

Abstract
The activation of dendritic cells (DCs) is necessary to initiate immune responses. Angiotensin II (Ang II) can enhance the maturation and activation of DCs, but the mechanisms are still unclear. Ubiquitin-activating enzyme (E1/Uba1) is the common first step in ubiquitylation, which decides whether or not the modified protein is ultimately degraded by the proteasome. This study aimed to investigate the role of E1 in Ang II-induced activation of DCs and the underlying mechanisms. First, we showed that Ang II stimulation significantly up-regulated E1 expression in DCs. Moreover, Ang II treatment markedly induced phenotypic maturation, the secretion of cytokines and the immunostimulatory capacity of DCs. In contrast, inhibition of E1 by a small molecule inhibitor, 4 [4-(5-nitro-furan-2-ylmethylene)-3, 5-dioxo-pyrazolidin-1-yl]-benzoic acid ethyl ester (PYR41), markedly attenuated these effects. Mechanistically, PYR41 treatment markedly decreased K63-linked ubiquitination of tumour necrosis factor receptor-associated factor 6 and nuclear factor-κB essential modulator, inhibited proteasomal degradation of nuclear factor-κB inhibitor α and mitogen-activated protein kinase phosphatase 1 thereby resulting in activation of nuclear factor-κB, extracellular signal-regulated kinase 1/2 and signal transducer and activator of transcription 1 signalling pathways in DCs induced by Ang II. Taken together, our results demonstrate a novel role of E1 in Ang II-induced activation of DCs, and inhibition of E1 activity might be a potential therapeutic target for DC-mediated autoimmune diseases.
Keywords: angiotensin II, cardiovascular disease, dendritic cells, inflammation, ubiquitin-activating enzyme E1
Introduction
Dendritic cells (DCs) are highly specialized antigen-presenting cells with a unique ability to activate naive T lymphocytes.1 The precursors originating from primary lymphoid tissues such as the bone marrow and the thymus can differentiate into DCs. Committed DCs enter the circulation and seed peripheral tissues, where they reside in an immature state and serve as sentinels – screening foreign antigens or signals that indicate infection or inflammation. Immature DCs actively capture and process antigens, and then enter a maturation/activation programme in response to maturation-inducing stimuli, such as bacterial products, inflammatory cytokines or hormones. During this process, DCs leave the peripheral tissues and migrate to the secondary lymphoid organs, where they activate the proliferation and differentiation of naive T cells associated with several coordinated events such as up-regulation of co-stimulatory molecules and secretion of cytokines.2,3
Angiotensin II (Ang II), as the major effector in the renin–angiotensin system, plays a critical role in the maintenance of haemodynamics and in the development of atherosclerosis, hypertension and cardiac remodelling.4,5 Studies have shown that Ang II also has a pro-inflammatory and immunomodulatory function. Ang II can regulate the normal differentiation of DCs from human monocytes by interacting with Ang II receptor type 1. Blockade of these receptors not only inhibits DC differentiation induced by granulocyte–macrophage colony-stimulating factor (GM-CSF) plus interleukin-4 (IL-4), but also switches the differentiation of monocytes into macrophage-like cells,6–8 indicating that Ang II plays an important role in the maturation and T-cell stimulatory activities of DCs. However, the molecular mechanism by which Ang II induces activation of DCs remains to be elucidated.
The ubiquitin–proteasome system represents the main mechanism of ubiquitin-dependent degradation of intracellular proteins, which participates in the regulation of signal transduction, apoptosis, the cell cycle and antigen presentation.9 In addition to protein degradation by proteasome, ubiquitinated proteins can play roles in DNA repair and the activation of protein kinases such as IκB kinase (IKK).10 Ubiquitin is conjugated to a substrate lysine in an enzymatic cascade involving a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2) and a ubiquitin–protein ligase (E3). E1 is the first step in ubiquitylation, which decides whether the modified protein is ultimately degraded by the proteasome. E1 exists as two isoforms, E1a (117 000 MW) and E1b (110 000 MW). Of these isoforms only E1a is localized in both the nucleus and the cytoplasm and may modulate nuclear protein ubiquitination.11 Recent studies suggest that E1 can be a novel target for the treatment of haematological malignancies. E1 inhibitor 4[4-(5-nitro-furan-2-ylmethylene)-3, 5-dioxo-pyrazolidin-1-yl]-benzoic acid ethyl ester (PYR41) attenuates cytokine-mediated nuclear factor-κB (NF-κB) activation and activates the transcriptional activity of this tumour suppressor through inhibition of p53 degradation.12,13 However, the role of E1 in the process of DC activation remains unclear.
In the present study, we applied the E1 inhibitor PYR41 to investigate the role of E1 in Ang II-induced activation of DCs and underlying mechanisms. These findings may provide a novel immunological mechanism of Ang II-induced cardiovascular disease and a potential therapeutic strategy for suppression of deregulated and unwanted immune responses.
Materials and methods
Antibodies and reagents
Fluorescein isothiocyanate-conjugated anti-CD40, anti-CD80, anti-CD86 and anti-MHCII antibodies were purchased from eBioscience (San Diego, CA). Anti-Uba1, mitogen-activated protein kinase phosphatase 1 (MKP-1), extracellular signal-regulated kinase 1/2 (ERK1/2), phospho-ERK1/2, signal transducer and activator of transcription 1 (STAT1), phospho-STAT1, IKKβ, phospho-IKKα/β, NF-κB inhibitor α (IκBα), phospho-IκBα, NF-κB/p65, phospho-NF-κB/p65, K63-linkage specific polyubiquitin (D7A11), β-actin and horseradish peroxidase-linked anti-mouse or rabbit IgG antibody were obtained from Cell Signaling Technology (Danvers, MA). The purified PYR41 (≥ 90% purity), anti-tumour necrosis factor (TNF) receptor-associated factor (TRAF6; H-274), NF-κB essential modulator (NEMO; FL-419) and protein A/G plus-agarose were purchased from Santa Cruz Biotechnology (Dallas, TX). RPMI-1640, fetal bovine serum and PBS were purchased from HyClone Laboratories Inc. (Logan, UT). Recombinant murine GM-CSF was purchased from PeproTech Inc. (Rocky Hill, NJ). Ang II and lipopolysaccharide (LPS) from Escherichia coli 0127:B8 were from Sigma-Aldrich (St Louis, MO). The ELISA kits for IL-6, TNF-α and IL-12 were from the Dakewe Biotech Company (Shenzhen, China). Carboxyfluorescein diacetate-succinimidyl ester (CFSE) was from Dojindo Laboratories (Kumamoto, Japan) and 8-to 10-week-old C57BL/6 and BALB/c mice raised in a pathogen-free animal house were purchased from the Vital River Laboratory Animal Technology Co. Ltd (Beijing, China). All other chemicals frequently used in our laboratory were purchased from either Sigma-Aldrich or BD Pharmingen (San Jose, CA).
Dendritic cell culture
The culture medium used throughout these studies was RPMI-1640 with 10% fetal bovine serum supplemented with 100 μg/ml streptomycin and 100 units/ml penicillin. DC2.4 cells are cultured and passaged as described elsewhere.14 Bone-marrow-derived dendritic cells (BMDCs) were generated by a modification of a previously described method.15 Briefly, C57BL/6 mice were anaesthetized by intraperitoneal injection and the femurs and tibiae were removed and isolated from the surrounding muscle tissue. Both metaphyses were removed to expose the marrow cavity and the cells were blown from the cavity using RPMI-1640 and a 1-ml syringe. Cells (2 × 106) were plated in sterile Petri dishes (100 × 15 mm) in 10 ml of culture medium supplemented with 20 ng/ml GM-CSF and cultured in a humidified atmosphere of 5% CO2 at 37° for 10 days. Finally, cells were resuspended (106 cells/ml) in fresh culture medium (supplemented with GM-CSF), and 2 ml (final volume) was seeded into six-well tissue culture plates for preparation. All procedures were approved by the Institutional Animal Care and Use Committee of Capital Medical University. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Treatment of DCs
Dendritic cells were pre-treated with PYR41 (0, 1 or 5 μm) for 30 min before being co-incubated with or without Ang II (100 nm). To observe the E1 expression, DCs were incubated with Ang II (100 nm) for different lengths of time (0, 2, 6, 16 and 24 hr). For the signalling pathway analysis, DCs were incubated with Ang II (100 nm) for different time-points (0, 5, 15, 30 and 60 min) or pre-treated with PYR41 (0, 1 or 5 μm) for 30 min before co-incubation with or without Ang II (100 nm) for a certain period of time. One group of DCs was incubated in fresh culture medium with an equivalent volume of vehicles as the blank control. One group of DCs was only incubated with PYR41 (5 μm) as the negative control. Another group of DCs stimulated with LPS (1 μg/ml) for a certain period of time was used as the positive control. DCs were removed by vigorous pipetting and were resuspended in 5% fetal bovine serum in PBS for analysis of membrane marker expression by flow cytometry. Supernatants were harvested and frozen at −80° before analysis for cytokines by ELISA kit. Cell viability was assessed using the methyl thiazolyl tetrazolium method. According to this criterion, cell viability was > 80% under all experimental conditions used.
Flow cytometry
Phenotypic analysis (the surface expression of antigen markers) was performed by flow cytometry. The BMDCs were collected and resuspended in PBS at a concentration of 2 × 105/ml. Cells were incubated with the following anti-mouse monoclonal antibodies (eBioscience): FITC-conjugated anti-CD40, anti-CD80, anti-CD86 or anti-MHCII for 30 min at room temperature in the dark. Appropriate isotype-matched immunoglobulins were used as negative controls. Then cells were analysed on a FACSCalibur flow cytometer with CellQuest software (Becton Dickinson, San Jose, CA). Results were expressed as the percentages of positive cells calculated as specific antibody minus the value obtained with the isotype control.
Cytokine assay by ELISA
Cytokine concentrations of IL-6, TNF-α and IL-12 in supernatants from DC cultures were measured with commercially available Enzyme-Linked Immuno-Sorbent Kits from the Dakewe Biotech Company according to the manufacturer's instructions.
Mixed lymphocyte reaction
Single-cell suspensions from spleens of BALB/c mice were obtained by grinding followed by filtration through a nylon mesh. The lymphocytes were enriched with Ficoll (TBDscience, Tianjin, China). Lymphocytes (1 × 106/ml) were stained with CFSE and then co-cultured with C57BL/6 BMDCs (1 × 105/ml) after mitomycin C treatment (Roche, Basel, Switzerland). After 5 days, harvested cells were stained with phycoerythrin-conjugated anti-CD3 (eBioscience) and T-cell proliferation was evaluated by flow cytometry.
Western blot and immunoprecipitation analysis
Total protein was isolated from different treated DCs with lysis buffer (20 mm Tris–HCl, 1% Triton X-100, 0·05% SDS, 5 mg of sodium deoxycholate, 150 mm NaCl and 1 mm PMSF) containing protease/phosphatase inhibitor cocktail (Roche Diagnostics, Basel, Switzerland) as described previously.16 Between 50 and 60 μg protein per sample was loaded on a 10% SDS–PAGE gel and transferred onto nitrocellulose membranes. The membranes were incubated with primary antibodies as indicated overnight and then incubated with horseradish peroxidase-conjugated secondary antibody for 1 hr. The blots were developed with an enhanced chemiluminescence detection system (Beijing Sage creation) and the bands were scanned, and densitometry analysis was performed with the software Image J 2x (National Institutes of Health, Bethesda, MD). For the phospho-specific protein, we calculated the relative intensity for target proteins through dividing the absolute intensity of phosphorylated proteins by the absolute intensity of total target proteins or β-actin. We then calculated the ratio of the relative intensity for target protein in treatment groups to that in the control group. For immunoprecipitation assays, cell lysates were pre-cleared with protein A/G plus agarose for 1 hr at 4° and then 2 μg of anti-TRAF6 or NEMO and 30 μl protein A/G agarose was added and incubated overnight at 4° to capture primary immune complexes. The beads were washed and fractionated by 10% SDS–PAGE followed by immunoblotting as previously described.17,18
RNA extraction and quantitative real-time PCR
Total RNA was isolated from DCs using Trizol reagent according to the manufacturer's instructions. First-strand cDNA synthesis was performed with 1 μg of total RNA in a reaction volume of 20 μl using PrimeScript® RT reagent Kit with gDNA Eraser (TaKaRa, Kyoto, Japan). The transcript levels of Uba1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were detected by quantitative real time-PCR (qPCR) analysis. The following gene-specific primers were used. Uba1: 5′-AAGAACCTGGTGCTCACTGG-3′ and 5′-TTGGCAACCTGAGCCTTTGA-3′; GAPDH: 5′-GGTTGTCTCCTGCGACTTCA-3′ and 5′-GGTGGTCCAGGGTTTCTTACTC-3′.
Statistical analysis
All data are expressed as mean ± SEM. Differences between groups were evaluated for statistical significance using a Student's t-test. P < 0·05 was considered statistically significant.
Results
Ang II up-regulates E1 expression in DCs
To investigate the role of E1 in Ang II-induced maturation and activation of DCs in vitro, we stimulated the BMDCs or DC2.4 cells with a dose of 100 nm Ang II for different times (0, 2, 6, 16, 24 hr). Quantitative PCR analysis showed that the mRNA levels of E1/Uba1 in BMDCs was significantly up-regulated during Ang II stimulation (Fig. 1a). Western blot further revealed that the protein levels of E1 (E1a) in BMDCs were also markedly increased after Ang II stimulation (Fig. 1b). The increased protein expression of E1 (E1a) was confirmed in DC2.4 cells (Fig. 1c). Hence, E1 in DCs may play a role in Ang II-induced DC maturation and activation.
Figure 1.
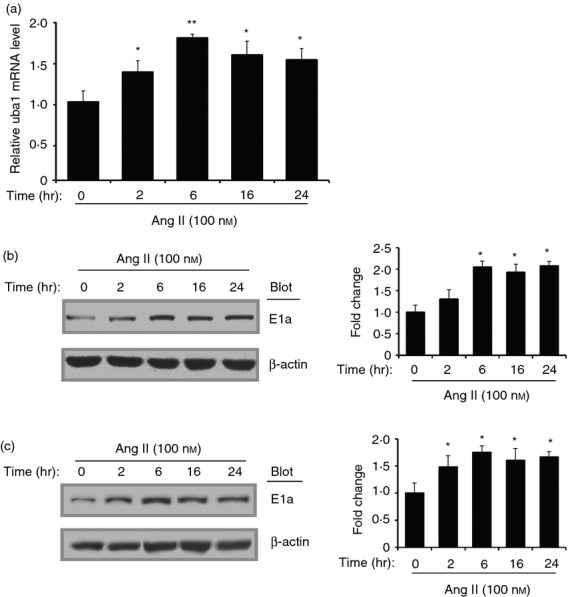
Angiotensin II (Ang II) up-regulates E1 expression in dendritic cells (DCs). Bone marrow-derived dendritic cells (BMDCs) were stimulated with Ang II (100 nm) for 0–24 hr. (a) Messenger RNA levels of E1 were determined with real-time PCR and shown as the ratio to control (0 hr). The protein levels of E1 (Uba1) in BMDCs (b) and DC2.4 cells (c) were detected with Western blot analysis. Representative Western blots were shown (left). The intensity of protein bands was quantified and shown as the ratio to control (0 hr) after normalization by β-actin (right). Results are expressed as mean ± SEM for three independent experiments. *P < 0·05, ** P < 0·01 versus control (0 hr).
PYR41 inhibits the phenotypic maturation of DCs induced by Ang II
To determine the effect of E1 in phenotypic maturation of DCs induced by Ang II, BMDCs were pre-treated with the E1 inhibitor PYR41 and then stimulated with Ang II (100 nm) for 24 hr. The DC maturation surface molecules, including CD40, CD80, CD86, or MHCII were detected by flow cytometry. The concentrations of PYR41 (1 and 5 μm) were determined in preliminary experiments (data not shown) and subsequently used throughout the experiments. As shown in Fig. 2, the majority of immature DCs generated in the control group expressed low levels of CD40 and CD80, moderate levels of CD86 and high levels of MHCII. The Ang II treatment significantly enhanced the expression levels of CD40 and CD80 but did not affect the expression of CD86 and MHCII, whereas treatment of cells with E1 inhibitor PYR41 significantly decreased the levels of CD40 and CD80 expression in a dose-dependent manner (Fig. 2). In addition, LPS (as a positive control) treatment also increased the levels of CD40, CD80, CD86 or MHCII expression in DCs compared with control (Fig. 2). These findings suggest that inhibition of E1 by PYR41 can attenuate Ang II-induced maturation of BMDCs.
Figure 2.
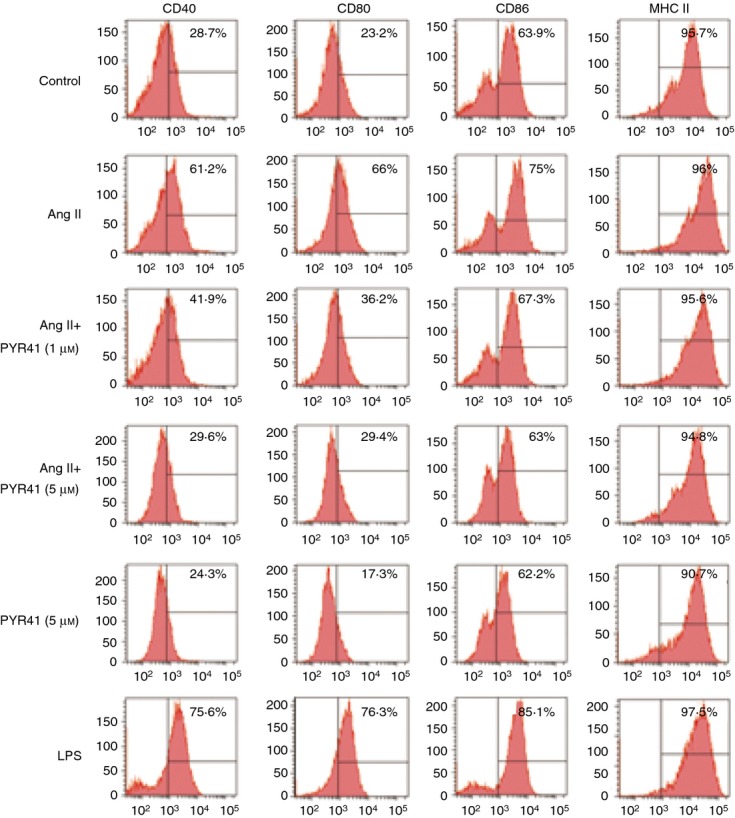
PYR41 inhibits the phenotypic maturation of dendritic cells (DCs) induced by Angiotensin II (Ang II). Bone marrow-derived DCs (BMDCs) were treated with PYR41 (1 or 5 μm) for 30 min and co-incubated with or without Ang II (100 nm) for another 24 hr. The expressions of surface markers on DCs were analysed with flow cytometry. The positive control was performed by addition of lipopolysaccharide (LPS; 1 μg/ml) for 24 hr. Indicated numbers are the percentages of positive cells. Histograms show the expression of CD40, CD80, CD86 or MHCII. A representative experiment is shown for each condition.
PYR41 inhibits Ang II-induced secretion of cytokines in DCs
Ang II has been shown to induce production of pro-inflammatory cytokines.19 We next analysed the effect of PYR41 on the function of BMDCs to produce inflammatory cytokines, including IL-12 (T-cell stimulatory cytokine), IL-6 and TNF-α. As shown in Fig. 3, LPS stimulation resulted in a significant increase of all these three cytokines in BMDCs. Similarly, Ang II treatment also enhanced the secretion of IL-12, TNF-α or IL-6, while PYR41 at high concentration (5 μm) significantly reduced the secretion of these cytokines compared with the Ang II treatment group. PYR41 alone had no obvious effect on the secretion of these cytokines. These data suggest that the E1 inhibitor PYR41 can suppress Ang II-induced activation of DCs.
Figure 3.
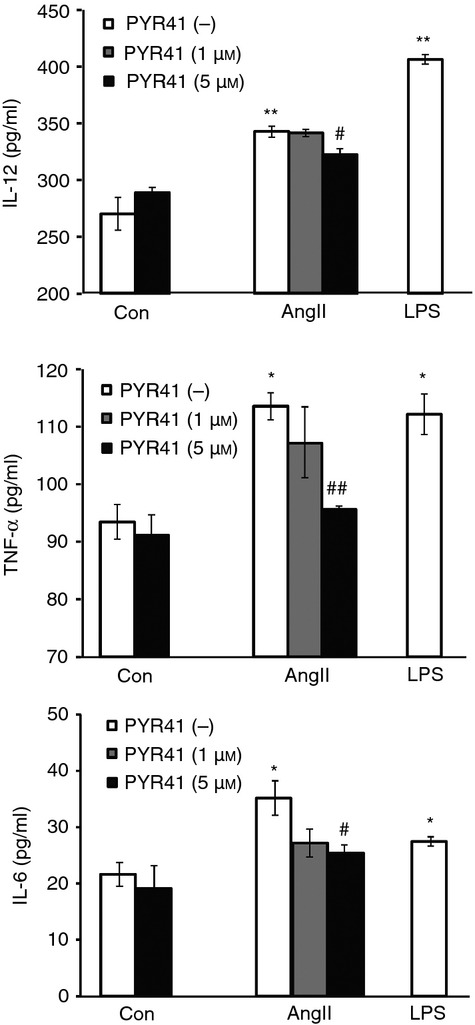
PYR41 reduces the secretion of inflammatory cytokines in dendritic cells (DCs) induced by Angiotensin II (Ang II). Bone marrow-derived DCs (BMDCs) were treated as in Fig. 2. The cytokine concentrations of interleukin-12 (IL-12), tumour necrosis factor-α (TNF-α) or IL-6 in supernatants from DC culture medium were analysed with ELISA. Data are presented as mean ± SEM from three independent experiments. *P < 0·05, **P < 0·01 versus control without PYR41 treatment. #P < 0·05, ##P < 0·01 versus Ang II without PYR41 treatment.
PYR41 inhibits the DCs initiating T-cell proliferation and activation induced by Ang II
To assess whether PYR41 inhibits Ang II-activated DCs to be fully functional antigen-presenting cells, we further analysed the effect of PYR41 on immunostimulatory capacity of DCs treated with Ang II in a mixed lymphocyte reaction. As shown in Fig. 4(a), lymphocytes alone had low proliferation ability (1·4%; No DCs). Immature DCs significantly enhanced T-cell proliferation (42·1%; Control). Ang II-treated DCs markedly increased T-cell proliferation compared with control (60·8% versus 42·1%), whereas PYR41 treatment (1 or 5 μm) dose-dependently inhibited this effect of DCs induced by Ang II on T-cell proliferation compared with Ang II-treated DCs (47·6% and 42·1% versus 60·8%). In addition, LPS-treated DCs also significantly initiated T-cell proliferation compared with control (70·8% versus 42·1%). PYR41 alone can inhibit the immunostimulatory capacity of DCs to some extent compared with control (31·5% versus 42·1%).
Figure 4.
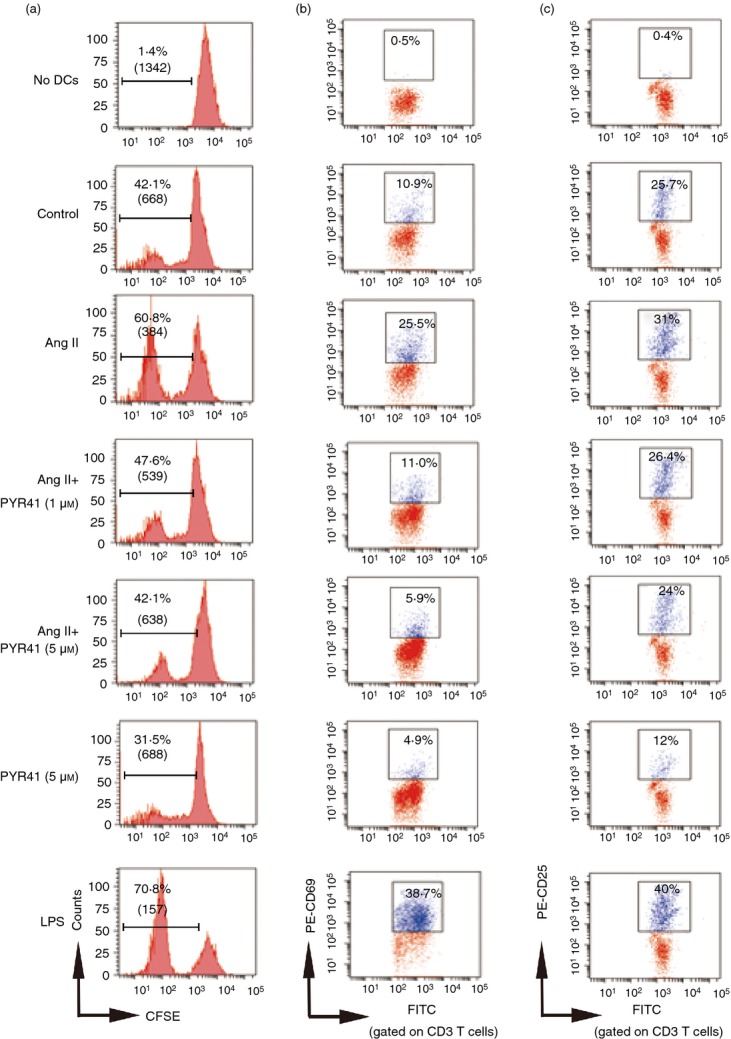
PYR41 attenuates the immunostimulatory capacity of dendritic cells (DCs) after Angiotensin II (Ang II) stimulation. Bone marrow-derived DCs (BMDCs) were treated as in Fig. 2, and then treated with Mitomycin C. Thereafter, cells were washed and 105 DCs/well were incubated with 106 allogeneic lymphocytes in 12-well plates for 5 days. (a) In vitro proliferation of lymphocytes assays were performed with the fluorescent dye CFSE and analysed by flow cytometry. Loss of CFSE fluorescence reflects cellular division. Indicated numbers are the percentages of dividing cells and mean fluorescence intensity (numbers in parentheses). At day 5, cells were harvested and the early activation markers of T cells CD69 (b) and CD25 (c) were analysed by flow cytometry by gating on the CD3 lymphocyte population. Indicated numbers are the percentages of positive cells. A representative experiment is shown for each condition.
Meanwhile, the activation markers (such as CD25 and CD69) expressed by allogeneic T cells were detected with flow cytometry. As shown in Fig. 4(b,c), FITC-CD3-positive T cells were previously gated. Lymphocytes alone had almost no expression of CD69 and CD25 (0·4% and 0·5%; No DCs). Immature DCs can enhance T-cell activation to some extent (10·9% and 25·7%, respectively; versus control). The LPS-treated DCs significantly promoted T-cell activation compared with control (38·7% versus 10·9%; 40% versus 25·7%). Similarly, Ang II-treated DCs also markedly induced doubled CD69 and CD25 on T cells compared with the control group (25·5% versus 10·9%; 31% versus 25·7%). In contrast, PYR41 treatment (1 or 5 μm) dose-dependently attenuated this effect (11% and 5·9% versus 25·5%; 26·4% and 24% versus 31%). PYR41 alone also inhibited the expression of CD69 and CD25 expression (4·9% versus 10·9%; 12% versus 25·7%). These results suggest that the E1 inhibitor PYR41 suppresses the Ang II-induced immunostimulatory capacity of DCs.
Ang II enhances activation of NF-κB, mitogen-activated protein kinase and STAT1 pathways in DC2.4 cells
Since NF-κB, mitogen-activated protein kinase (MAPK) and STAT1 signalling pathways play critical roles in DC antigen presentation and T-cell-dependent immune responses.20–23 To determine whether these signalling pathways are involved in Ang II-induced DC activation, we first analysed the phosphorylation of NF-κB, MAPKs [ERK1/2, Jun N-terminal kinase 1/2 (JNK1/2) and p38 MAPK] or STAT1 in DC2.4 cells treated with Ang II for different time-points (0, 5, 15 or 60 min). We found that Ang II significantly increased the level of NF-κB/p65 phosphorylation at 15min and increased the levels of ERK1/2 and STAT1 phosphorylation at 30 min, but did not markedly affect JNK and p38 phosphorylation compared with control (0 min) (Fig. 5).
Figure 5.
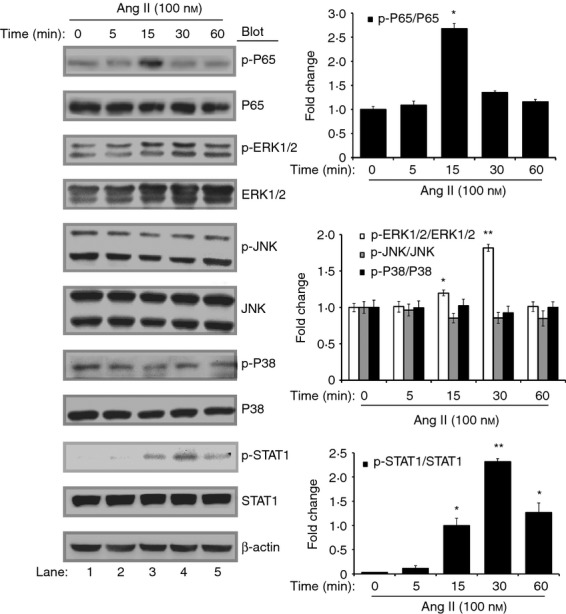
Effects of Angiotensin II (Ang II) on the nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways in dendritic cells (DCs). DC2.4 cells were treated with Ang II (100 nm) for different time-points. Representative Western blots showed expression levels of total and phospho-p65, total and phospho-ERK1/2, total and phospho-JNK1/2, total and phospho-p38 (left). The intensity of protein bands was quantified and shown as the ratio of phosphorylated protein/total protein to control (0 min) after normalization by β-actin (right). Data are expressed as mean ± SEM for three independent experiments. *P < 0·05, **P < 0·01 versus control (0 min).
PYR41 inhibits Ang II-induced activation of NF-κB and ERK/STAT1 in DC2.4 cells and BMDCs
It has been reported that IκBα and MKP-1 (the upstream protein of MAPKs) are the representative protein substrate of the ubiquitin–proteasome system.24,25 To elucidate the molecular mechanisms of PYR41 to inhibit DC maturation and function induced by Ang II, DC2.4 cells were treated with PYR41 (1 or 5 μm) and then stimulated with Ang II. The protein levels of IκBα and p65 were detected by Western blot analysis at 15 min. At 30 min, MKP-1, ERK1/2 and STAT1 were detected by Western blot analysis. As shown in Figs 6(a) and 7(a), Ang II significantly decreased the protein levels of IκBα and MKP-1, but markedly increased the levels of phosphorylated p65, ERK1/2 or STAT1 compared with control (lane 4 versus lane 1). In contrast, PYR41 significantly reversed these effects in a dose-dependent manner (lanes 5 and 6 versus lane 4). Furthermore, PYR41 treatment alone also increased the protein levels of IκBα and MKP-1 and decreased the levels of phosphorylated p65, ERK1/2 or STAT1 (lanes 2 and 3 versus lane 1). These results were further confirmed in BMDCs treated with or without Ang II and PYR41 by Western blot analysis at 15 or 30 min, respectively (Figs 6b and 7b). Collectively, these results suggest that PYR41 inhibits activation of NF-κB and ERK/STAT1 signalling pathways by preventing degradation of IκBα and MKP-1 in DCs in response to Ang II.
Figure 6.
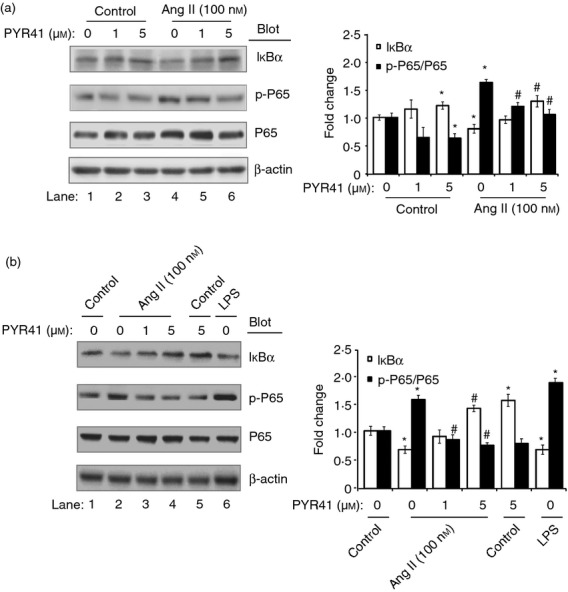
PYR41 attenuates Angiotensin II (Ang II)-induced activation of inhibitor of nuclear factor-κB (IκBα)/nuclear factor-κB (NF-κB) pathway in dendritic cells (DCs). DC2.4 cells (a) and bone marrow-derived dendritic cells (BMDCs) (b) were treated with PYR41 (1 or 5 μm) for 30 min and co-incubated with or without Ang II (100 nm) for 15 min. The positive control was performed by addition of lipopolysaccharide (LPS; 1 μg/ml) for 15 min. Representative Western blots showed expression levels of IκBα, total and phospho-p65, in each group (left). The intensity of protein bands was quantified and shown as the ratio to control after normalization by β-actin (right). Results are expressed as mean ± SEM for three independent experiments. *P < 0·05, **P < 0·01 versus control without PYR41 treatment. #P < 0·05, ##P < 0·01 versus Ang II without PYR41 treatment.
Figure 7.
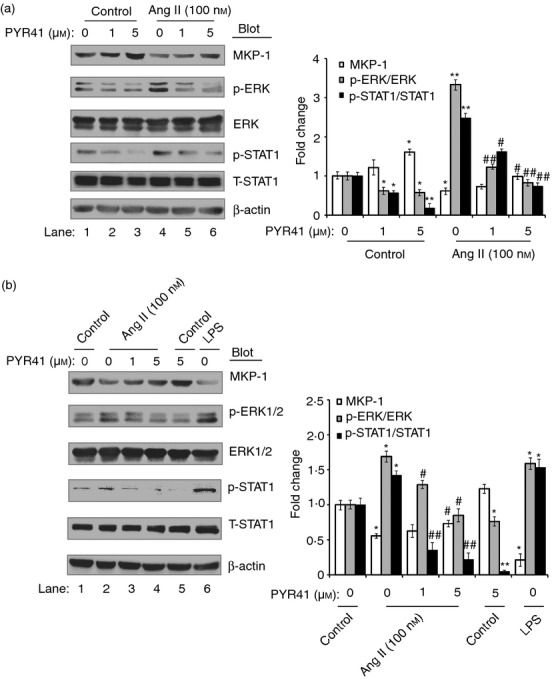
PYR41 suppresses the activation of the mitogen-activated protein kinase phosphatase 1 (MKP-1)/ extracellular signal-regulated kinase (ERK)/ signal transducer and activator of transcription 1 (STAT1) pathway in dendritic cells (DCs) induced by Angiotensin II (Ang II). DC2.4 cells (a) and bone marrow-derived DCs (BMDCs) (b) were treated with PYR41 (1 or 5 μm) for 30 min and co-incubated with or without Ang II (100 nm) for 30 min. The positive control was performed by addition of lipopolysaccharide (LPS; 1 μg/ml) for 30 min. Representative Western blots showed expression levels of MKP-1, total and phospho-ERK1/2 and STAT1 in each group (left). The intensity of protein bands was quantified and shown as the ratio to control after normalization by β-actin (right). Results are expressed as mean ± SEM for three independent experiments. *P < 0·05, **P < 0·01 versus control without PYR41 treatment. #P < 0·05, ##P < 0·01 versus Ang II without PYR41 treatment.
PYR41 suppresses Ang II-induced degradation of IκBα through inhibition of K63-link ubiquitination of TRAF6 and NEMO
Previous studies have demonstrated that modification by a K63-linked polyubiquitin chain on TRAF6 and NEMO can regulate the NF-κB signalling pathway through IKK activation.26,27 We then analysed the effect of the E1 inhibitor PYR41 on the phosphorylation of IKKα/β and IkBα, and the ubiquitination of TRAF6 and NEMO in DC2.4 cells. As shown in Fig. 8(a), Ang II significantly increased the levels of phosphorylated IKKα/β and IkBα compared with control (lane 4 versus lane 1), whereas PYR41 treatment markedly attenuated these effects in a dose-dependent manner (lanes 5 and 6 versus lane 4). To further explore the mechanism for PYR41 to inhibit the activation of IKKα/β, we examined K63-linked ubiquitination of TRAF6 and NEMO, which are the upstream regulators of IKKα/β. As shown in Fig. 8(b), Ang II significantly increased the levels of K63-linked ubiquitination of TRAF6 and NEMO compared with control (lane 3 versus lane 1). In contrast, PYR41 markedly suppressed these effects (lane 4 versus lane 3). Furthermore, PYR41 treatment alone also decreased the levels of K63-linked ubiquitination of TRAF6 and NEMO (lane 2 versus lane 1). Together, these results suggest that PYR41 inhibits activation of the NF-κB signalling pathway by preventing K63-linked ubiquitination of TRAF6 and NEMO in DCs in response to Ang II.
Figure 8.
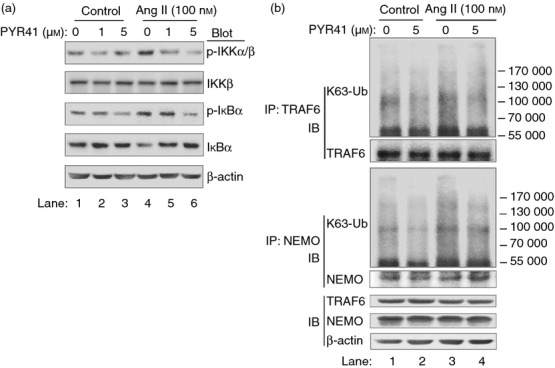
PYR41 inhibits Angiotensin II (Ang II)-induced degradation of inhibitor of nuclear factor-κB (IκBα) via inhibition of K63-link ubiquitination of tumour necrosis factor receptor-associated factor 6 (TRAF6) and nuclear factor-κB essential modulator (NEMO). (a) DC2.4 cells were treated with PYR41 (1 or 5 μm) for 30 min and co-incubated with or without Ang II (100 nm) for 15 min. Representative Western blots showed expression levels of total and phospho-IκB kinase (IKK) α/β and IκBα in each group. (b) DC2.4 cells were treated with PYR41 (5 μm) for 30 min and co-incubated with or without Ang II (100 nm) for 15 min. Representative Western blots showed K63-linked ubiquitination of TRAF6 and NEMO immunoprecipitated from DCs in each group (top four blots), and TRAF6 and NEMO in those same cells without immunoprecipitation (bottom).
Discussion
This study demonstrates for the first time that E1 expression was significantly up-regulated in DCs in response to Ang II. Furthermore, Ang II treatment markedly induced phenotypic maturation, the secretion of cytokines and immunostimulatory capacity of DCs. In contrast, PYR41 as an E1-specific inhibitor markedly attenuated Ang II-induced effects on DCs. These inhibitory effects were associated with the inhibition of K63-linked ubiquitination of TRAF6 and NEMO and proteasomal degradation of IκBα and MKP-1, leading to inactivation of p65, ERK1/2 and STAT1 pathways in DCs. Collectively, our data indicate that the E1 inhibitor PYR41 can prevent Ang II-induced activation of DCs through the modulation of multiple signalling pathways.
Dendritic cells are currently known as the most powerful antigen-presenting cells.28 The majority of DCs in vivo are in an immature state with high expression levels of phagocytosis-associated receptors, MHC II and low expression of co-stimulatory molecules, including CD40, CD80 or CD86. Therefore, immature DCs have weak ability to initiate the T-cell activation. However, DCs will undergo a series of phenotypic and functional maturations in response to various stimuli, including the products of pathogenic microorganisms (LPS, CpG), inflammatory cytokines (TNF-α, interferon-γ), and others.29,30 Ang II is an octapeptide hormone that plays a central role in cardiovascular homeostasis. Recent studies demonstrate that Ang II can mediate several key events of the inflammatory processes in several immune cells, such as macrophages, T cells and NK cells.31–35 It is reported that DCs express Ang II receptors,36 and Ang II can induce maturation and activation of human and murine DCs in vivo and in vitro.6,7 Consistent with these observations, our results reported here also support that Ang II has a profound regulatory effect on functional activation of DCs.
Several signalling pathways are known to play critical roles in regulating the maturation and activation of DCs, including NF-κB, MAPKs and STAT1.20,21,37–40 For examples, NF-κB coordinately regulates the phenotypic and functional maturation.20 STAT1 signalling is required for phenotypic maturation and T helper type 1 cell priming of mouse BMDCs,22,23 and the phosphorylation of STAT1 requires the activation of MAPK kinase/ERK cascades in human acute myeloid leukaemia cells and macrophages.39,40 Recently, studies indicate that Ang II stimulation results in activation of ERK, but not p38 MAPK and JNK in mouse BMDCs, whereas in human monocyte-derived DCs, Ang II activates ERK, p38 and NF-κB, but not JNK.21,37 Here our data indicated that Ang II treatment only increased the levels of NF-κB, ERK1/2 or STAT1 phosphorylation, but not p38 MAPK and JNK1/2 in DC2.4 cells (Fig. 5), indicating that these signalling pathways may be involved in DC activation in response to Ang II.
Activation of DCs is accompanied by changes of morphology, surface markers and function, which are related to alterations in the intracellular proteins. The ubiquitin–proteasome system is the major way to degrade the intracellular protein, which participates in the maturation and activation of human DCs.41,42 Studies have shown that proteasome inhibitors play an important role in immunosuppression and various diseases,43 probably through the inhibition of LPS and TNF-α-induced maturation and activation of DCs.44 E1 as the first enzyme in ubiquitination is a rate-limiting step in the degradation of ubiquitinated protein. Recently, several studies have indicated that E1 plays an important role in the development of cancers.12,13 However, it is unknown whether E1 has a role in the activation of DCs induced by Ang II. In the present study, we demonstrated that PYR41 significantly attenuated Ang II-induced phenotypic maturation, the secretion of cytokines and immunostimulatory capacity of DCs (Figs 4), indicating that E1 is required for Ang II-induced activation of DCs.
Accumulating evidence indicates that maturation and activation of human DCs is accompanied by the alterations of ubiquitin–proteasome system substrates.41 For examples, both IκBα and MKP-1, the upstream proteins of NF-κB and ERK1/2, are the representative protein substrates of the ubiquitin–proteasome system. Several E3 ligases, including β-TrCP, atrogin-1 and Skp2, can promote IκBα and MKP-1 ubiquitination and subsequent degradation by the 26 S proteasome.45–47 More importantly, the ubiquitin-mediated degradation of IκBα and MKP-1 is associated with activation of NF-κB and ERK1/2 signalling pathways.24,25 In the present study, we found that Ang II significantly down-regulated IκBα and MKP-1 protein levels and resulted in activation of NF-κB and ERK1/2/STAT1. In contrast, the E1 inhibitor PYR41 markedly reversed these effect (Figs 6 and 7). Taken together, we concluded that the E1 inhibitor PYR41 attenuates the E1-mediated proteasomal degradation of IκBα and MKP-1, which leads to inactivation of NF-κB and ERK/STAT1 pathways.
Recent studies have demonstrated that ubiquitination has functions in addition to its role in targeting protein degradation. A polyubiquitin chain that targets a protein for proteasomal degradation is linked through lysine-48 (K48) of ubiquitin. However, K63-linked polyubiquitin chains have been found to regulate protein kinase such as IκBα activation through a degradation-independent mechanism.10,27 The kinase complex IKK is composed of two related catalytic subunits IKKα/β, of regulatory subunit NEMO (IKK-γ). Genetic studies have demonstrated that the IKKβ and NEMO subunits of IKK are required for activation of NF-κB by most stimuli.48 Moreover, NEMO is post-translationally modified through TRAF6-dependent K63-linked polyubiquitination. TRAF6 is an E3 ubiquitin ligase and self-ubiquitinated in a K63 manner; it plays an essential role in the activation of IKK, the MAPK cascade and TAK1.49 However, the mechanism by which the E1 inhibitor PYR41 attenuates IKK and NF-κB activation remains unclear. Our results demonstrated that PYR41 markedly decreased the levels of phosphorylated IKKα/β and IκkBα and the K63-linked ubiquitination of TRAF6 and NEMO (Fig. 8). Hence, these results indicate that PYR41 inhibits activation of the IKK/NF-κB signalling pathway through inhibition of K63-link ubiquitination of TRAF6 and NEMO in DCs in response to Ang II.
In conclusion, the present study revealed a novel role for the ubiquitin-activating enzyme E1 in Ang II-induced maturation and activation of DCs and the underlying mechanisms. E1 promotes the K63-linked ubiquitination of TRAF6/NEMO and proteasomal degradation of IκBα and MKP-1, thereby leading to sustained activation of NF-κB and ERK/STAT1 pathways, which are necessary for the maturation and activation of DCs, and this effect of Ang II on DCs was markedly attenuated by the E1 inhibitor PYR41. The proposed pathway is presented in Fig. 9. Collectively, these data suggest that the E1 inhibitor PYR41 could represent a novel therapeutic drug for DC-mediated autoimmune responses and cardiovascular diseases.
Figure 9.
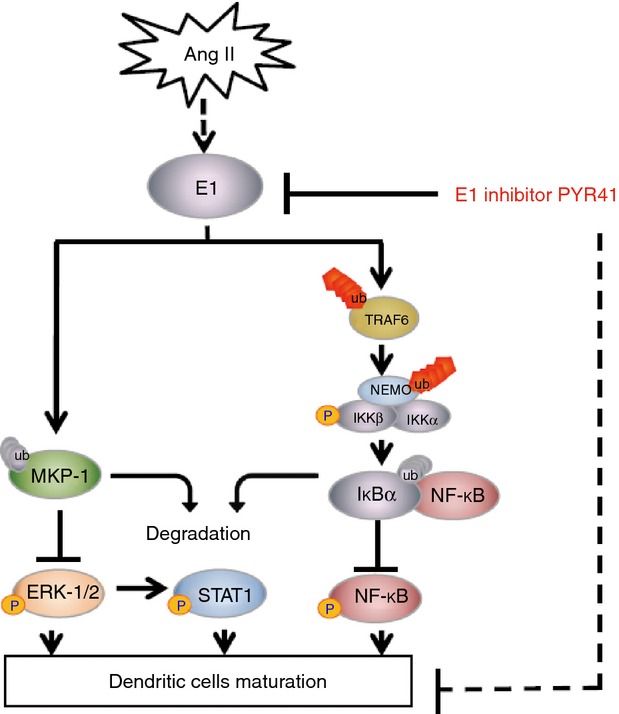
Proposed pathways of Angiotensin II (Ang II)-induced maturation and activation of dendritic cells (DCs) mediated by E1. Our data show that Ang II increases E1 activation. On the one hand, E1 promotes degradation of mitogen-activated protein kinase phosphatase 1 (MKP-1) through the ubiquitin-mediated proteolytic pathway, which in turn enhances the activation of extracellular signal-regulated kinase (ERK)/ signal transducer and activator of transcription 1 (STAT1) pathways; On the other hand, E1 enhances K63-linked ubiquitination of tumour necrosis factor receptor-associated factor 6 (TRAF6) and nuclear factor-κB (NF-κB) essential modulator (NEMO), which results in activation of inhibitor of NF-κB kinase (IKK), degradation of inhibitor of NF-κB (IκBα) and activation of NF-κB leading to the maturation and activation of DCs in response to Ang II stimulation. While the E1 inhibitor PYR41 suppresses Ang II-induced actions on DCs.
Acknowledgments
HHL and JD conceived and designed the experiments. CC and YM performed the experiments and LW, HXW, CT and GDP contributed reagents/materials/analysis tools. HHL, JD and CC wrote the paper. This work was supported by the 973 programme (No. 2012CB517802), China National Natural Science Funds (No. 81025001, 81330003), Chang Jiang Scholar Programme, the Beijing high-level talents programme (PHR20110507).
Glossary
- Ang II
Angiotensin II
- APCs
antigen-presenting cells
- BMDCs
bone marrow derived dendritic cells
- CFSE
Carboxyfluorescein diacetate-succinimidyl ester
- DCs
dendritic cells
- E1/Uba1
ubiquitin activating enzyme
- E2
ubiquitin conjugating enzyme
- E3
ubiquitin-protein ligase
- ERK
extracellular signal-regulated kinase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- IKK-γ
IκB kinase γ
- IL-4
interleukin-4
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MLR
mixed lymphocyte reaction
- NEMO
NF-κB essential modulator
- NF-κB
nuclear factor-κB
- PYR41
4[4-(5-nitro-furan-2-ylmethylene)-3,5-dioxo-pyrazolidin-1-yl]-benzoic acid ethyl ester
- STAT
signal regulated transducer and activator of transcription
- TNF
tumour necrosis factor
- TRAF6
TNF receptor-associated factor
- UPS
ubiquitin-proteasome system
Disclosures
The authors have no conflicts of interest to declare.
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez D, Vollmann EH, von Andrian UH. Mechanisms and consequences of dendritic cell migration. Immunity. 2008;29:325–42. doi: 10.1016/j.immuni.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukuda D, Sata M, Ishizaka N, Nagai R. Critical role of bone marrow angiotensin II type 1 receptor in the pathogenesis of atherosclerosis in apolipoprotein E deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:90–6. doi: 10.1161/ATVBAHA.107.152363. [DOI] [PubMed] [Google Scholar]
- 5.Guzik TJ, Hoch NE, Brown KA, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–60. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nahmod KA, Vermeulen ME, Raiden S, et al. Control of dendritic cell differentiation by angiotensin II. FASEB J. 2003;17:491–3. doi: 10.1096/fj.02-0755fje. [DOI] [PubMed] [Google Scholar]
- 7.Nahmod K, Gentilini C, Vermeulen M, et al. Impaired function of dendritic cells deficient in angiotensin II type 1 receptors. J Pharmacol Exp Ther. 2010;334:854–62. doi: 10.1124/jpet.109.161760. [DOI] [PubMed] [Google Scholar]
- 8.Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010;2:247–57. doi: 10.1002/emmm.201000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mani A, Gelmann EP. The ubiquitin–proteasome pathway and its role in cancer. J Clin Oncol. 2005;23:4776–89. doi: 10.1200/JCO.2005.05.081. [DOI] [PubMed] [Google Scholar]
- 10.Sun L, Chen ZJ. The novel functions of ubiquitination in signaling. Curr Opin Cell Biol. 2004;16:119–26. doi: 10.1016/j.ceb.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Stephen AG, Trausch-Azar JS, Ciechanover A, Schwartz AL. The ubiquitin-activating enzyme E1 is phosphorylated and localized to the nucleus in a cell cycle-dependent manner. J Biol Chem. 1996;271:15608–14. doi: 10.1074/jbc.271.26.15608. [DOI] [PubMed] [Google Scholar]
- 12.Yang Y, Kitagaki J, Dai RM, et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007;67:9472–81. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- 13.Xu GW, Ali M, Wood TE, et al. The ubiquitin-activating enzyme E1 as a therapeutic target for the treatment of leukemia and multiple myeloma. Blood. 2010;115:2251–9. doi: 10.1182/blood-2009-07-231191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158:2723–30. [PubMed] [Google Scholar]
- 15.Lutz MB, Kukutsch N, Ogilvie AL, Rössner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 16.Wang HX, Yang H, Han QY, Li N, Jiang X, Tian C, Du J, Li HH. NADPH oxidases mediate a cellular ‘memory’ of angiotensin II stress in hypertensive cardiac hypertrophy. Free Radic Biol Med. 2013;65:897–907. doi: 10.1016/j.freeradbiomed.2013.08.179. [DOI] [PubMed] [Google Scholar]
- 17.Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, Patterson C. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117:3211–23. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng Y, Chen C, Wang L, Wang X, Tian C, Du J, Li HH. Toll-like receptor-2 ligand peptidoglycan upregulates expression and ubiquitin ligase activity of CHIP through JNK pathway. Cell Physiol Biochem. 2013;32:1097–105. doi: 10.1159/000354509. [DOI] [PubMed] [Google Scholar]
- 19.Nabah YN, Mateo T, Estelles R, et al. Angiotensin II induces neutrophil accumulation in vivo through generation and release of CXC chemokines. Circulation. 2004;110:3581–6. doi: 10.1161/01.CIR.0000148824.93600.F3. [DOI] [PubMed] [Google Scholar]
- 20.Yoshimura S, Bondeson J, Foxwell BM, Brennan FM, Feldmann M. Effective antigen presentation by dendritic cells is NF-κB dependent: coordinate regulation of MHC, co-stimulatory molecules and cytokines. Int Immunol. 2001;13:675–83. doi: 10.1093/intimm/13.5.675. [DOI] [PubMed] [Google Scholar]
- 21.Wei-guo Z, Hui Y, Shan L, et al. PPAR-γ agonist inhibits Ang II-induced activation of dendritic cells via the MAPK and NF-κB pathways. Immunol Cell Biol. 2010;88:305–12. doi: 10.1038/icb.2009.100. [DOI] [PubMed] [Google Scholar]
- 22.Jackson SH, Yu CR, Mahdi RM, Ebong S, Egwuagu CE. Dendritic cell maturation requires STAT1 and is under feedback regulation by suppressors of cytokine signaling. J Immunol. 2004;172:2307–15. doi: 10.4049/jimmunol.172.4.2307. [DOI] [PubMed] [Google Scholar]
- 23.Johnson LM, Scott P. STAT1 expression in dendritic cells, but not T cells, is required for immunity to Leishmania major. J Immunol. 2007;178:7259–66. doi: 10.4049/jimmunol.178.11.7259. [DOI] [PubMed] [Google Scholar]
- 24.Magnani M, Crinelli R, Bianchi M, Antonelli A. The ubiquitin-dependent proteolytic system and other potential targets for the modulation of nuclear factor-κB (NF-κB) Curr Drug Targets. 2000;1:387–99. doi: 10.2174/1389450003349056. [DOI] [PubMed] [Google Scholar]
- 25.Lin YW, Chuang SM, Yang JL. ERK1/2 achieves sustained activation by stimulating MAPK phosphatase-1 degradation via the ubiquitin–proteasome pathway. J Biol Chem. 2003;278:21534–41. doi: 10.1074/jbc.M301854200. [DOI] [PubMed] [Google Scholar]
- 26.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 27.Chen ZJ. Ubiquitin signalling in the NF-κB pathway. Nat Cell Biol. 2005;7:758–65. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moberg CL. An appreciation of Ralph Marvin Steinman (1943–2011) J Exp Med. 2011;208:2337–42. doi: 10.1084/jem.20112294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hackstein H, Thomson AW. Dendritic cells: emerging pharmacological targets of immunosuppressive drugs. Nat Rev Immunol. 2004;4:24–34. doi: 10.1038/nri1256. [DOI] [PubMed] [Google Scholar]
- 30.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–26. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 31.Marchesi C, Paradis P, Schiffrin EL. Role of the renin–angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–74. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 32.Bush E, Maeda N, Kuziel WA, Dawson TC, Wilcox JN, DeLeon H, Taylor WR. CC chemokine receptor 2 is required for macrophage infiltration and vascular hypertrophy in angiotensin II-induced hypertension. Hypertension. 2000;36:360–3. doi: 10.1161/01.hyp.36.3.360. [DOI] [PubMed] [Google Scholar]
- 33.Dai Q, Xu M, Yao M, Sun B. Angiotensin AT1 receptor antagonists exert anti-inflammatory effects in spontaneously hypertensive rats. Br J Pharmacol. 2007;152:1042–8. doi: 10.1038/sj.bjp.0707454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jurewicz M, McDermott DH, Sechler JM, Tinckam K, Takakura A, Carpenter CB, Milford E, Abdi R. Human T and natural killer cells possess a functional renin–angiotensin system: further mechanisms of angiotensin II-induced inflammation. J Am Soc Nephrol. 2007;18:1093–102. doi: 10.1681/ASN.2006070707. [DOI] [PubMed] [Google Scholar]
- 35.Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2009;296:R208–16. doi: 10.1152/ajpregu.90521.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lapteva N, Nieda M, Ando Y, et al. Expression of renin-angiotensin system genes in immature and mature dendritic cells identified using human cDNA microarray. Biochem Biophys Res Commun. 2001;285:1059–65. doi: 10.1006/bbrc.2001.5215. [DOI] [PubMed] [Google Scholar]
- 37.Nie W, Yan H, Li S, Zhang Y, Yu F, Zhu W, Fan F, Zhu J. Angiotensin-(1-7) enhances angiotensin II induced phosphorylation of ERK1/2 in mouse bone marrow-derived dendritic cells. Mol Immunol. 2009;46:355–61. doi: 10.1016/j.molimm.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 38.Nakahara T, Moroi Y, Uchi H, Furue M. Differential role of MAPK signaling in human dendritic cell maturation and Th1/Th2 engagement. J Dermatol Sci. 2006;42:1–11. doi: 10.1016/j.jdermsci.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 39.Fang Y, Zhong L, Lin M, et al. MEK/ERK dependent activation of STAT1 mediates dasatinib-induced differentiation of acute myeloid leukemia. PLoS One. 2013;8:e66915. doi: 10.1371/journal.pone.0066915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li N, McLaren JE, Michael DR, Clement M, Fielding CA, Ramji DP. ERK is integral to the IFN-γ-mediated activation of STAT1, the expression of key genes implicated in atherosclerosis, and the uptake of modified lipoproteins by human macrophages. J Immunol. 2010;185:3041–8. doi: 10.4049/jimmunol.1000993. [DOI] [PubMed] [Google Scholar]
- 41.Ebstein F, Lange N, Urban S, Seifert U, Kruger E, Kloetzel PM. Maturation of human dendritic cells is accompanied by functional remodelling of the ubiquitin-proteasome system. Int J Biochem Cell Biol. 2009;41:1205–15. doi: 10.1016/j.biocel.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 42.Herrmann J, Ciechanover A, Lerman LO, Lerman A. The ubiquitin-proteasome system in cardiovascular diseases – a hypothesis extended. Cardiovasc Res. 2004;61:11–21. doi: 10.1016/j.cardiores.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 43.Nencioni A, Grunebach F, Patrone F, Ballestrero A, Brossart P. The proteasome and its inhibitors in immune regulation and immune disorders. Crit Rev Immunol. 2006;26:487–98. doi: 10.1615/critrevimmunol.v26.i6.20. [DOI] [PubMed] [Google Scholar]
- 44.Nencioni A, Schwarzenberg K, Brauer KM, Schmidt SM, Ballestrero A, Grunebach F, Brossart P. Proteasome inhibitor bortezomib modulates TLR4-induced dendritic cell activation. Blood. 2006;108:551–8. doi: 10.1182/blood-2005-08-3494. [DOI] [PubMed] [Google Scholar]
- 45.Banerjee S, Zmijewski JW, Lorne E, Liu G, Sha Y, Abraham E. Modulation of SCF β-TrCP-dependent IκBα ubiquitination by hydrogen peroxide. J Biol Chem. 2010;285:2665–75. doi: 10.1074/jbc.M109.060822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie P, Guo S, Fan Y, Zhang H, Gu D, Li H. Atrogin-1/MAFbx enhances simulated ischemia/reperfusion-induced apoptosis in cardiomyocytes through degradation of MAPK phosphatase-1 and sustained JNK activation. J Biol Chem. 2009;284:5488–96. doi: 10.1074/jbc.M806487200. [DOI] [PubMed] [Google Scholar]
- 47.Lin YW, Yang JL. Cooperation of ERK and SCFSkp2 for MKP-1 destruction provides a positive feedback regulation of proliferating signaling. J Biol Chem. 2006;281:915–26. doi: 10.1074/jbc.M508720200. [DOI] [PubMed] [Google Scholar]
- 48.Sebban-Benin H, Pescatore A, Fusco F, et al. Identification of TRAF6-dependent NEMO polyubiquitination sites through analysis of a new NEMO mutation causing incontinentia pigmenti. Hum Mol Genet. 2007;16:2805–15. doi: 10.1093/hmg/ddm237. [DOI] [PubMed] [Google Scholar]
- 49.Kishida S, Sanjo H, Akira S, Matsumoto K, Ninomiya-Tsuji J. TAK1-binding protein 2 facilitates ubiquitination of TRAF6 and assembly of TRAF6 with IKK in the IL-1 signaling pathway. Genes Cells. 2005;10:447–54. doi: 10.1111/j.1365-2443.2005.00852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]