein 1 (HMGB-1), fibrinogen and aggregated and modified or misfolded proteins. They tend to appear as the result of stress or tissue damage. Through expression of conserved pattern-recognition receptors (PRRs), local CNS cells are triggered by the appearance of such PAMPS and DAMPS. The PRRs include C-type lectins, Toll-like receptors (TLRs), retinoic acid inducible gene I-like receptors, nucleotide-binding domain, leucine-rich repeat-containing proteins (NLRs), HIN200/PYHIN family members designated as in the ‘Absent-In-Melanoma’ receptors, as well as interferon-induced proteins called IFITs.39,40 For many of these PPRs, their expression pattern and role in neurodegenerative disorders are still under investigation. The best studied PRRs are the TLRs, which are expressed by many cells in the CNS. Activation of TLR induces several signalling pathways via the intracellular adaptor proteins Myd88 and Toll/interleukin-1 receptor-domain-containing adapter-inducing interferon-β, and production of a wide range of immunoregulatory mediators. The involvement of TLRs in neurodegenerative diseases is evidenced by pathology studies of human disorders, as well as by data from experimental animal models. For example, TLR9-deficient mice produce less pro-inflammatory cytokines and exhibit reduced neuronal damage, whereas TLR4-knockout mice are resistant to the induction of experimental autoimmune encephalomyelitis (EAE), an animal model of MS (Table 2). Activation of TLR3 suppresses EAE and leads to the production of several neuroprotective mediators in vitro.41 Several TLR-deficient mice also exhibit milder clinical disease following brain injury, and middle cerebral artery occlusion, indicating a pathogenic role for TLR activation in these models. On the other hand, TLRs aid uptake of Aβ indicative of beneficial effects in other situations. As is so often the case in the context of immune regulation, TLR-mediated responses can both aggravate inflammation and contribute to its control.
Another group of PRRs, NLRs initiate the assembly of inflammasomes that activate caspases and in this way, promote production and secretion of mature interleukin-1β and interleukin-18.51 Aβ and prion proteins are potent activators of the NLRP3 inflammasome. NLRP1 and NLRP5 are known to contribute to neuronal death while Nlrp3 or ASC (apoptosis-associated speck-like protein containing a CARD)-deficient microglia secrete significantly reduced levels of pro-inflammatory cytokines following Aβ phagocytosis. Consistent with these findings, the presence of ASC and NLRP3 are necessary for progression of EAE in mice. Yet other receptors important in neuron–glia interactions are the purinergic receptors P1 (adenosine) and P2 (ATP) receptors.51 Adenosine receptors can mediate both potentially neuroprotective and potentially neurotoxic effects. Their roles in different neurodegenerative diseases are not elucidated yet. In Parkinson's disease (PD), AD and ALS, the expression of both P1 and P2 receptors is altered. In attempts to clarify the functional significance of these changes, much attention has been devoted to the role of the adenosine A2A receptorin PD because antagonists for this receptor improve clinical symptoms as well as protection against toxin-induced neuronal degeneration.52 Hence, PRRs play an ambivalent role in neurodegenerative diseases, depending on the mode of activation.
Complement, a major effector arm of the innate immune system, is often considered a link between the innate and adaptive immune responses. During development, synapses destined for removal are tagged with complement proteins and so may be eliminated by microglial cells expressing complement. Such expression in neurodegenerative diseases is important for elimination of aggregated proteins that are found for example in AD, or necrotic cells in neurological complications of systemic lupus erythematous. However, the broad and often profoundly unregulated expression of complement components indicates that complement activation may equally contribute to the demise of neurons and axons. That neurons and oligodendrocytes express low levels of complement regulatory proteins renders these cells particularly vulnerable to complement-associated death.53
As emphasized above, the anti-inflammatory environment afforded by the immune privilege status is very effective in dampening any adaptive immune response within the CNS. Despite the microenvironment being hostile to T cells, large numbers of T cells still routinely patrol the perivascular space in a healthy CNS, and activated lymphocytes readily cross the blood–brain barrier. The extent and nature of any adaptive T-cell-mediated immune response that may occur in neurodegenerative diseases is largely determined by the type of local innate immune response. This, in turn, is determined by the initial trigger that activates such responses: the collection of PAMPs and DAMPs simultaneously acting on innate immune cells. In CNS infections, adaptive immunity is primarily directed to the infectious agent. If the infected cell is a neuron, direct cytolysis and neuronal damage may ensue.
Evidence that T cells may be involved in neurodegenerative disorders comes from studies on alterations of T-cell subsets in the periphery of patients during disease and evidence for a direct association between the presence of T cells and neurodegeneration in the CNS. For example, the decline in T-cell responses to Aβ during ageing, and the absence of such responses in AD suggests that in AD, T cells may play a role in clearing plaques. Evidence for T-cell mediated plaque removal in mice supports this notion.54 In PD, CD4+ T cells are observed in the substantia nigra whereas in MS, T cells are clearly associated with demyelinating lesions and neuronal damage. T-cell derived granzyme B and perforin, as well as apoptosis-inducing signals, may be operational. Although the major histocompatibility recognition elements for T cells are generally down-regulated within the healthy nervous system, MHC class II antigens become up-regulated on microglia and perivascular microglia/pericytes during inflammation and can serve to facilitate antigen presentation to T cells. In some instances neural cells can express MHC class I antigens, opening the potential for CD8-mediated killing. Direct neuronal killing of CD4-expressing T helper type 17 cells has been noted, suggesting the potential of MHC class II independent killing.55
That the adaptive immune system can contribute to neuronal damage in the CNS is shown by paraneoplastic neurological disorders. Such disorders are associated with the emergence of both T cells and antibodies against antigens expressed on neuronal tumours. For example, limbic encephalitis is associated with autoantibodies to the N-methyl d-aspartate receptor and voltage-gated potassium channel, whereas in stiff person syndrome, antibodies are directed to GAD65 and amphiphysin.10,11 Removing the tumour or performing plasmapheresis is often beneficial, particularly when the autoantibodies contribute to neuronal dysfunction.
Autoantibodies to neuronal proteins are also present in other neurodegenerative diseases such as MS, during which antibodies are produced intrathecally. That at least some of these anti-neuronal antibodies may be pathogenic is suggested by the finding that in animal models, antibodies against neuronal antigens can induce neuronal damage, or are associated with progression of neurological disease.56–59 That some of these antibodies are pathogenic is supported by the findings that plasma exchange is effective in a subgroup of patients. In neuromyelitis optica (NMO), the findings are clearer in that direct evidence for pathogenicity has also been obtained for serum antibodies to aquaporin-4, an abundant water channel expressed by astrocytes. Such antibodies are pathogenic in animals. Although these antibodies may not trigger a direct attack on neurons, the loss of trophic support afforded by the astrocytes that are targeted, may well contribute to neurodegeneration in NMO patients. In both MS and NMO the exact mechanism of antibody-mediated neuronal damage, or the origin of autoreactive antibodies, remains to be fully clarified. Although it has been suggested that in MS, activation of an intrathecal B-cell pool may be caused by Epstein–Barr virus infection, this explanation remains controversial.60,61 In some movement disorders, antibodies to group A β-haemolytic streptococcal infections are known to cross-react with basal ganglia tissue, inducing motor and psychiatric symptoms. In these cases, treatment with antibiotics is very effective. In addition to pathogenic responses both play critical roles in mediating damage to neurons. Yet, and as stated above already, both innate and adaptive immune responses also play critical roles in down-regulating inflammatory processes and actively participate in the repair process. These beneficial roles are discussed further below.
Infectious agents and peripheral infections
Infections with viruses, bacteria and prions induce neurodegeneration by a variety of mechanisms. As well as inducing a direct cytolytic effect, neurotrophic viruses may trigger apoptosis, necrosis or autophagy of infected neurons because of virus-specific immunity. Infections may also induce the dying back of axons and secondary myelin damage, induce protein aggregation, or trigger neurotoxic pathways. Although not a direct target themselves, neurons can be subject to collateral damage through an immune-mediated attack on nearby, infected oligodendrocytes or astrocytes, or as a result of released pro-inflammatory factors (Fig. 2).
Figure 2.
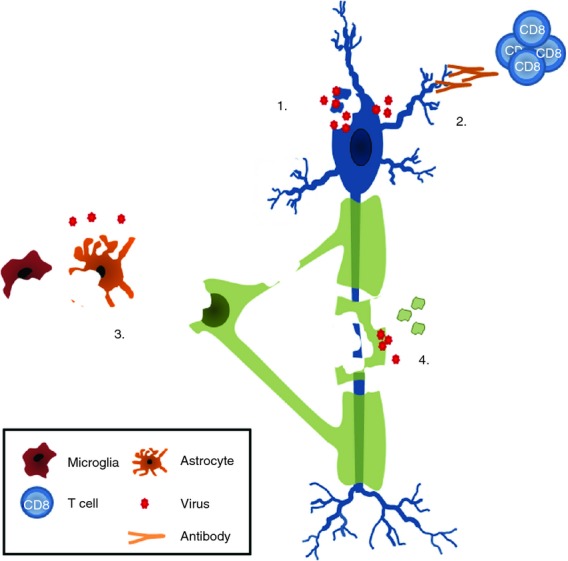
Proposed mechanisms of viral-induced neuronal damage. Infection of neurons with viruses leads to apoptosis, necrosis, autophagy or dying back of axons (1). Immune-mediated attack of neurons by virus-specific CD8+ T cells or autoantibodies induces damage (2). Infection of endothelial cells, ependymal cells, glia (astrocytes, oligodendrocytes, microglia) leads to so-called bystander damage as the result of release of cytokines or reactive oxygen species that damage neurons in a variety of ways (see text for details) (3). Secondary neuronal degeneration as a consequence of trophic support, when oligodendrocytes and myelin are infected with viruses (4).
Serological and epidemiological studies have revealed marked associations between infections and many neurodegenerative diseases. In some cases, viruses are considered to act as trigger, or to contribute to progression of disease by exacerbating clinical symptoms, for example in MS (Table 1). The association of viruses with neurodegenerative diseases in humans is supported by studies in animal models (Table 2). Not only can viruses or microbial components like lipopolysaccharide directly cause neuronal damage, they also influence the severity of disease. Peripheral infections are also known to precipitate as well as exacerbate ongoing neurodegeneration.62 Evidence for this comes from studies in experimental models in which peripheral inflammation is associated with exacerbation of experimental AD, MS, PD, prion disease and stroke. For example, an increase in Aβ in the CNS of animals that survive sepsis correlates with a decrease in synaptophysin levels and cognitive impairment. Transgenic mice susceptible to neurodegeneration develop less severe disease when raised under pathogen-free conditions.63 That such associations also exists in humans is supported by the fact that upper respiratory tract infections are associated with the occurrence of clinical relapses during MS while AD has been associated with spirochetes and other pathogens such as Chlamydia pneumoniae and herpes simplex virus type 1; whether this is cause or effect remains to be determined (Table 4).
Table 4.
Infectious agents linked to neurodegenerative disorders
| Disease | Infectious agent | Association | References |
|---|---|---|---|
| AD | HSV-1 | HSV-1 genome present in brain tissues. Genes related to HSV-1 reactivation in familial AD. Virus associated with protein aggregates. HSV-1 induces protein aggregation | 64 |
| Chlamydia pneumoniae | 90% of AD brains infected with C. pneumoniae in plaques Infection of mice induces AD pathology with Aβ aggregation | 65 | |
| Borrelia burgdorferi | Organism isolated from plaques | 66 | |
| ALS | Retroviruses | Analysis of serum and brain tissues reveals increased expression of HERV-K | 67 |
| PD | Influenza | Risk of developing Parkinsonian symptoms increases with number of influenza attacks. Outbreaks of encephalitis lethargica (von Ecomo's disease) in 1918 influenza pandemic caused by type A H1N1. In PD influenza infections associated with transient tremor and gait disturbances. H5N1 induces α-synuclein deposits | 68,69 |
| MS | EBV HHV6 Pseudomonas aeruginosa, Retroviruses | Infectious mononucleosis associated with 2·3-fold higher risk to develop MS. Heightened antibody responses to EBV. Controversy over expression of EBV in the brain Antibodies to acinetobacteria cross-react with myelin Relapses associated with upper respiratory tract infections HHV6 present in MS lesions | 60,61,70 |
| HIV infection | HIV | HIV-associated neurocognitive disorder; HIV mild neurocognitive disorder; HIV-associated dementia. Replication of HIV and degeneration of synapses and neurons. Activation of microglia, macrophages and astrocytes. HIV increases amyloid plaque and Aβ accumulation | 71 |
AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; EBV, Epstein–Barr virus; HIV, human immunodeficiency virus; HSV, herpes simplex virus; HERV-K, human endogenous retrovirus type K; HHV6, human herpes virus 6; MS, multiple sclerosis.
Metals trigger neurodegeneration
Exposure to metals and environmental pollutants has been postulated as a risk factor for neurodegenerative disorders.22 Also, as several of these agents can trigger or modulate immune responses, their possible involvement in inflammatory neurodegeneration should be considered. Indeed, an animal model of PD uses the neurotoxin 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) to selectively injure nigrostriatal dopaminergic neurons. The experimental use of MPTP was originally inspired by the PD-like syndrome in drug addicts who accidentally injected themselves with MPTP as a contaminant of pethidine. MPTP is metabolized by astrocytes to form the active metabolite 1-methyl-4-phenylpyridinium (MPP+) that enters cells via the dopamine transporter and inhibits complex 1 of the mitochondrial electron transport chain. As a result, depletion of striatal dopamine neurons occurs, and recruitment and activation of microglia ensues. Similar effects are observed following administration of rotenone, or 6-hydroxydopamine infusion. Likewise, the deleterious effects on the CNS of psychostimulant use,72 exposure to pesticides,73 mercury intoxication74 and diesel exhaust particles involve at least in part neuroinflammatory mechanisms. One mechanism by which toxic agents could augment neurodegenerative diseases is through the induction of protein aggregation, or by activation of pathogenic innate immune responses, both common findings in neurodegenerative diseases of the peripheral and central nervous systems.
Ageing and immune responses in the CNS
Ageing is a major risk factor for many neurodegenerative disorders. In addition to the environmental factors discussed above, genetic mutations, oxidative or metabolic stress including endoplasmic reticulum and mitochondrial dysfunction are known to act as triggers for protein misfolding and aggregation. It is also known that these triggers accumulate with age. Another characteristic of ageing is the accumulation of advanced glycation end-products (AGE), which is accelerated in MS, AD and PD.75 This is mirrored by an increase in the receptor for AGE (RAGE), which induces pro-inflammatory cytokines and free radicals following engagement of RAGE, so perpetuating a cycle of damage. RAGE is increased in the AD-affected CNS, where it is expressed on neurons and astrocytes. During MS, RAGE is expressed on oligodendrocytes in response to stress, and levels of soluble RAGE are now under examination as a potential biomarker of disease.75 One known ligand of RAGE is HMGB1, a DNA-binding protein with pro-inflammatory properties. Levels of HMGB1 are increased in AD and PD as well as in MS, and accordingly HMGB1 has been suggested to amplify the inflammatory response that causes the disease, possibly as a consequence of its ability to trigger TLR4-mediated inflammatory responses76 in contrast to Huntington's disease where HMGB1 has been reported to protect neurons.77
Several factors contribute to an age-related increase in the risk of neurodegeneration,78 In demyelinating diseases such as MS, it has become apparent that the ability to repair damage in the CNS declines with age. Concomitant with this is a reduction in the anti-oxidant decline, antioxidant defence mechanisms weaken, leaving axons and neurons more vulnerable to damage, part of which is due to the capacity of the innate immune system to facilitate repair by glia.79 In addition, immune senescence the process by which immune responses become less efficient with age. This phenomenon manifests itself by an increased susceptibility to infections, a reduced ability to mount adaptive immune responses following vaccinations, and an increased susceptibility to autoimmune diseases. These phenomena all reflect a reduced ability to adequately control inflammatory responses.80 Aging is associated with altered innate immune responses such as dysregulation of TLR responses and production of cytokines and other inflammatory mediators. In aged mice, dysmorphic microglia appear, which display a more reactive and activated phenotype. In addition, the levels of immune suppressive neuronal factors decrease, such as CX3CL1, its receptor CX3CR1 and CD200. This suggests that the ability of neurons to maintain microglia in a quiescent state becomes gradually less effective in an aged brain.29
Much of what is known about the impact of ageing on neuroinflammatory processes has been derived from experimental intervention in aged animals such as the senescence-accelerated mouse.43 In addition, systemic injection of lipopolysaccharide in aged mice leads to higher levels of microglial cytokines compared with young mice, reflecting a more sensitized state or ‘primed’ state of these aged microglia. That age impacts neuroinflammation is shown by studies using parabiosis between old and young mice, showing the importance of immune senescence in repair and synaptogenesis.79,81,82 However, it is still unclear which factors in humans lead to such priming. As discussed above systemic infections may fulfil that role.
Immune-mediated neurodegeneration
Necrosis of neurons is observed following acute brain injury and neurotrophic infections, or as a result of the release of damaging chemicals such as glutamate, nitric oxide and reactive oxygen species (Fig. 3). Damaged neurons themselves may also exacerbate immune-mediated disease by release of chemokines such as CCL21,83 a ligand of the chemokine receptor CCR7 present on T cells, macrophages and microglia.84,85 Consequences of neuronal loss also include a reduction in the inhibitory signals that control production of pro-inflammatory cytokines and neurotoxic substances including nitric oxide, oxidative radicals and proteolytic enzymes by activated microglia. Disturbance in glutamate homeostasis as a result of astrocyte damage, for example, leads to excitotoxicity and is observed in acute viral encephalomyelitis, AD, PD and acute brain injury.
Figure 3.

Immune-mediated neurodegeneration. Misfolded and aggregated proteins such as Aβ and α-synuclein aggregates (1), activate microglia and suppress brain derived neuronal growth factor production by astrocytes. Neuronal specific antibodies (2) activate the complement system or FcR-mediated damage. Natural killer and T cells damage neurons either via MHC class-I or non-classical MHC molecules (3). Damage to neurons reduces the levels of neuroimmunoregulatory molecules, decreasing the immune suppressive environment (4). Excessive production of glutamate by, for example, stressed neurons and activated immune cells, together with reduced glutamate uptake causes excitotoxic damage of neurons (5). Macrophage and microglia activation triggers release of reactive oxygen and nitrogen species, matrix metalloproteinases, chemokines and cytokines known to damage axons and neurons and to cause mitochondrial dysfunction. Neurons (i) and oligodendrocytes (ii) release exosomes that may trigger pathogenic immune responses (6). NO, nitric oxide; ROS, reactive oxygen species.
As explained above, activation of macrophages and microglia is observed during infections, trauma, following exposure to toxic compounds or, indeed, upon ageing. Aggregated, altered or misfolded proteins that accumulate during ageing or as a cause or consequence of pathology are known to represent DAMPs and bind to PRRs, key molecules that drive the innate immune responses. Although not all of these pathways are acutely detrimental to neurons, chronic microglia activation may compromise blood–brain barrier integrity, and lead to a markedly increased influx of peripheral immune cells as well as potentially neuron-reactive antibodies.
Cytotoxic CD8+ and CD4+ T cells can contribute to neuronal damage or destruction by directly targeting neurons. This is for example observed in Rasmussen syndrome. Under pathological conditions, neurons express MHC class I antigens, which makes them susceptible to CD8+ T-cell-mediated damage. Neurons and axons also express non-classical MHC molecules, and are susceptible to perforin- and granzyme B-mediated damage and Fas–FasL interactions. Neuronal damage may also occur as a consequence of an immune-mediated attack on myelin, oligodendrocytes or astrocytes, as observed during NMO.
Antibodies to neurons correlate with CNS injury in systemic lupus erythematous, ALS, MS and paraneoplastic neurological disorders. These antibodies may well be directed to receptors such as N-methyl d-aspartate receptors, inducing neuronal excitotoxicity, or to intracellular antigens such as neurofilaments. Autoantibodies to neurofascin, KIR4, contactin-2 and neurofilaments are present in MS patients, and such antibodies can mediate axonal injury in mice.10,86 The autoantibodies could activate the complement system, inducing damage as a result of membrane attack complex formation. In AD, one risk factor is the presence of polymorphisms in complement receptor 1 that may reduce the ability of microglia to clear Aβ.87 In vitro, aggregated amyloid and prion proteins bind C1q directly and activate complement in the absence of antibodies, thereby aiding clearance. The role of complement in ALS is somewhat different and depends on the model.88,89 In addition, deficiency of the complement regulator exacerbates Wallerian degeneration, clearly showing a role for complement in neurodegeneration.90
Apart from being pathogenic in some cases, immune responses can also be helpful in controlling and limiting pathogenic responses (Fig. 4). As discussed above, the CNS is equipped with an arsenal of neuroimmunoregulatory molecules that help to maintain immune privilege. Despite the well-known anti-inflammatory properties of regulatory microglia in several neurodegenerative disorders,7,91 astrocytes also contribute to protection from immune-mediated damage by preferentially inducing apoptosis of infiltrating T cells by Fas–FasL interactions. Astrocytes also produce interleukin-1 and encourage influx of regulatory T cells by producing interleukin-27. In addition, astrocytes are the major source of nerve growth factor and glial cell line-derived neurotrophic factor in the CNS, thereby aiding neuronal regeneration.92,93 Such neuronal growth factors are also secreted by T cells94 that have been shown to aid neurite outgrowth in vitro, reduce secondary neurodegeneration in vivo, and contribute to learning behaviour. These findings indicate a likely neuroreparative role at least for some T cells during neurodegenerative disorders.95
Figure 4.

Neuroprotection and regeneration by cells of the immune system. T and B cells secrete neuroprotective factors and suppress pro-inflammatory responses (1), since macrophages/microglia that phagocytose myelin stimulate growth and repair via glial cell-derived neuronal growth factors (2). Antibodies aid remyelination thereby restoring trophic support of neurons (3) and help to remove the aggregated proteins (4). Reparative microglia and astrocytes produce neuronal growth factors (5) neurons (i) and oligodendrocytes (ii) release exosomes that dampen the pathogenic immune responses. BBB, blood–brain barrier; BDNF, brain derived neural growth factor; IL-4, interleukin-4; SP-1, sphingosine-1-phosphate; VLA-4, very-late antigen 4.
Cross-talk between the immune system and the CNS is known to occur via exosomes, small vesicles released from multiple cell types that contain protein, lipids or RNA species including viral RNA.96 Following their secretion, exosomes are taken up by neighbouring cells and deliver their cargo to those recipient cells. It has been well established that neurons, microglia, astrocytes and oligodendrocytes secrete exosomes, and stressed oligodendrocytes do so even more. In the latter case, myelinating oligodendrocytes transport RNA and proteins to axons, and such support helps to maintain neuronal integrity.97 In PD for example, the protein α-synuclein, a mediator of neurodegeneration, is released by cultured neurons via exosomes. Addition of such exosomes to cultured neurons induces neuronal cell death, again pointing to a role of exosomes in disease.98 Given these emerging examples of how exosomes participate in the communication between neural cells under neurodegenerative conditions, it seems rather likely that they will turn out to have an important role in many neurodegenerative diseases by providing trophic support, by transferring pathogenic aggregated proteins or aiding repair by providing growth factors. They are also likely to play an important role in antigen presentation within the CNS by transferring antigens in the CNS parenchyma to perivascular antigen-presenting cells that meet up with T cells engaged in immune surveillance. Exosome-mediated signalling is clearly an intriguing new field of study in neurodegenerative disorders.
Immunotherapy in neurodegenerative disorders
Evidence for the involvement of immunity in the development and progression of neurodegenerative disorders has inspired immunotherapeutic approaches to prevent neuronal loss and to aid neuronal growth. In paraneoplastic neurological disorders, for example, the reduction of autoantibodies by plasmapheresis is usually beneficial in suppressing the immune response and improving the neurological status, at least in the short term. For long-term treatment combined therapies targeting the tumour as well are often necessary.
The use of systemic anti-inflammatory pharmacological agents may contribute to the management of neurodegenerative diseases. Epidemiological studies have shown a lower prevalence of AD in people who have regularly taken non-steroidal anti-inflammatory drugs99 and in MS there is a prominent role for glucocorticosteroids during the treatment of exacerbations.
Inhibiting T-cell trafficking into the CNS appears a rational approach for some neurodegenerative diseases. However, such approaches not only block the entry of potentially pathogenic T cells, but also of reparative regulatory T cells as well as T cells required for control of tumours and infections. Inhibition with antibodies directed to T cells and or monocytes (trafficking) is one of the therapeutic targets in neurodegenerative diseases.
Natalizumab, a humanized monoclonal antibody against very late activating antigen-4 is an example of a therapeutic agent that acts by blocking lymphocytes going into the CNS. Alternative agents include Fingolimod, which is designed to entrap lymphocytes in lymphoid tissue. Another mechanism of action of Fingolimod is promoting oligodendrocyte proliferation and differentiation and therefore repair. Rituximab, a chimeric monoclonal antibody against the CD20 antigen, present on B cells, induces a transient depletion of B cells, resulting in a substantial effect on the inflammatory disease activity in MS.100 Due to the susceptibility of the CNS for infections, long-term treatment with these therapies needs to be seriously considered.
Supplementation with intravenous immunoglobulins has gained increasing interest as a therapeutic approach to neuroinflammatory disorders. Although their actions remain to be fully clarified, intravenous immunoglobulins are likely to promote the development of regulatory T cells with beneficial reparative effects when recruited into the CNS. Active and passive immunotherapies to provide naturally occurring antibodies to remove Aβ plaques have also been used. Such approaches have also been applied to manage prion diseases and PD, in which antibodies target α-synuclein that accumulates as Lewy bodies in dopaminergic neurons.
The idea that innate immune responses, in particular by microglia, are inextricably involved in neurodegeneration has inspired approaches to inhibit or modulate microglial activation for therapeutic purposes. Certain agents that inhibit microglial responses, such as minocycline or nicergoline, have been tested in several models of human disorders101 (Fig. 4). However, many have not yet been screened in patients, or have been found to be less effective in humans than in the corresponding animal models. Other ways to modulate the intimate interactions between microglia and neurons have been exploited for therapeutic approaches as well. For example, application of exogenous CXCL1 (generally expressed on neurons) has been shown to be neuroprotective, probably by controlling microglial activation. Likewise, the heat-shock protein hsp B5 exerts neuroprotective effects. hsp B5 is a chaperokine produced by oligodendrocytes, and it activates regulatory microglial responses40 during MS. In several animal models of neurodegeneration, hsp B5 has been found to be neuroprotective, indicating that modulating innate microglial responses within the CNS can be an effective approach in neurodegenerative diseases. Other approaches targeting innate responses include blocking RAGE, supplying glutamate antagonists, and targeting oxidative stress or the complement pathway.2
Sodium-channel-blocking drugs have been reported to inhibit microglial activity and may have neuroprotective activity,102,103 during neuroinflammation. Finally, CB-derived drugs are promising neuroprotective strategies and have been shown to not only exert neuroprotective, but also some immunosuppressive, activity.104 When activated, neuronal CB1 receptors have been shown to attenuate excitotoxic glutamatergic neurotransmission,104 this triggers signalling pathways that are effective in inhibiting signs of degeneration, although benefit was noted in only a subset of people with progressive MS105 neurodegeneration in animal models.106,107
Of particular interest is the use of neural stem and progenitor cell replacement therapy. These cells are no longer believed to primarily replace damaged cells, but rather to modulate immune responses and repair processes in the CNS108 by secreting a variety of soluble factors. However, endogenous, and transplanted neural stem and progenitor cells are also influenced by the immune environment, which may either augment migration and proliferation, or inhibit their effects.109 As a result, their impact is influenced by the nature of the inflammatory microenvironment in which they end up.
Conclusions
The nervous and immune systems are inextricably interlinked. In several cases, mediators used by the immune system to combat infections and promote tissue repair are also used by the CNS for growth and development. This common language is exemplified by the role of microglia in synaptic pruning, and the role of cytokines during development of the brain. Despite the fact that the CNS is an immune-privileged site, innate and adaptive immune responses do regularly take place in the CNS. They are essential to eliminate infectious agents and tumours, as well as for clearing debris and promoting tissue repair. Despite these beneficial roles of immune responses, such responses must remain under tight control to prevent any damage to the CNS. The blood–brain barrier and the powerful immune-regulatory functions of all types of neural cells act together to secure such tight control. Microglia are actively maintained in a quiescent state, and the influx and local activation of peripheral immune cells is severely limited. Despite these measures, chronic immune activation within the CNS is a pathological hallmark of a wide number of neurodegenerative disorders. The factors that drive chronic inflammation include the accumulation of misfolded and aggregated proteins, heat-shock proteins and other DAMPs that are all known to trigger local innate responses. Viral and bacterial infections clearly play a role as well, as do dietary substances, or the lack of them, and toxic compounds in the environment. It appears that with aging the threshold for immune activation within the CNS goes down. This so-called priming of immune responses may play a major role in the increased risk of neurodegenerative diseases in the elderly. To develop novel therapeutic approaches to control these diseases, a closer look at the way immune regulation is organized in the CNS is useful. A better understanding of the endogenous protective pathways will undoubtedly reveal ways to harness reparative processes, and so improve control over chronic inflammatory neurodegenerative disorders.
Acknowledgments
For our research discussed in this review the authors gratefully acknowledge the financial support of the Dutch Multiple Sclerosis (MS) Research Foundation and the MS Society of Great Britain and Northern Ireland. We are also very grateful to all our colleagues for their views and contributions to the topic during many discussions.
Disclosures
JMvN holds equity in Delta Crystallon BV. The other authors have no conflicts of interest to report.
References
- 1.Geneva: World Health Organization; 2012. World Health Organization Dementia a public health priority. http://site.ebrary.com/lib/ucmerced/Doc?id=10718026 [accessed 14 November 2013] [Google Scholar]
- 2.Amor S, Puentes F, Baker D, et al. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–69. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Town T, Tan J, Flavell RA, et al. T-cells in Alzheimer's disease. Neuromolecular Med. 2005;7:255–64. doi: 10.1385/NMM:7:3:255. [DOI] [PubMed] [Google Scholar]
- 4.Bellucci A, Bugiani O, Ghetti B, et al. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener Dis. 2011;8:221–9. doi: 10.1159/000322228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosley RL, Hutter-Saunders JA, Stone DK, et al. Inflammation and adaptive immunity in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009381. doi: 10.1101/cshperspect.a009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maetzler W, Berg D, Synofzik M, et al. Autoantibodies against amyloid and glial-derived antigens are increased in serum and cerebrospinal fluid of Lewy body-associated dementias. J Alzheimers Dis. 2011;26:171–9. doi: 10.3233/JAD-2011-110221. [DOI] [PubMed] [Google Scholar]
- 7.Ellrichmann G, Reick C, Saft C, et al. The role of the immune system in Huntington's disease. Clin Dev Immunol. 2013;2013:541259. doi: 10.1155/2013/541259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papadimitriou D, Le Verche V, Jacquier A, et al. Inflammation in ALS and SMA: sorting out the good from the evil. Neurobiol Dis. 2010;37:493–502. doi: 10.1016/j.nbd.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci. 2007;10:1355–60. doi: 10.1038/nn1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melzer N, Meuth SG, Wiendl H. Neuron-directed autoimmunity in the central nervous system: entities, mechanisms, diagnostic clues, and therapeutic options. Curr Opin Neurol. 2012;25:341–8. doi: 10.1097/WCO.0b013e3283531efb. [DOI] [PubMed] [Google Scholar]
- 11.Zuliani L, Graus F, Giometto B, et al. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatr. 2012;83:638–45. doi: 10.1136/jnnp-2011-301237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Archer LD, Langford-Smith KJ, Bigger BW, et al. Mucopolysaccharide diseases: a complex interplay between neuroinflammation, microglial activation and adaptive immunity. J Inherit Metab Dis. 2014;37:1–12. doi: 10.1007/s10545-013-9613-3. [DOI] [PubMed] [Google Scholar]
- 13.Sta M, Sylva-Steenland RMR, Casula M, et al. Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis. 2011;42:211–20. doi: 10.1016/j.nbd.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Puentes F, Topping J, Kuhle J, et al. Immune reactivity to neurofilament proteins in the clinical staging of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatr. 2013 doi: 10.1136/jnnp-2013-305494. doi: 10.1136/jnnp-2013-305494. [DOI] [PubMed] [Google Scholar]
- 15.Podda G, Nyirenda M, Crooks J, et al. Innate immune responses in the CNS: role of toll-like receptors, mechanisms, and therapeutic opportunities in multiple sclerosis. J Neuroimmune Pharmacol. 2013;8:791–806. doi: 10.1007/s11481-013-9483-3. [DOI] [PubMed] [Google Scholar]
- 16.Van Noort JM, Baker D, Amor S. Mechanisms in the development of multiple sclerosis lesions: reconciling autoimmune and neurodegenerative factors. CNS Neurol Disord Drug Targets. 2012;11:556–69. doi: 10.2174/187152712801661293. [DOI] [PubMed] [Google Scholar]
- 17.Van der Valk P, Amor S. Preactive lesions in multiple sclerosis. Curr Opin Neurol. 2009;22:207–13. doi: 10.1097/WCO.0b013e32832b4c76. [DOI] [PubMed] [Google Scholar]
- 18.Van Noort JM, Bsibsi M, Gerritsen WH, et al. α-B-crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol. 2010;69:694–703. doi: 10.1097/NEN.0b013e3181e4939c. [DOI] [PubMed] [Google Scholar]
- 19.Saji E, Arakawa M, Yanagawa K, et al. Cognitive impairment and cortical degeneration in neuromyelitis optica. Ann Neurol. 2013;73:65–76. doi: 10.1002/ana.23721. [DOI] [PubMed] [Google Scholar]
- 20.Mitsdoerffer M, Kuchroo V, Korn T. Immunology of neuromyelitis optica: a T cell–B cell collaboration. Ann N Y Acad Sci. 2013;1283:57–66. doi: 10.1111/nyas.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradford BM, Mabbott NA. Prion disease and the innate immune system. Viruses. 2012;4:3389–419. doi: 10.3390/v4123389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kraft AD, Harry GJ. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int J Environ Res Public Health. 2011;8:2980–3018. doi: 10.3390/ijerph8072980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chamorro Á, Meisel A, Planas AM, et al. The immunology of acute stroke. Nat Rev Neurol. 2012;8:401–10. doi: 10.1038/nrneurol.2012.98. [DOI] [PubMed] [Google Scholar]
- 24.Brait VH, Arumugam TV, Drummond GR, et al. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab. 2012;32:598–611. doi: 10.1038/jcbfm.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bauer J, Vezzani A, Bien CG. Epileptic encephalitis: the role of the innate and adaptive immune system. Brain Pathol. 2012;22:412–21. doi: 10.1111/j.1750-3639.2012.00580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das M, Mohapatra S, Mohapatra SS. New perspectives on central and peripheral immune responses to acute traumatic brain injury. J Neuroinflammation. 2012;9:236. doi: 10.1186/1742-2094-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Locatelli G, Wörtge S, Buch T, et al. Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat Neurosci. 2012;15:543–50. doi: 10.1038/nn.3062. [DOI] [PubMed] [Google Scholar]
- 28.Hoarau J-J, Krejbich-Trotot P, Jaffar-Bandjee M-C, et al. Activation and control of CNS innate immune responses in health and diseases: a balancing act finely tuned by neuroimmune regulators (NIReg) CNS Neurol Disord Drug Targets. 2011;10:25–43. doi: 10.2174/187152711794488601. [DOI] [PubMed] [Google Scholar]
- 29.Kierdorf K, Prinz M. Factors regulating microglia activation. Front Cell Neurosci. 2013;7:44. doi: 10.3389/fncel.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peferoen L, Kipp M, van der Valk P, et al. Oligodendrocyte–microglia cross-talk in the CNS. Immunology. 2013;141:302–13. doi: 10.1111/imm.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldbaum O, Richter-Landsberg C. Stress proteins in oligodendrocytes: differential effects of heat shock and oxidative stress. J Neurochem. 2001;78:1233–42. doi: 10.1046/j.1471-4159.2001.00507.x. [DOI] [PubMed] [Google Scholar]
- 32.Toguchi M, Gonzalez D, Furukawa S, et al. Involvement of Sema4D in the control of microglia activation. Neurochem Int. 2009;55:573–80. doi: 10.1016/j.neuint.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 33.Okada A, Tominaga M, Horiuchi M, et al. Plexin-A4 is expressed in oligodendrocyte precursor cells and acts as a mediator of semaphorin signals. Biochem Biophys Res Commun. 2007;352:158–63. doi: 10.1016/j.bbrc.2006.10.176. [DOI] [PubMed] [Google Scholar]
- 34.Tian L, Lappalainen J, Autero M, et al. Shedded neuronal ICAM-5 suppresses T-cell activation. Blood. 2008;111:3615–25. doi: 10.1182/blood-2007-09-111179. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Szretter KJ, Vermi W, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13:753–60. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mott RT, Ait-Ghezala G, Town T, et al. Neuronal expression of CD22: novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia. 2004;46:369–79. doi: 10.1002/glia.20009. [DOI] [PubMed] [Google Scholar]
- 37.Chavarría A, Cárdenas G. Neuronal influence behind the central nervous system regulation of the immune cells. Front Integr Neurosci. 2013;7:64. doi: 10.3389/fnint.2013.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skaper SD, Facci L, Giusti P. Mast cells, glia and neuroinflammation: partners in crime? Immunology. 2013;141:314–27. doi: 10.1111/imm.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol. 2013;13:551–65. doi: 10.1038/nri3479. [DOI] [PubMed] [Google Scholar]
- 40.Bsibsi M, Holtman IR, Gerritsen WH, et al. α-B-crystallin induces an immune-regulatory and antiviral microglial response in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol. 2013;72:970–9. doi: 10.1097/NEN.0b013e3182a776bf. [DOI] [PubMed] [Google Scholar]
- 41.Bsibsi M, Persoon-Deen C, Verwer RWH, et al. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia. 2006;53:688–95. doi: 10.1002/glia.20328. [DOI] [PubMed] [Google Scholar]
- 42.Lee J-E, Han P-L. An update of animal models of Alzheimer disease with a reevaluation of plaque depositions. Exp Neurobiol. 2013;22:84–95. doi: 10.5607/en.2013.22.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimada A, Hasegawa-Ishii S. Senescence-accelerated mice (SAMs) as a model for brain aging and immunosenescence. Aging Dis. 2011;2:414–35. [PMC free article] [PubMed] [Google Scholar]
- 44.Moser JM, Bigini P, Schmitt-John T. The wobbler mouse, an ALS animal model. Mol Genet Genomics. 2013;288:207–29. doi: 10.1007/s00438-013-0741-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Figiel M, Szlachcic WJ, Switonski PM, et al. Mouse models of polyglutamine diseases: review and data table. Part I. Mol Neurobiol. 2012;46:393–429. doi: 10.1007/s12035-012-8315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blesa J, Phani S, Jackson-Lewis V, et al. Classic and new animal models of Parkinson's disease. J Biomed Biotechnol. 2012;2012:845618. doi: 10.1155/2012/845618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wadsworth JDF, Asante EA, Collinge J. Review: contribution of transgenic models to understanding human prion disease. Neuropathol Appl Neurobiol. 2010;36:576–97. doi: 10.1111/j.1365-2990.2010.01129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Macrae IM. Preclinical stroke research–advantages and disadvantages of the most common rodent models of focal ischaemia. Br J Pharmacol. 2011;164:1062–78. doi: 10.1111/j.1476-5381.2011.01398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van der Star BJ, Vogel DYS, Kipp M, et al. In vitro and in vivo models of multiple sclerosis. CNS Neurol Disord Drug Targets. 2012;11:570–88. doi: 10.2174/187152712801661284. [DOI] [PubMed] [Google Scholar]
- 50.Amor S, Smith PA, Hart B', et al. Biozzi mice: of mice and human neurological diseases. J Neuroimmunol. 2005;165:1–10. doi: 10.1016/j.jneuroim.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 51.Bajramovic JJ. Regulation of innate immune responses in the central nervous system. CNS Neurol Disord Drug Targets. 2011;10:4–24. doi: 10.2174/187152711794488610. [DOI] [PubMed] [Google Scholar]
- 52.Shook BC, Jackson PF. Adenosine A(2A) receptor antagonists and Parkinson's disease. ACS Chem Neurosci. 2011;2:555–67. doi: 10.1021/cn2000537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brennan FH, Anderson AJ, Taylor SM, et al. Complement activation in the injured central nervous system: another dual-edged sword? J Neuroinflammation. 2012;9:137. doi: 10.1186/1742-2094-9-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fisher Y, Nemirovsky A, Baron R, et al. T cells specifically targeted to amyloid plaques enhance plaque clearance in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e10830. doi: 10.1371/journal.pone.0010830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang J, Ke K-F, Liu Z, et al. Th17 cell-mediated neuroinflammation is involved in neurodegeneration of aβ1-42-induced Alzheimer's disease model rats. PLoS One. 2013;8:e75786. doi: 10.1371/journal.pone.0075786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Puentes F, van der Star BJ, Victor M, et al. Characterization of immune response to neurofilament light in experimental autoimmune encephalomyelitis. J Neuroinflammation. 2013;10:118. doi: 10.1186/1742-2094-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huizinga R, Gerritsen W, Heijmans N, et al. Axonal loss and gray matter pathology as a direct result of autoimmunity to neurofilaments. Neurobiol Dis. 2008;32:461–70. doi: 10.1016/j.nbd.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 58.Srivastava R, Aslam M, Kalluri SR, et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N Engl J Med. 2012;367:115–23. doi: 10.1056/NEJMoa1110740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kusunoki S. Autoantibodies in neuroimmunological diseases; relevance of fine specificity. Exp Neurol. 2013;250C:219–20. doi: 10.1016/j.expneurol.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 60.Lassmann H, Niedobitek G, Aloisi F, et al. Epstein–Barr virus in the multiple sclerosis brain: a controversial issue – report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain. 2011;134:2772–86. doi: 10.1093/brain/awr197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peferoen LAN, Lamers F, Lodder LNR, et al. Epstein–Barr virus is not a characteristic feature in the central nervous system in established multiple sclerosis. Brain. 2010;133:e137. doi: 10.1093/brain/awp296. [DOI] [PubMed] [Google Scholar]
- 62.Perry VH. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. 2010;120:277–86. doi: 10.1007/s00401-010-0722-x. [DOI] [PubMed] [Google Scholar]
- 63.Capsoni S, Carucci NM, Cattaneo A. Pathogen free conditions slow the onset of neurodegeneration in a mouse model of nerve growth factor deprivation. J Alzheimers Dis. 2012;31:1–6. doi: 10.3233/JAD-2012-120427. [DOI] [PubMed] [Google Scholar]
- 64.Bearer EL. HSV, axonal transport and Alzheimer's disease: in vitro and in vivo evidence for causal relationships. Future Virol. 2012;7:885–99. doi: 10.2217/fvl.12.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Honjo K, van Reekum R, Verhoeff NPLG. Alzheimer's disease and infection: do infectious agents contribute to progression of Alzheimer's disease? Alzheimers Dement. 2009;5:348–60. doi: 10.1016/j.jalz.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 66.Miklossy J. Alzheimer's disease – a neurospirochetosis. Analysis of the evidence following Koch's and Hill's criteria. J Neuroinflammation. 2011;8:90. doi: 10.1186/1742-2094-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alfahad T, Nath A. Retroviruses and amyotrophic lateral sclerosis. Antiviral Res. 2013;99:180–7. doi: 10.1016/j.antiviral.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hack N, Jicha GA, Abell A, et al. Substantia nigra depigmentation and exposure to encephalitis lethargica. Ann Neurol. 2012;72:912–7. doi: 10.1002/ana.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De Chiara G, Marcocci ME, Sgarbanti R, et al. Infectious agents and neurodegeneration. Mol Neurobiol. 2012;46:614–38. doi: 10.1007/s12035-012-8320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Virtanen JO, Jacobson S. Viruses and multiple sclerosis. CNS Neurol Disord Drug Targets. 2012;11:528–44. doi: 10.2174/187152712801661220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Del Palacio M, Alvarez S, Muñoz-Fernández MÁ. HIV-1 infection and neurocognitive impairment in the current era. Rev Med Virol. 2012;22:33–45. doi: 10.1002/rmv.711. [DOI] [PubMed] [Google Scholar]
- 72.Clark KH, Wiley CA, Bradberry CW. Psychostimulant abuse and neuroinflammation: emerging evidence of their interconnection. Neurotox Res. 2013;23:174–88. doi: 10.1007/s12640-012-9334-7. [DOI] [PubMed] [Google Scholar]
- 73.Lewis JA, Gehman EA, Baer CE, et al. Alterations in gene expression in Caenorhabditis elegans associated with organophosphate pesticide intoxication and recovery. BMC Genomics. 2013;14:291. doi: 10.1186/1471-2164-14-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kern JK, Geier DA, Audhya T, et al. Evidence of parallels between mercury intoxication and the brain pathology in autism. Acta Neurobiol Exp (Wars) 2012;72:113–53. doi: 10.55782/ane-2012-1887. [DOI] [PubMed] [Google Scholar]
- 75.Harris RA, Amor S. Sweet and sour – oxidative and carbonyl stress in neurological disorders. CNS Neurol Disord Drug Targets. 2011;10:82–107. doi: 10.2174/187152711794488656. [DOI] [PubMed] [Google Scholar]
- 76.Laird MD, Shields JS, Sukumari-Ramesh S, et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia. 2014;62:26–38. doi: 10.1002/glia.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Min HJ, Ko EA, Wu J, et al. Chaperone-like activity of high-mobility group box 1 protein and its role in reducing the formation of polyglutamine aggregates. J Immunol. 2013;190:1797–806. doi: 10.4049/jimmunol.1202472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Streit WJ, Xue Q-S. The brain's aging immune system. Aging Dis. 2010;1:254–61. [PMC free article] [PubMed] [Google Scholar]
- 79.Miron VE, Boyd A, Zhao J-W, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–8. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goronzy JJ, Li G, Yang Z, et al. The janus head of T cell aging – autoimmunity and immunodeficiency. Front Immunol. 2013;4:131. doi: 10.3389/fimmu.2013.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ajami B, Bennett JL, Krieger C, et al. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142–9. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 82.Villeda SA, Luo J, Mosher KI, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–4. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Biber K, Tsuda M, Tozaki-Saitoh H, et al. Neuronal CCL21 up-regulates microglia P2X4 expression and initiates neuropathic pain development. EMBO J. 2011;30:1864–73. doi: 10.1038/emboj.2011.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Durafourt BA, Moore CS, Zammit DA, et al. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012;60:717–27. doi: 10.1002/glia.22298. [DOI] [PubMed] [Google Scholar]
- 85.Melief J, Koning N, Schuurman KG, et al. Phenotyping primary human microglia: tight regulation of LPS responsiveness. Glia. 2012;60:1506–17. doi: 10.1002/glia.22370. [DOI] [PubMed] [Google Scholar]
- 86.Elliott C, Lindner M, Arthur A, et al. Functional identification of pathogenic autoantibody responses in patients with multiple sclerosis. Brain. 2012;135:1819–33. doi: 10.1093/brain/aws105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Crehan H, Hardy J, Pocock J. Microglia, Alzheimer's disease, and complement. Int J Alzheimers Dis. 2012;2012:983640. doi: 10.1155/2012/983640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee JD, Kamaruzaman NA, Fung JN, et al. Dysregulation of the complement cascade in the hsod1G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. 2013;10:119. doi: 10.1186/1742-2094-10-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lobsiger CS, Boillée S, Pozniak C, et al. C1q induction and global complement pathway activation do not contribute to ALS toxicity in mutant SOD1 mice. Proc Natl Acad Sci USA. 2013;110:E4385–92. doi: 10.1073/pnas.1318309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ramaglia V, King RHM, Morgan BP, et al. Deficiency of the complement regulator CD59a exacerbates Wallerian degeneration. Mol Immunol. 2009;46:1892–6. doi: 10.1016/j.molimm.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 91.Schwartz M, Kipnis J, Rivest S, et al. How do immune cells support and shape the brain in health, disease, and aging? J Neurosci. 2013;33:17587–96. doi: 10.1523/JNEUROSCI.3241-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gleichman AJ, Carmichael ST. Astrocytic therapies for neuronal repair in stroke. Neurosci Lett. 2013 doi: 10.1016/j.neulet.2013.10.055. doi: 10.1016/j.neulet.2013.10.055. [DOI] [PubMed] [Google Scholar]
- 93.White RE, Jakeman LB. Don't fence me in: harnessing the beneficial roles of astrocytes for spinal cord repair. Restor Neurol Neurosci. 2008;26:197–214. [PMC free article] [PubMed] [Google Scholar]
- 94.Chisholm SP, Cervi AL, Nagpal S, et al. Interleukin-17A increases neurite outgrowth from adult postganglionic sympathetic neurons. J Neurosci. 2012;32:1146–55. doi: 10.1523/JNEUROSCI.5343-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kipnis J, Gadani S, Derecki NC. Pro-cognitive properties of T cells. Nat Rev Immunol. 2012;12:663–9. doi: 10.1038/nri3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carson JH, Gao Y, Tatavarty V, et al. Multiplexed RNA trafficking in oligodendrocytes and neurons. Biochim Biophys Acta. 2008;1779:453–8. doi: 10.1016/j.bbagrm.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Frühbeis C, Fröhlich D, Kuo WP, et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte–neuron communication. PLoS Biol. 2013;11:e1001604. doi: 10.1371/journal.pbio.1001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rajendran L, Honsho M, Zahn TR, et al. Alzheimer's disease β-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci USA. 2006;103:11172–7. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jaturapatporn D, Isaac MG, McCleery J, et al. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer's disease. Cochrane Database Syst Rev. 2012;2:CD006378. doi: 10.1002/14651858.CD006378.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Castillo-Trivino T, Braithwaite D, Bacchetti P, et al. Rituximab in relapsing and progressive forms of multiple sclerosis: a systematic review. PLoS One. 2013;8:e66308. doi: 10.1371/journal.pone.0066308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li C, Yuan K, Schluesener H. Impact of minocycline on neurodegenerative diseases in rodents: a meta-analysis. Rev Neurosci. 2013;24:553–62. doi: 10.1515/revneuro-2013-0040. [DOI] [PubMed] [Google Scholar]
- 102.Morsali D, Bechtold D, Lee W, et al. Safinamide and flecainide protect axons and reduce microglial activation in models of multiple sclerosis. Brain. 2013;136:1067–82. doi: 10.1093/brain/awt041. [DOI] [PubMed] [Google Scholar]
- 103.Raftopoulos RE, Kapoor R. Neuroprotection for acute optic neuritis—Can it work? Mult Scler Relat Disord. 2013;2:307–11. doi: 10.1016/j.msard.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 104.Pryce G, Ahmed Z, Hankey DJR, et al. Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain. 2003;126:2191–202. doi: 10.1093/brain/awg224. [DOI] [PubMed] [Google Scholar]
- 105.Zajicek J, Ball S, Wright D, et al. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol. 2013;12:857–65. doi: 10.1016/S1474-4422(13)70159-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Butti E, Bacigaluppi M, Rossi S, et al. Subventricular zone neural progenitors protect striatal neurons from glutamatergic excitotoxicity. Brain. 2012;135:3320–35. doi: 10.1093/brain/aws194. [DOI] [PubMed] [Google Scholar]
- 107.Pryce G, Baker D. Potential control of multiple sclerosis by cannabis and the endocannabinoid system. CNS Neurol Disord Drug Targets. 2012;11:624–41. doi: 10.2174/187152712801661310. [DOI] [PubMed] [Google Scholar]
- 108.Laterza C, Merlini A, De Feo D, et al. iPSC-derived neural precursors exert a neuroprotective role in immune-mediated demyelination via the secretion of LIF. Nat Commun. 2013;4:2597. doi: 10.1038/ncomms3597. [DOI] [PubMed] [Google Scholar]
- 109.Kokaia Z, Martino G, Schwartz M, et al. Cross-talk between neural stem cells and immune cells: the key to better brain repair? Nat Neurosci. 2012;15:1078–87. doi: 10.1038/nn.3163. [DOI] [PubMed] [Google Scholar]