

Abstract
Kallistatin, a plasma protein, has been shown to exert multi-factorial functions including inhibition of inflammation, oxidative stress and apoptosis in animal models and cultured cells. Kallistatin levels are reduced in patients with sepsis and in lipopolysaccharide (LPS)-induced septic mice. Moreover, transgenic mice expressing kallistatin are more resistant to LPS-induced mortality. Here, we investigated the effects of human kallistatin on organ injury and survival in a mouse model of polymicrobial sepsis. In this study, mice were injected intravenously with recombinant kallistatin (KS3, 3 mg/kg; or KS10, 10 mg/kg body weight) and then rendered septic by caecal ligation and puncture 30 min later. Kallistatin administration resulted in a > 10-fold reduction of peritoneal bacterial counts, and significantly decreased serum tumour necrosis factor-α, interleukin-6 and high mobility group box-1 (HMGB1) levels. Kallistatin also inhibited HMGB1 and toll-like receptor-4 gene expression in the lung and kidney. Administration of kallistatin attenuated renal damage and decreased blood urea nitrogen and serum creatinine levels, but increased endothelial nitric oxide synthase and nitric oxide levels in the kidney. In cultured endothelial cells, human kallistatin via its heparin-binding site inhibited HMGB1-induced nuclear factor-κB activation and inflammatory gene expression. Moreover, kallistatin significantly reduced apoptosis and caspase-3 activity in the spleen. Furthermore, kallistatin treatment markedly improved the survival of septic mice by 23% (KS3) and 41% (KS10). These results indicate that kallistatin is a unique protecting agent in sepsis-induced organ damage and mortality by inhibiting inflammation and apoptosis, as well as enhancing bacterial clearance in a mouse model of polymicrobial sepsis.
Keywords: high mobility group box-1, kallistatin, organ injury, sepsis, survival
Introduction
Sepsis is one of the leading causes of mortality in intensive care units and continues to be a challenge for clinical investigators.1 Sepsis is a systemic inflammatory response to infection that can lead to multi-organ dysfunction. Since sepsis syndrome is the result of excessive inflammation, anti-inflammatory strategies have been investigated with favourable effects in animal models of sepsis but with marginal results in humans.2 Although numerous signalling cascades are triggered during septic shock, selective blocking of inflammatory mediators is not sufficient to arrest this process. Indeed, clinical trials for sepsis have been referred to as the ‘graveyard for pharmaceutical companies’.2 Therefore, the development of novel strategies is crucial to provide an effective treatment of severe sepsis and septic shock in humans.
Kallistatin, a human plasma protein, was first identified as a tissue kallikrein-binding protein, kallikrein inhibitor and a unique serine proteinase inhibitor.3–5 Kallistatin exerts multi-factorial properties, such as anti-inflammatory, anti-angiogenic, anti-apoptotic and anti-fibrotic actions, in experimental animal models and cultured cells, independent of its interaction with tissue kallikrein.6–12 Kallistatin administration by gene delivery has been shown to protect against cardiovascular and renal injury, inflammation and apoptosis, whereas kallistatin depletion by antibody injection exacerbates organ damage and inflammation in hypertensive rats.10,13,14 Importantly, transgenic mice over-expressing kallistatin are more resistant to lipopolysaccharide (LPS) -induced mortality than their control littermates.15 Kallistatin gene transfer also attenuates Gram-positive streptococcus-induced mortality, inflammation and liver damage in mice.16 Furthermore, human kallistatin levels are markedly reduced in patients with sepsis and liver diseases, inflammatory bowel diseases and in community-acquired pneumonia.17–19 These findings indicate that kallistatin, via its pleiotropic properties, may provide a promising approach for prevention and treatment of septic shock. In this study, we investigated the effects of human kallistatin administration on organ injury, inflammation, apoptosis and survival in a mouse model of polymicrobial sepsis.
Materials and methods
Purification and characterization of recombinant human kallistatin
Recombinant human kallistatin was secreted into the serum-free medium of cultured human embryonic kidney cells (HEK293T), and cultured medium was concentrated by ammonium sulphate precipitation followed by nickel-affinity chromatography as described previously.20 Recombinant wild-type kallistatin, heparin-mutant and active-site mutant were expressed in Escherichia coli and purified as described elsewhere.20 The purity and identity of human kallistatin were verified by SDS–PAGE and Western blot using a specific monoclonal antibody.17
Caecal ligation and puncture, and animal treatment
CD-1 mice (male, 7–8 weeks old; Harlan, Indianapolis, IN) were housed in a germ-free environment. All procedures complied with the standards for care and use of animal subjects as stated in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Resources, National Academy of Sciences, Bethesda, MD). The protocol for all animal studies was approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina. Caecal ligation and puncture (CLP) was performed as described previously.21 Briefly, mice were anaesthetized with isoflurane and a 2-cm midline incision was made below the diaphragm to expose the caecum. The caecum was first ligated at the colon juncture with a 5-0 silk ligature suture without interrupting intestinal continuity, punctured twice with a 21-gauge needle, then laced back in the abdomen, and the incision was closed in two layers with a 5-0 silk ligature suture. All animals were then fluid-resuscitated with 0·8 ml normal sterile saline subcutaneously. Sham operation was performed in the same way as CLP, but without ligation and puncture of the caecum. Mice were randomly assigned to one of four groups (n = 8 per group): (i) Sham group receiving PBS; (ii) CLP control group receiving PBS; (iii) CLP + KS3 group receiving 3 mg/kg body weight of kallistatin; and (iv) CLP + KS10 group receiving 10 mg/kg body weight of kallistatin. Mice were injected intravenously with 0·1 ml PBS or kallistatin in 0·1 ml PBS 30 min before surgery. Mice were killed 24 hr after CLP, and peritoneal fluid, serum and tissues were collected for biochemical and histological analyses. Human kallistatin levels in the serum were determined by an ELISA specific for human kallistatin.17
For the survival study, mice were randomly assigned to one of four groups (n = 16 per group): (i) Sham group receiving PBS; (ii) CLP control group receiving PBS; (iii) CLP + KS3 group receiving 3 mg/kg body weight of kallistatin; and (iv) CLP + KS10 group receiving 10 mg/kg body weight of kallistatin. Mice were injected intravenously with 0·1 ml PBS or kallistatin in 0·1 ml PBS 30 min before surgery. Mouse survival was monitored every 24 hr for a total of 144 hr.
Bacterial load
Bacterial load in the peritoneal cavity was determined 24 hr after Sham or CLP surgery (n = 5 per group). The peritoneal cavity was lavaged with 5 ml sterile PBS and then diluted with sterile PBS. One hundred microlitres of each dilution was then cultured on chocolate agar plates (Fisher Scientific, Pittsburgh, PA) at 37° under aerobic conditions. Colony-forming units were counted 24 hr later.
Blood urea nitrogen, serum creatinine, tumour necrosis factor-α, interleukin-6 and high mobility group box-1 measurements
Blood urea nitrogen and serum creatinine were measured with a QuantiChrom Urea Assay Kit and a QuantiChrom Creatinine Assay Kit (BioAssay Systems, Hayward, CA), respectively, according to the manufacturer's instructions. Serum tumour necrosis factor-α (TNF-α) levels were determined by a mouse TNF-α ELISA kit (EMD Millipore, Billerica, MA). Interleukin-6 (IL-6) levels in serum were measured using a mouse IL-6 ELISA kit (eBioscience, San Diego, CA). High mobility group box-1 (HMGB1) levels in serum were examined by a mouse HMGB1 ELISA kit (Antibodies-online Inc., Atlanta, GA).
Tissue staining
For histological studies, tissues were fixed with 4% paraformaldehyde, dehydrated, embedded and cut into 4-μm sections. Periodic Acid–Schiff reagent (PAS) staining was performed as described previously.14 Primary antibody against endothelial nitric oxide synthase (eNOS, 1 : 200; Cell Signaling, Boston, MA) was used for immunostaining. For apoptosis analysis, terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) staining was performed in spleen sections using a TUNEL kit according to the manufacturer's instructions (Roche, Indianapolis, IN).
Cell culture studies
Human umbilical vein endothelial cells (HUVECs) were maintained in endothelial basal medium-2 (EBM-2; Lonza, Walkersville, MD) supplemented with EBM-2 SingleQuots (Lonza). Cells were maintained at 37° in 5% CO2, 95% air. Passages 3–6 were used. Cells were pre-treated with 0·5 μm kallistatin for 30 min followed by stimulation with recombinant human HMGB1 (rhHMGB1; 400 ng/ml; R&D Systems, Minneapolis, MN) or medium alone for 24 hr. Total RNA was extracted and the mRNA levels of TNF-α, vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) were measured by real-time RT-PCR. To determine the role of kallistatin's heparin-binding domain in rhHMGB1-induced phosphorylated nuclear factor-κB (NF-κB) expression in HUVECs, cells were incubated with wild-type kallistatin (2 μm), heparin-mutant kallistatin (2 μm) or active site-mutant kallistatin (2 μm) for 30 min and then stimulated with HMGB1 (1 μg/ml) for another 16 hr. Phosphorylated p65 NF-κB activities in nuclear lysates were determined using ELISA (Cell Signaling Technology, Danvers, MA). To determine the role of kallistatin's heparin-binding domain in rhHMGB1-induced inflammatory gene expression in HUVECs cells were incubated with wild-type kallistatin (2 μm), heparin-mutant kallistatin (2 μm) or active site-mutant kallistatin (2 μm) for 30 min and then stimulated with HMGB1 (400 ng/ml) for another 24 hr. Total RNA was extracted and the mRNA levels of TNF-α and VCAM-1 were measured by real-time RT-PCR.
Caspase-3 activity assay and nitrate/nitrite (NOX) measurement
Caspase-3 activity was determined with a caspase-3 colorimetric activity assay kit (EMD Millipore Corporation, Temecula, CA) according to the manufacturer's instructions. Briefly, tissues were extracted in lysis buffer (50 mm HEPES, pH 7·4, 100 mm NaCl, 0·1% CHAPS, 1 mm dithiothreitol, 0·1 mm EDTA) on ice for 10 min, then centrifuged for collection of the supernatant. Supernatants were transferred to a fresh tube and the protein concentration for each sample was measured. Equal volumes of the lysates were incubated with assay mixture (including colorimetric substrate) at 37° for 1 hr. The fluorescence intensity (405 nm) was recorded at room temperature. NOX levels, an index of nitric oxide (NO) levels, in kidney tissue were determined by a fluorometric assay as previously described.11
Western blot analysis
Kidney tissues were minced and lysed with ice-cold RIPA lysis buffer (10 mm Tris–HCl, pH 7·4, 1% Triton X-100, 150 mm NaCl, 1 mm EGTA, 1 mm EDTA, 1 mm phenylmethylsulphonyl fluoride, 1 μg/ml aprotinin, 1 μg/ml leupeptin and 1 μg/ml pepstatin A). All lysed samples were kept on ice for 30 min, and centrifuged for 10 min at 4° at 12 000 g. The supernatant was collected and stored at −20° until further analysis.
For Western blotting, lysates were subjected to 10% SDS–PAGE and transferred onto a polyvinylidene difluoride membrane. The membranes were blocked with 7% milk in TBST (20 mm Tris–HCl, 500 mm NaCl, and 0·1% Tween-20) for 1 hr. After washing with TBST twice, membranes were incubated with primary antibody overnight at 4°. The following primary antibodies were used: monoclonal anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (BioAbchem, Ladson, SC); monoclonal anti-HMGB1 antibody (Cell Signaling, Boston, MA), and polyclonal anti-Toll-like receptor-4 (TLR4) antibody (Santa Cruz, Dallas, TX). The membranes were washed twice with TBST and incubated with horseradish peroxidase-conjugated secondary antibody in blocking buffer for 1 hr. After washing three times with TBST, immunoreactive bands were visualized by incubation with ECL plus detection reagents (GE Healthcare, Waukesha, WI) for 5 min and exposure to BioMax light film (Thermo Scientific, Waltham, MA). The densitometry of bands was quantified with imagej2× software (National Institutes of Health, Bethesda, MD).
Real-time RT-PCR
Total RNA was extracted from lung tissues, kidney tissues and cultured endothelial cells with TRIzol Reagent (Invitrogen, Carlsbad, CA) following the manufacturer's protocol. The cDNA was synthesized using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. Quantitative real-time PCR was performed by Prism 7300 Real Time PCR System (Applied Biosystems) using TaqMan Gene Expression Master Mix (Applied Biosystems) in a final reaction volume of 20 μl with each primer. The following primers were purchased from Applied Biosystems: human 18S (Hs 99999901_sl), human TNF-α (Hs 00174128_ml), human VCAM-1 (Hs 00365486_ml), human ICAM-1 (Hs 00277001_ml), mouse GAPDH (Mm 99999915_gl), mouse VCAM-1 (Mm01320970_ml), mouse ICAM-1 (Mm00516024_g1), mouse HMGB1 (Mm 00849805_gl) and mouse TLR4 (Mm 00445274_m1). A negative control without cDNA did not produce any amplicons. Data were analysed with 2−ΔΔCt value calculation using 18S or GAPDH for normalization.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SE). Statistical significance was determined by analysis of variance with Fisher's probable least-squares difference test or log-rank (Mantel–Cox) test using graphpad prism software (GraphPad Software, Inc., La Jolla, CA). A value of P < 0·05 was considered statistically significant.
Results
Human kallistatin administration reduces peritoneal bacterial counts in CLP mice
Purified human kallistatin was identified by Coomassie blue staining and Western blotting using a specific monoclonal antibody against human kallistatin (Fig. 1a). The lower band (52 000 molecular weight) is mature kallistatin, and the upper band (104 000) is the kallistatin dimer. Human kallistatin was injected intravenously into CLP mice 30 min before surgery. Human kallistatin was detected in mouse serum at 24 hr after injection in the kallistatin-treated groups, but not in the Sham or CLP control groups (CLP + KS3: 16·2 ± 5·3 ng/ml, CLP + KS10: 56·8 ± 9·9 ng/ml; n = 6, Fig. 1b). The bacterial load in the peritoneal cavity was determined by colony-forming unit count by plating of serially diluted samples on chocolate agar plates. Mice receiving kallistatin treatment exhibited a > 10-fold reduction in bacterial counts compared with CLP control mice (CLP + KS3: 89·7 ± 4·9% reduction, CLP + KS10: 92·5 ± 1·7% reduction; n = 5, P < 0·05; Fig. 1c).
Figure 1.
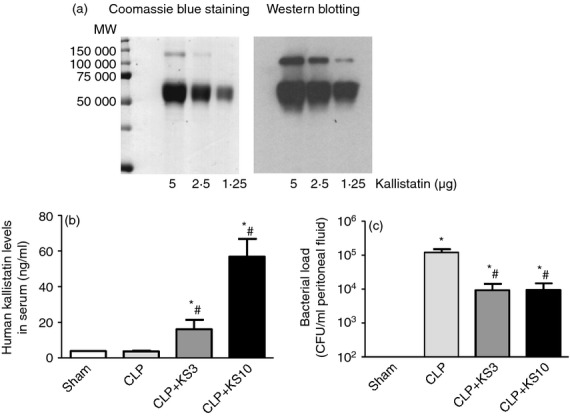
Kallistatin treatment reduces bacterial load in caecal ligation and puncture (CLP) mice. (a) Purified recombinant human kallistatin was identified by Coomassie blue staining and Western blot. Lower band: mature kallistatin at 52 000 molecular weight; upper band: kallistatin dimer at 104 000 molecular weight. (b) Recombinant human kallistatin was detected in the serum of CLP mice after injection of kallistatin at 3 mg/kg (KS3) or 10 mg/kg (KS10), but not in the sham-operated or CLP control mice (n = 6). (c) Bacterial load in peritoneal fluid (n = 5) of kallistatin-treated mice was significantly lower than that of the CLP control mice. Data are expressed as means ± standard error of the mean (SE). *P < 0·05 compared with the Sham group. # P < 0·05 compared with the CLP control group.
Kallistatin reduces serum cytokine levels and inhibits inflammatory gene expression in the lung of CLP mice
Kallistatin administration significantly reduced TNF-α levels (CLP control: 29·2 ± 3·4 pg/ml, CLP + KS3: 13·7 ± 3·9 pg/ml, CLP + KS10: 13·5 ± 1·6 pg/ml, n = 3 or n = 4, P < 0·05; Fig. 2a) and IL-6 levels (CLP control: 28·4 ± 1·8 ng/ml, CLP + KS3: 15·1 ± 3·2 ng/ml, CLP + KS10: 7·7 ± 3·3 ng/ml, n = 3 or n = 4, P < 0·05; Fig. 2b) in mouse serum, compared with the CLP control group. Kallistatin treatment also reduced serum HMGB1 levels, compared with the CLP control group (CLP control: 44·91 ± 8·7 ng/ml, CLP + KS3: 18·1 ± 2·5 ng/ml, CLP + KS10: 20·1 ± 2·9 ng/ml, n = 4, P < 0·05; Fig. 2c). As shown in Fig. 2(d,e), lung mRNA levels of HMGB1 and TLR4 were markedly increased after CLP, but significantly inhibited by kallistatin treatment (n = 4, P < 0·05).
Figure 2.
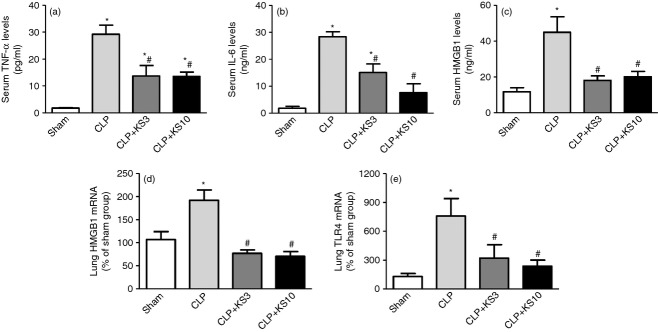
Kallistatin treatment reduces serum tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6) and high mobility group box-1 (HMGB1) levels, and decreases lung HMGB1 and toll-like receptor-4 (TLR4) mRNA levels in caecal ligation and puncture (CLP) mice. (a) Serum TNF-α levels (n = 3 or n = 4), (b) serum IL-6 levels (n = 3 or n = 4) and (c) serum HMGB1 levels (n = 4) were significantly decreased in kallistatin-administered mice compared with that of the CLP control mice. Lung mRNA levels of HMGB1 (d) and TLR4 (e) were significantly reduced by kallistatin treatment (n = 4). Data are expressed as means ± standard error of the mean (SE). * P < 0·05 compared with the Sham group. # P < 0·05 compared with the CLP control group.
Kallistatin attenuates renal injury and dysfunction, but increases eNOS and NOx levels in CLP mice
Periodic Acid–Schiff reagent staining was performed to evaluate the effect of kallistatin on CLP-induced renal injury 24 hr after CLP surgery. Histological findings in the Sham group showed renal tubules with no damage. Dilated renal tubules and swollen tubular cells were observed in the CLP control group, and slightly thickened basement membranes of renal tubules were found in CLP mice with kallistatin administration. Mice in the Sham and kallistatin-treated groups exhibited greater PAS-positive and eNOS-positive intensity than the CLP control group (Fig. 3a). Blood urea nitrogen levels were significantly increased in the CLP control group (39·8 ± 6·1 mg/dl, n = 8, P < 0·05) compared with the Sham group (17·7 ± 2·4 mg/dl, n = 3), but significantly reduced by kallistatin administration (CLP + KS3: 18·6 ± 3·3 mg/dl, CLP + KS10: 21·5 ± 2·1 mg/dl; n = 5, P < 0·05; Fig. 3b). Kallistatin treatment also decreased CLP-induced serum creatintine levels (CLP control: 0·57 ± 0·03 mg/dl, CLP + KS3: 0·38 ± 0·04 mg/dl, CLP + KS10: 0·37 ± 0·05 mg/dl; n = 3 to n = 6, P < 0·05; Fig. 3c). Mice receiving kallistatin treatment, but not the CLP control mice, exhibited significantly increased NO formation compared with the Sham group (CLP + KS3: 2·6 ± 0·5 fold increase, CLP + KS10: 3·1 ± 0·4 fold increase; n = 4, P < 0·05; Fig. 3d).
Figure 3.

Kallistatin treatment ameliorates renal tubular injury and reduces blood urea nitrogen (BUN) and serum creatinine in caecal ligation and puncture (CLP) mice, but increases endothelial nitric oxide synthase (eNOS) expression and nitrate/nitrite (NOx) levels. (a) Periodic Acid–Schiff reagent (PAS) staining and eNOS immunohistochemistry was performed on kidney sections to examine renal histology and eNOS expression (n = 6). The representative sections are shown at ×200 magnification. (b) BUN (n = 3 to n = 8) and (c) serum creatinine (n = 3 to n = 8) levels were significantly decreased in mice receiving human kallistatin compared with the CLP control group. (d) NOx levels were significantly increased by kallistatin administration in CLP mice (n = 4). ‘→’ represents irregular membrane and brush loss in renal PAS staining or eNOS-positive cells in renal eNOS immunohistochemistry. Data represent means ± standard error of the mean (SE) from two independent experiments. *P < 0·05 compared with the Sham group. # P < 0·05 compared with the CLP control group.
Kallistatin inhibits the expression of pro-inflammatory genes in the kidney of CLP mice
Kallistatin treatment markedly decreased CLP-induced renal HMGB1 and TLR4 mRNA and protein levels compared with the CLP control group as determined by real-time PCR and Western blot (n = 3 to n = 6, P < 0·05; Fig. 4a–c). Kallistatin treatment also significantly inhibited CLP-induced renal VCAM-1 and ICAM-1 mRNA levels compared with the CLP control group (n = 3, P < 0·05; Fig. 4d,e).
Figure 4.

Kallistatin treatment reduces high mobility group box-1 (HMGB1), toll-like receptor-4 (TLR4), vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1) expression in kidney tissue of caecal ligation and puncture mice. Levels of (a, c) HMGB1, (b, c) TLR4, (d) VCAM-1 and (e) ICAM-1 were significantly reduced in kallistatin-treated mice (n = 3 to n = 6). Data are expressed as means ± standard error of the mean (SE). * P < 0·05 compared with the other groups.
Kallistatin antagonizes HMGB1-induced NF-κB activation and inflammatory gene expression in cultured endothelial cells
High levels of HMGB1 are associated with severe systemic inflammation, organ damage and high rate of mortality in sepsis.22 Our in vivo studies showed that kallistatin treatment decreased HMGB1 expression in the serum, lung and kidney of CLP-treated mice. Therefore, we further examined the effect of kallistatin on HMGB1-induced inflammatory response in cultured endothelial cells. TNF-α, VCAM-1 and ICAM-1 mRNA levels in HUVECs were increased upon stimulation with HMGB1, but were significantly suppressed by recombinant human kallistatin (n = 6, P < 0·05; Fig. 5a–c). Kallistatin contains two important structural elements: an active site for binding to tissue kallikrein and a heparin-binding site.23,24 Kallistatin, via its heparin-binding site, has been shown to inhibit TNF-α-mediated inflammation by competing with TNF-α binding to its receptor through heparan sulphate proteoglycans.12 Like TNF-α, HMGB1 binding to its receptor is largely dependent on heparan sulphate.25 Therefore, we further investigated the role of kallistatin's structural elements in HMGB1-mediated NF-κB activation and inflammatory gene expression in cultured endothelial cells. Wild-type kallistatin alone had no effect on activation of NF-κB (Fig. 5d), and neither did heparin-mutant or active-site mutant kallistatin (data not shown). Treatment with HMGB1 significantly increased NF-κB activation, and the increase was inhibited by wild-type kallistatin and active-site mutant kallistatin, but not the heparin-mutant kallistatin (n = 3, P < 0·05). In addition, wild-type kallistatin and active-site mutant kallistatin, but not heparin-mutant kallistatin, significantly reduced HMGB1-induced TNF-α and VCAM-1 mRNA levels in endothelial cells (n = 4, P < 0·05; Fig. 5e,f). These new findings indicate that kallistatin via its heparin-binding site exerts anti-inflammatory effects by inhibiting HMGB1-mediated inflammation response in a mouse model of polymicrobial sepsis.
Figure 5.
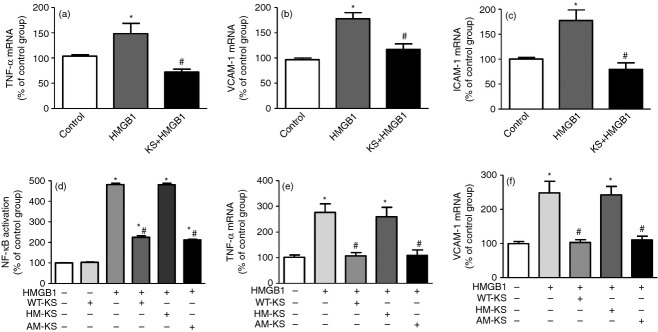
Kallistatin inhibits recombinant human high mobility group box-1 (HMGB1) -induced nuclear factor-κB (NF-κB) activation and inflammatory gene expression via its heparin-binding site in human endothelial cells. Endothelial cells were incubated with or without kallistatin (0·5 μm) for 30 min and then stimulated with HMGB1 (400 ng/ml) for another 24 hr. Kallistatin treatment significantly decreased HMGB1-induced (a) tumour necrosis factor-α (TNF-α), (b) vascular cell adhesion molecule 1 (VCAM-1) and (c) intercellular adhesion molecule 1 (ICAM-1) expression (n = 6). In subsequent studies, endothelial cells were incubated with wild-type kallistatin (WT-KS; 2 μm), heparin-mutant kallistatin (HM-KS; 2 μm) or active site-mutant kallistatin (AM-KS; 2 μm) for 30 min, and then stimulated with HMGB1 (1 μg/ml for another 16 hr or 400 ng/ml for another 24 hr). The wild-type or active site-mutant kallistatin, but not the heparin-mutant kallistatin, significantly inhibited HMGB1-induced (d) NF-κB activation, (e) TNF-α and (f) VCAM-1 expression (n = 3 to n = 4). Data are expressed as means ± standard error of the mean (SE) from two independent experiments. * P < 0·05 compared with the control group. # P < 0·05 compared with the HMGB1 alone group.
Kallistatin inhibits apoptosis and caspase-3 activity in the spleen of CLP mice
Caecal ligation and puncture-treated mice exhibited a marked increase in TUNEL-positive cells in the spleen as shown by representative TUNEL staining images (Fig. 6a). However, cell apoptosis in the spleen was attenuated by kallistatin administration at both the 3 mg/kg and 10 mg/kg doses. Quantitative analysis confirmed this finding (Fig. 6b). Our results further demonstrated that kallistatin significantly decreased CLP-induced caspase-3 activity in the spleen (CLP + KS3: 46·6 ± 11·3% reduction, CLP + KS10: 47 ± 4·9% reduction; n = 4, P < 0·05; Fig. 6c).
Figure 6.
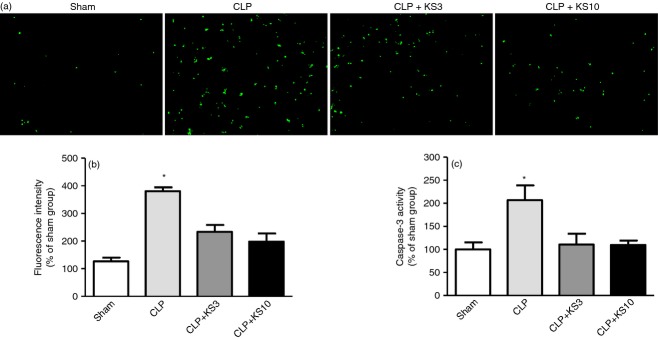
Kallistatin inhibits caecal ligation and puncture (CLP)-induced apoptosis in white pulp of the spleen. (a) Representative images of terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) staining are shown at ×200 magnification (n = 3, TUNEL stains green). (b) Quantitative analysis showed that kallistatin treatment significantly inhibited CLP-induced apoptosis. (c) Caspase-3 activity of kallistatin-treated mice was significantly decreased compared with that of CLP control mice (n = 4). Data are expressed as means ± standard error of the mean (SE). * P < 0·05 compared with the other groups.
Kallistatin reduces CLP-induced mortality
To determine the effect of kallistatin on CLP-induced mortality, PBS or kallistatin (3 mg/kg or 10 mg/kg) was injected intravenously into mice 30 min before CLP surgery. Mouse survival was monitored every 24 hr for a total of 6 days. Sham-operated mice (n = 16) exhibited 100% survival. All CLP control mice died within 6 days after CLP, whereas treatment with kallistatin increased the mean survival rate by 23% (KS3) and 41% (KS10) compared with the CLP control mice (n = 16, P < 0·05; Fig. 7).
Figure 7.
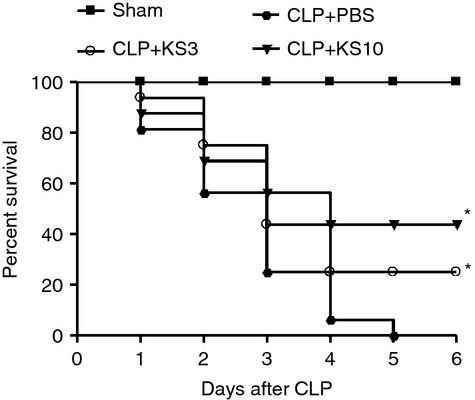
Kallistatin treatment improves survival in septic mice. Mice receiving 3 mg/kg (KS3) or 10 mg/kg (KS10) of human kallistatin presented significantly reduced mortality rate during the observation period compared with caecal ligation and puncture (CLP) control mice (n = 16); Sham-operated mice exhibited 100% survival (n = 16). * P < 0·05 compared with the CLP control group.
Discussion
This study demonstrated that administration of human kallistatin protein exerts protective effects against organ damage and mortality in mice with polymicrobial sepsis. Kallistatin administration in CLP mice significantly reduced the serum levels of several inflammatory cytokines (TNF-α, IL-6 and HMGB1). Kallistatin treatment also reduced CLP-induced renal damage, dysfunction and inflammatory gene expression. The renal protective effects of kallistatin is, in part, attributed to NO bioavailability, as kallistatin administration increased eNOS and NO levels in CLP mice. Kallistatin attenuated sepsis-induced apoptosis and caspase-3 activity in the spleen. Importantly, human kallistatin treatment improved the survival of CLP mice. Moreover, kallistatin administration in CLP mice resulted in a > 10-fold decrease of bacterial counts in the peritoneal fluid. Likewise, we recently demonstrated that human kallistatin expression leads to significant bacterial clearance and antimicrobial activity in Group A streptococcus-infected mice by increased superoxide production in immune cells.16 Kallistatin also enhanced immune cell viability and reduced their apoptosis.16 Therefore, kallistatin may reduce bacterial counts by enhancing immune cell survival and promoting immune cell antimicrobial activity in septic mice. These findings indicate that kallistatin plays a promising role in protection against sepsis-induced lethality and organ damage by inhibition of systemic inflammation and apoptosis, as well as enhancement of anti-microbial activity in CLP septic mice.
This is the first study to demonstrate that kallistatin via its heparin-binding site antagonizes HMGB1-induced NF-κB activation and pro-inflammatory gene expression in cultured endothelial cells. Systemic inflammation, organ damage and high rate of mortality are characteristics of severe sepsis, and correlate with the expression of HMGB1, a late pro-inflammatory cytokine.22 High levels of HMGB1 are associated with severe sepsis in animals and humans. Under pathological conditions, HMGB1 interacts with receptors like TLR4 and TLR2, resulting in signal transduction events that elicit inflammatory responses.26 TLR4 is considered to be the critical receptor mediating the inflammatory activity of HMGB1. Administration of anti-HMGB1 antibody was found to markedly attenuate lethality of mice with established sepsis.27 The present study revealed that kallistatin treatment reduced serum HMGB1 levels, and decreased HMGB1 and TLR4 gene expression in the lung and kidney of CLP mice. We also showed that kallistatin reduced HMGB1-induced NF-κB activation and pro-inflammatory gene expression via its heparin-binding site in cultured endothelial cells. HMGB1 expression has been shown to be up-regulated by NF-κB activation through an autocrine mechanism.28 Hence, kallistatin not only antagonizes HMGB1's inflammatory response, but may also down-regulate HMGB1 expression through a positive feedback loop. These new findings uncover a novel role of kallistatin in protection against sepsis-induced organ damage by suppressing HMGB1 activation and expression. Kallistatin is therefore a unique protective agent in CLP-induced organ damage and mortality through inhibition of HMGB1-mediated activity.
Sepsis leads to multi-organ dysfunction via a systemic inflammatory response to infection. Acute kidney injury occurs in sepsis and is associated with sepsis-induced mortality.29 We have demonstrated that kallistatin exerts multifunctional activity in protection against cardiovascular and renal injury by inhibiting inflammation and apoptosis in animal models and in cultured cells.10,12,13 A recent study found that polymorphism of the kallistatin gene (SERPINA4) is strongly associated with a decreased risk of developing acute kidney injury in patients with septic shock.30 Moreover, plasma kallistatin levels are markedly reduced in patients with septic shock.17 Furthermore, transgenic mice expressing kallistatin are more resistant to Gram-negative E. coli LPS-induced mortality compared with control mice.15 Our results showed that recombinant kallistatin administration reduced CLP-induced kidney damage and dysfunction as evidenced by PAS staining and decreased blood urea nitrogen and serum creatinine levels. Kallistatin treatment also reduced serum cytokine levels and renal expression of the pro-inflammatory genes HMGB1, TLR4, ICAM-1 and VCAM-1, but increased renal eNOS expression and NO levels in CLP mice. Kallistatin was previously shown to inhibit vascular inflammation by interacting with the transcription factor Kruppel-like factor 4 and increasing eNOS expression and NO levels.31 These findings suggest that kallistatin prevents kidney injury through suppression of renal inflammation via NO formation.
The endothelium is a key organ in the pathogenesis of sepsis, as endothelial dysfunction in sepsis contributes to multi-organ failure.32 Kallistatin is a unique endogenous molecule as it has pleiotropic properties by both direct and indirect effects. Structural and functional analysis revealed that kallistatin contains two important structural elements: an active site for binding to tissue kallikrein and a heparin-binding site.23,24 We previously showed that kallistatin via its heparin-binding site inhibits VEGF's effects by competing with VEGF binding to heparin sulphate proteoglycans on endothelial cell surfaces, thereby suppressing VEGF-induced endothelial cell proliferation, migration, tube formation and angiogenesis.7 Likewise, kallistatin through its heparin-binding domain competes with the binding of TNF-α to endothelial cells and inhibits TNF-α-mediated NF-κB activation and inflammatory gene expression.12 Moreover, kallistatin increases NO formation by stimulating eNOS expression and activity in endothelial cells.9,10 Hence, kallistatin is a novel inhibitor of endothelial inflammation via dual mechanisms: (i) inhibiting TNF-α-induced NF-κB activation and pro-inflammatory gene expression, and (ii) stimulating endothelial eNOS expression and activation, so increasing NO formation in endothelial cells.12,31 It has recently been demonstrated that HMGB1 binds to heparan sulphate.25 We therefore examined whether kallistatin could inhibit HMGB1-induced NF-κB activation and inflammatory gene expression. Our results showed that kallistatin, via its heparin-binding site, prevented NF-κB activation, TNF-α and VCAM-1 gene expression in endothelial cells after HMGB1 stimulation. Through its heparin-binding site, kallistatin may inhibit HMGB1-induced NF-κB activation and inflammatory gene expression by competing with HMGB1 binding to its receptor. Hence, the protective effect of kallistatin administration against organ damage and mortality after sepsis is most likely attributed to its reduction on inflammation and apoptosis via antagonizing HMGB1- and TNF-α-mediated effects through its heparin-binding site (see Supporting information, Figure S1).
Increasing evidence suggests that apoptosis significantly contributes to the immune hypo-responsiveness and organ injury during sepsis.33,34 Recent studies demonstrate that the apoptotic pathway may play a significant role in septic shock complicated by acute lung and kidney injury.30,35 Indeed, spleens from septic patients exhibited apoptotic characteristics, active caspase-3 immunostaining and lymphocyte depletion in the white pulp.33 It has also been reported that adoptive transfer of apoptotic splenocytes in septic mice increased mortality, whereas blocking apoptosis improved survival after sepsis.34,36 Our previous studies showed that kallistatin decreases mortality in LPS-induced septic mice, and protects against vascular injury and oxidative stress-induced endothelial apoptosis via activation of Akt-dependent eNOS signalling.9,15 In the present study, we showed that kallistatin treatment attenuated CLP-induced apoptosis in the splenic white pulp as determined by TUNEL staining and caspase-3 activity assay. During sepsis, apoptosis occurs primarily in lymphoid organs, such as the spleen and thymus.37,38 It has been reported that apoptotic cells can activate macrophages to release HMGB1, and that apoptosis in the spleen is essential for the release of HMGB1 in mouse models of sepsis.39,40 HMGB1 was shown to contribute to splenocyte inflammatory priming and expansion in mouse sepsis, and anti-HMGB1 monoclonal antibody significantly attenuated splenocyte priming.41,42 Although antibodies against HMGB1 conferred protection against organ damage, they did not prevent the accumulation of apoptotic cells in the spleen.39 Our previous study showed that kallistatin protects against vascular injury and TNF-α-induced apoptosis in endothelial cells via activation of Akt-dependent eNOS signalling.9 In this study, we showed that kallistatin treatment significantly reduced circulating levels of several inflammatory cytokines (HMGB1, TNF-α and IL-6) in association with decreased apoptotic cells in the spleen. Kallistatin may attenuate organ injury via inhibiting TNF-α- and HMGB1-mediated effects, so reducing apoptosis in the spleen, leading to a further inhibition of HMGB1 release during sepsis. Therefore, the anti-apoptotic property of kallistatin may play a role in preventing lymphocyte loss and inhibiting HMGB1 release, thereby improving organ function and survival during sepsis.
Kallistatin is a serine protein with novel advantages in the treatment of sepsis as it exhibits pleiotropic actions in protection against sepsis-induced lethality and organ damage. First, kallistatin inhibits the effects of the early and late inflammatory cytokines, TNF-α and HMGB1, leading to a decreased inflammatory response in sepsis. This is the first study to demonstrate that kallistatin, through its heparin-binding site, inhibits HMGB1-induced pro-inflammatory gene expression. Second, our previous reports showed that kallistatin antagonizes TNF-α-induced apoptosis and oxidative stress in cardiac, renal and vascular cells.9,11 Third, we showed that kallistatin administration markedly reduces bacterial counts in septic mice. This finding is consistent with the previous study showing that kallistatin enhances immune cell viability and bacterial clearance by increasing superoxide production in immune cells.16 Hence, kallistatin appears to have a double-edged role in modulating superoxide formation, dependent on cellular and pathological conditions. Therefore, kallistatin is a unique serum protein in protection against sepsis due to its anti-inflammatory, anti-apoptotic and bacterial killing actions.
In summary, human kallistatin administration exerts beneficial effects on sepsis-induced organ damage, inflammation, apoptosis and lethality in CLP-induced septic mice. Kallistatin, via its heparin-binding site, inhibits HMGB1-induced inflammatory responses, and so could be a promising new agent for treatment of sepsis. As an endogenous molecule, kallistatin therapy should have minimal side effects in protection and/or reversal of sepsis-induced injury. The protective action of kallistatin on sepsis needs to be further explored as a novel potential therapeutic approach for sepsis.
Acknowledgments
Julie Chao, Lee Chao and Pengfei Li designed this study. Pengfei Li, Grant Bledsoe, Zhi-Rong Yang and Hongkuan Fan performed the experiments. Pengfei Li, Juie Chao and Grant Bledsoe wrote this paper. This work was supported by the National Institutes of Health grants HL 29397, HL 44083 and C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources. We thank Dr. Jian-xing Ma of the University of Oklahoma Health Science Center for providing HEK293T cells expressing recombinant human kallistatin.
Disclosures
The authors declare having no competing interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Kallistatin reduces organ injury and improves survival of mice with polymicrobial sepsis.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med. 2003;9:517–24. doi: 10.1038/nm0503-517. [DOI] [PubMed] [Google Scholar]
- 3.Chao J, Chai KX, Chen LM, et al. Tissue kallikrein-binding protein is a serpin. I. Purification, characterization, and distribution in normotensive and spontaneously hypertensive rats. J Biol Chem. 1990;265:16394–401. [PubMed] [Google Scholar]
- 4.Chao J, Tillman DM, Wang MY, Margolius HS, Chao L. Identification of a new tissue-kallikrein-binding protein. Biochem J. 1986;239:325–31. doi: 10.1042/bj2390325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou GX, Chao L, Chao J. Kallistatin: a novel human tissue kallikrein inhibitor. Purification, characterization, and reactive center sequence. J Biol Chem. 1992;267:25873–80. [PubMed] [Google Scholar]
- 6.Miao RQ, Agata J, Chao L, Chao J. Kallistatin is a new inhibitor of angiogenesis and tumor growth. Blood. 2002;100:3245–52. doi: 10.1182/blood-2002-01-0185. [DOI] [PubMed] [Google Scholar]
- 7.Miao RQ, Chen V, Chao L, Chao J. Structural elements of kallistatin required for inhibition of angiogenesis. Am J Physiol Cell Physiol. 2003;284:C1604–13. doi: 10.1152/ajpcell.00524.2002. [DOI] [PubMed] [Google Scholar]
- 8.Wang CR, Chen SY, Wu CL, Liu MF, Jin YT, Chao L, Chao J. Prophylactic adenovirus-mediated human kallistatin gene therapy suppresses rat arthritis by inhibiting angiogenesis and inflammation. Arthritis Rheum. 2005;52:1319–24. doi: 10.1002/art.20991. [DOI] [PubMed] [Google Scholar]
- 9.Shen B, Gao L, Hsu YT, Bledsoe G, Hagiwara M, Chao L, Chao J. Kallistatin attenuates endothelial apoptosis through inhibition of oxidative stress and activation of Akt-eNOS signaling. Am J Physiol Heart Circ Physiol. 2010;299:H1419–27. doi: 10.1152/ajpheart.00591.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen B, Hagiwara M, Yao YY, Chao L, Chao J. Salutary effect of kallistatin in salt- induced renal injury, inflammation, and fibrosis via antioxidative stress. Hypertension. 2008;51:1358–65. doi: 10.1161/HYPERTENSIONAHA.107.108514. [DOI] [PubMed] [Google Scholar]
- 11.Gao L, Yin HS, Smith RJ, Chao L, Chao J. Role of kallistatin in prevention of cardiac remodeling after chronic myocardial infarction. Lab Invest. 2008;88:1157–66. doi: 10.1038/labinvest.2008.85. [DOI] [PubMed] [Google Scholar]
- 12.Yin H, Gao L, Shen B, Chao L, Chao J. Kallistatin inhibits vascular inflammation by antagonizing tumor necrosis factor-α-induced nuclear factor κB activation. Hypertension. 2010;56:260–7. doi: 10.1161/HYPERTENSIONAHA.110.152330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao J, Yin H, Yao YY, Shen B, Smith RS, Jr, Chao L. Novel role of kallistatin in protection against myocardial ischemia–reperfusion injury by preventing apoptosis and inflammation. Hum Gene Ther. 2006;17:1201–13. doi: 10.1089/hum.2006.17.1201. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Bledsoe G, Hagiwara M, Shen B, Chao L, Chao J. Depletion of endogenous kallistatin exacerbates renal and cardiovascular oxidative stress, inflammation, and organ remodeling. Am J Physiol Renal Physiol. 2012;303:F1230–8. doi: 10.1152/ajprenal.00257.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen LM, Chao L, Chao J. Beneficial effects of kallikrein-binding protein in transgenic mice during endotoxic shock. Life Sci. 1997;60:1431–5. doi: 10.1016/s0024-3205(97)00094-5. [DOI] [PubMed] [Google Scholar]
- 16.Lu SL, Tsai CY, Luo YH, et al. Kallistatin modulates immune cells and confers anti-inflammatory response to protect mice from group A streptococcal infection. Antimicrob Agents Chemother. 2013;57:5366–72. doi: 10.1128/AAC.00322-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chao J, Schmaier A, Chen LM, Yang Z, Chao L. Kallistatin, a novel human tissue kallikrein inhibitor: levels in body fluids, blood cells, and tissues in health and disease. J Lab Clin Med. 1996;127:612–20. doi: 10.1016/s0022-2143(96)90152-3. [DOI] [PubMed] [Google Scholar]
- 18.Stadnicki A, Mazurek U, Gonciarz M, et al. Immunolocalization and expression of kallistatin and tissue kallikrein in human inflammatory bowel disease. Dig Dis Sci. 2003;48:615–23. doi: 10.1023/a:1022569623350. [DOI] [PubMed] [Google Scholar]
- 19.Lin WC, Lu SL, Lin CF, Chen CW, Chao L, Chao J, Lin YS. Plasma kallistatin levels in patients with severe community-acquired pneumonia. Crit Care. 2013;17:R27. doi: 10.1186/cc12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen VC, Chao L, Chao J. Reactive-site specificity of human kallistatin toward tissue kallikrein probed by site-directed mutagenesis. Biochim Biophys Acta. 2000;1479:237–46. doi: 10.1016/s0167-4838(00)00044-3. [DOI] [PubMed] [Google Scholar]
- 21.Fan H, Bitto A, Zingarelli B, Luttrell LM, Borg K, Halushka PV, Cook JA. Beta-arrestin 2 negatively regulates sepsis-induced inflammation. Immunology. 2010;130:344–51. doi: 10.1111/j.1365-2567.2009.03185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–91. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 23.Chen VC, Chao L, Chao J. A positively charged loop on the surface of kallistatin functions to enhance tissue kallikrein inhibition by acting as a secondary binding site for kallikrein. J Biol Chem. 2000;275:40371–7. doi: 10.1074/jbc.M005691200. [DOI] [PubMed] [Google Scholar]
- 24.Chen VC, Chao L, Pimenta DC, Bledsoe G, Juliano L, Chao J. Identification of a major heparin-binding site in kallistatin. J Biol Chem. 2001;276:1276–84. doi: 10.1074/jbc.M005791200. [DOI] [PubMed] [Google Scholar]
- 25.Xu D, Young J, Song D, Esko JD. Heparan sulfate is essential for high mobility group protein 1 (HMGB1) signaling by the receptor for advanced glycation end products (RAGE) J Biol Chem. 2011;286:41736–44. doi: 10.1074/jbc.M111.299685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park JS, Gamboni-Robertson F, He Q, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–24. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 27.Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 28.Wang G, Han D, Zhang Y, Xie X, Wu Y, Li S, Li M. A novel hypothesis: up-regulation of HO-1 by activation of PPARγ inhibits HMGB1-RAGE signaling pathway and ameliorates the development of ALI/ARDS. J Thorac Dis. 2013;5:706–10. doi: 10.3978/j.issn.2072-1439.2013.08.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehta RL, Bouchard J, Soroko SB, Ikizler TA, Paganini EP, Chertow GM, Himmelfarb J. Sepsis as a cause and consequence of acute kidney injury: sepsis as a cause and consequence of acute kidney injury. Intensive Care Med. 2011;37:241–8. doi: 10.1007/s00134-010-2089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frank AJ, Sheu CC, Zhao Y, et al. BCL2 genetic variants are associated with acute kidney injury in septic shock. Crit Care Med. 2012;40:2116–23. doi: 10.1097/CCM.0b013e3182514bca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen B, Smith RS, Jr, Hsu YT, Chao L, Chao J. Kruppel-like factor 4 is a novel mediator of Kallistatin in inhibiting endothelial inflammation via increased endothelial nitric-oxide synthase expression. J Biol Chem. 2009;284:35471–8. doi: 10.1074/jbc.M109.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iba T, Kidokoro A, Yagi Y. The role of the endothelium in changes in procoagulant activity in sepsis. J Am Coll Surg. 1998;187:321–9. doi: 10.1016/s1072-7515(98)00177-x. [DOI] [PubMed] [Google Scholar]
- 33.Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–51. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 34.Wesche-Soldato DE, Lomas-Neira JL, Perl M, Jones L, Chung CS, Ayala A. The role and regulation of apoptosis in sepsis. J Endotoxin Res. 2005;11:375–82. doi: 10.1179/096805105X76904. [DOI] [PubMed] [Google Scholar]
- 35.Chopra M, Reuben JS, Sharma AC. Acute lung injury: apoptosis and signaling mechanisms. Exp Biol Med (Maywood) 2009;234:361–71. doi: 10.3181/0811-MR-318. [DOI] [PubMed] [Google Scholar]
- 36.Chung CS, Song GY, Lomas J, Simms HH, Chaudry IH, Ayala A. Inhibition of Fas/Fas ligand signaling improves septic survival: differential effects on macrophage apoptotic and functional capacity. J Leukoc Biol. 2003;74:344–51. doi: 10.1189/jlb.0102006. [DOI] [PubMed] [Google Scholar]
- 37.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A. 1999;96:14541–6. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–50. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 39.Qin S, Wang H, Yuan R, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006;203:1637–42. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huston JM, Wang H, Ochani M, et al. Splenectomy protects against sepsis lethality and reduces serum HMGB1 levels. J Immunol. 2008;181:3535–9. doi: 10.4049/jimmunol.181.5.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valdes-Ferrer SI, Rosas-Ballina M, Olofsson PS, et al. HMGB1 mediates splenomegaly and expansion of splenic CD11b+ Ly-6Chigh inflammatory monocytes in murine sepsis survivors. J Intern Med. 2013;274:381–90. doi: 10.1111/joim.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valdes-Ferrer SI, Rosas-Ballina M, Olofsson PS, et al. High-mobility group box 1 mediates persistent splenocyte priming in sepsis survivors: evidence from a murine model. Shock. 2013;40:492–5. doi: 10.1097/SHK.0000000000000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Kallistatin reduces organ injury and improves survival of mice with polymicrobial sepsis.