

Abstract
A number of recently developed and approved therapeutic agents based on highly specific and potent antibodies have shown the potential of antibody therapy. As the next step, antibody-based therapeutics will be bioengineered in a way that they not only bind pathogenic targets but also address other issues, including drug targeting and delivery. For antibodies that are expected to act within brain tissue, like those that are directed against the pathogenic prion protein isoform, one of the major obstacles is the blood-brain barrier which prevents efficient transfer of the antibody, even of the engineered single-chain variants. We recently demonstrated that a specific prion-specific antibody construct which was injected into the murine tail vein can be efficiently transported into brain tissue. The novelty of the work was in that the cell penetrating peptide was used as a linker connecting both specificity-determining domains of the antibody peptide, thus eliminating the need for the standard flexible linker, composed of an arrangement of three consecutive (Gly4Ser) repeats. This paves the road toward improved bioengineered antibody variants that target brain antigens.
Keywords: prion protein, single chain antibody fragment (scFv), blood-brain barrier, cell-penetrating peptides, Creutzfeldt-Jakob disease (CJD)
Introduction
The causative agent of several brain disorders, the prion protein, is still far from being fully understood. Although little doubt remains about its essential involvement in the pathogenicity, we still miss a detailed understanding of the process when we consider prion’s biochemical properties and perturbances that result in the development of diseases such as the Creutzfeldt-Jakob disease (CJD). Despite its rare occurrence, CJD and related disorders are important because of their uniqueness and because we still have no therapy for the diseases once they are diagnosed. We have recently shown a novel bioengineering approach to antibody engineering in which the bioactive antibody fragment is equipped with a peptide that enables penetration across the blood-brain barrier and which replaces the standard linker used in joining the two variable domains of the single-chain antibody fragment (scFv).1 Here we describe the bioengineering aspects of the work and briefly explain the potential of antibody constructs that are developed to act in human or animal brain where specific targets that need to be inactivated in vivo are found.
Prions and Prion Therapy Approaches
Prion diseases or transmissible spongiform encephalopathies (TSEs) are fatal neurodegenerative disorders caused by a pathogenic form of the host-encoded prion protein (PrPC), named PrPSc or shortly prion.2 The conversion of the PrPC into the PrPSc is believed to involve post-translation modification from mainly α-helical to β-sheet protein arrangement.3 Disorders affect not only humans but also a wide variety of animals; the most common diseases are Creutzfeldt-Jakob disease (CJD) in humans, scrapie in sheep, and bovine spongiform encephalopathy (BSE) in cattle (reviewed in ref. 4). No effective prophylactic or therapeutic treatment is currently available.
Prion diseases in humans were first described almost 100 y ago,5 but did not attract widespread interest until the European BSE crisis occurred in 1990s.6 First suggestions that BSE is transmissible to humans lead to huge investments in research of prion diseases and more important, finding an effective cure. Further research revealed that clinical symptoms of prion diseases manifest themselves when most of the brain damage has already taken the place. Besides, the pathogenic prion can unfortunately only be detected by postmortem brain histology to confirm the diagnosis. Numerous compounds and different therapeutic strategies have been tested to overcome the obstacles and to find not only an appropriate drug but also an early premortem diagnostics of asymptomatic prion-infected individuals.7 Results show that anti-prion antibodies and their variants represent one of the most promising compounds that could be used in TSE treatment. Since the first successful production of high affinity anti-PrP antibodies,8 several antibodies against prion protein have been produced. Anti-prion activity in vitro9-12 as well as in vivo13-15 has been described for different antibodies. Antibodies that are able to distinguish between cellular and pathological PrP conformation were suggested to be a possible key to success.16
Benefits of Engineered Antibodies
Antibodies per se, despite their specificity and other immunological properties, are rather inconvenient for therapeutic use. They are difficult to produce in a recombinant form and require large doses due to their quaternary structure and size. Murine monoclonal antibodies that laboratories develop and characterize cannot be used on humans due to the immune response against non-human epitopes on these antibodies. Solutions went in two directions: reducing the size and structural complexity was achieved by development of single-chain antibody fragments (scFvs), while the response against murine epitopes was tackled by preparation of humanized forms.
Single-chain antibodies are about 25 kDa (less than a sixth of immunoglobulin G size) bioengineered molecules that combine the heavy and the light chain variable domain of an antibody by a flexible linker. The specificity thus remains unchanged, although this reduction in size results in absence of complement response that is present with regular antibodies. Due to its smaller size, scFvs are believed to better penetrate tissues. In addition, scFvs can be produced in prokaryotic systems as these antibody fragments lack glycosylation sites (reviewed in ref. 17). Bioengineered scFvs can also be designed as bispecific agents and for a range of applications other than clinical use.
Antibody Humanization
Therapeutic antibodies have become an important class of modern medicines. At the moment, monoclonal antibodies (mAbs) represent more than 30% of therapeutic proteins in clinical trials whereas 30 mAbs have already reached the market (as reviewed by Gilliland et al.).18
With a discovery of rodent antibodies and hybridoma technology in 1975 it became possible to prepare highly specific monoclonal antibodies for the first time.19 Further research soon revealed the biggest disadvantages of nonhuman antibodies: immunogenicity (see ref. 20 for review). Therefore their conversion into more human-like antibodies was introduced (Fig. 1). The first try to reduce immunogenicity of nonhuman antibodies was by chimerization, where constant domains were completely replaced with the human ones and connected with the nonhuman variable domains.21 A further step was humanization, where also nonhuman amino acid residues in variable domains were replaced with those more often found in human antibodies. Technically, humanization methods can be classified in two groups: rational methods and empirical methods.22 The most popular among the rational methods are CDR grafting,23 resurfacing,24 and SDR grafting,25 where the design of humanized version is based on sequence and structure analysis and assessing of a few humanized variants for desired properties. The antibody with the best results is chosen as a final humanized variant. Empirical methods on the other hand rely on selection rather than on design cycle with the invention of phage display (reviewed in ref. 22).
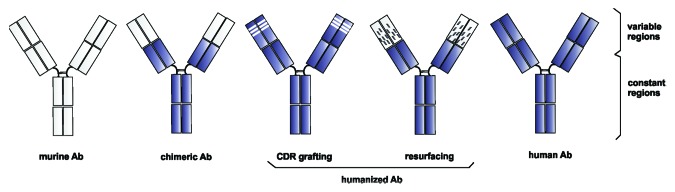
Figure 1. An overview of hybrid molecules between murine and human antibodies, developed for reducing the HAMA response. Humanized antibodies are represented by CDR grafting and resurfacing approaches. Each box corresponds to one immunoglobulin domain. Shaded: human sequences, empty: murine sequences.
By early 2012, 28 therapeutic antibodies were marketed in Europe or USA, of which 36% were prepared by humanization and 32% were human monoclonal antibodies,26 prepared in transgenic mice such as the XenoMouse (presented in a review in ref. 27). Still, immunogenicity is one of the major drawbacks of therapeutic antibodies. Approximately 40% of approved chimeric antibodies and 9% of humanized antibodies induce antidrug antibody response in vivo,28 and even fully human therapeutic antibodies can provoke patient’s immune system, e.g., 12% of patients treated with the fully human antibody drug Humira developed anti-drug antibodies,29 which decreases the efficiency of the drug. Unfortunately, there is no reliable method or test for prediction of antibodies immunogenicity. Prediction and testing immunogenicity is therefore one of the biggest challenges in biotherapeutics development, but greater investments and cooperation of industry and academia should result in new discoveries and development of more efficient drugs for the future.30
Targeting to Brain
Neurodegenerative diseases, such as Alzheimer disease, Parkinson disease and stroke, are rapidly increasing as population ages. However, successful treatment is limited to efficient drug delivery through the blood-brain barrier to the brain.
Blood-brain barrier (BBB) is almost impermeable, highly selective, and well-coordinated barrier that limits drug delivery by allowing to enter the brain from the bloodstream only to lipophilic molecules with low molecular mass (less than 400 Da).31 Therefore, nearly all large therapeutic molecules, such as antibodies, oligonucleotides and viral vectors, cannot pass through the BBB. Less than 10% of therapeutic agents for neurodegenerative diseases enter into clinical trial just because of poor brain penetration.32
In recent years, various strategies and several routes for delivering drugs into the brain have been developed and tested for their advantages and disadvantages. The approaches generally include drug manipulation, disrupting of the BBB or finding of alternative routes for drug delivery. Macromolecules have been tried to smuggle across the BBB as lipophilic precursors, inside liposomes, in the forms of produgs, using carrier or receptor mediated transport, disrupting the BBB by X-radiation, using intraventricular or intranasal route and many other ways (reviewed in ref. 33), but unfortunately none of these methods has so far achieved the desired safety and efficiency.
The opportunity of successful delivering biologically active cargoes into various tissues, cells and compartments including the brain was raised after discovery of natural polycationic cell penetrating peptides (CPPs). The first proof of protein transduction with CPP was described in 1988 when HIV-1 transactivating protein TAT was found to enter mammalian cells.34,35 A few years later the same behavior was described for Antennapedia homeodomain of Drosophila melanogaster,36 also called penetratin, followed by over 100 other peptides over past 20 y. They have been successfully used to improve the intracellular delivery of various biomolecules in different cell types including intracellular compartment and the brain both in vitro and in vivo and over 2000 articles have been published on a topic of CPP. Besides numerous preclinical and clinical studies have been conducted or are underway, although no CPP conjugate has been approved by FDA yet.37
Unfortunately, CPPs have also a few drawbacks. One of them is definitely their nonspecificity, so that they enter every cell that they come in contact with. The lack of selectivity enhances the risk of toxic effects of the compound on the healthy tissues.37
Brain Targeting Prion-Specific scFv
Our recent results1 show that antibody fragments with proven specificity against the pathogenic form of the prion protein38 can be modified in such a way that the standard linker connecting the variable domains of a specific antibody is replaced by the penetratin linker (Fig. 2). Although adding a CPP to an antibody fragment is not a novel approach, such peptides were standardly linked to the N- or C-terminus. Since scFvs contain the biotechnologically “unexploited” linker, we tested the idea that the linker could be replaced by the Trojan peptide. We faced two essential questions: (1) would the CPP enable correct folding that is essential for scFv function and (2) will the CPP truly enable transport of the scFv into brain tissue.
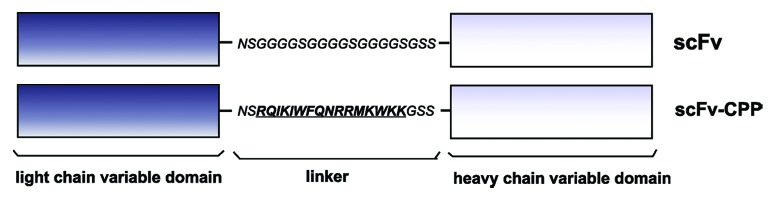
Figure 2. A schematic representation of the parent (top) and bioengineered scFv (bottom) in which the standard linker was replaced by the penetratin sequence (underlined).
Molecular modeling did not predict major conformational changes of the linker region due to replacement of the standard arrangement, as shown in Figure 3. Models were calculated by I-Tasser39-41 and visualized using Polyview-3D.42 The linker was predicted to retain a rather unordered structure, although the space-filled model (not shown) revealed a possible tighter interaction with the immunoglobulin fold via several bulkier residues in the penetratin sequence.

Figure 3. A side-by-side comparison of the two scFvs used in the study. Left: parent scFv; right: penetratin-scFv. Models were prepared as described in the text. Green: immunoglobulin folds of the heavy and light chain variable domains, including the C-terminal His-tag: red: linker region.
Experiments were started both with TAT and penetratin linkers, but we could successfully produce only the penetratin-scFv combination in E. coli, essentially as published before for the “wild-type” linker construct38 and using the in-house expression vector.43 No significant differences were observed in yield, protein purification and stability. The penetratin scFv retained the specificity for the parent antigen, although with a slightly weaker binding than the non-modified scFv. Using freshly isolated recombinant penetratin-scFv with the standard scFv as a control we clearly demonstrated that the penetratin construct efficiently crossed the BBB, while the control didn’t. Penetratin also guided scFv transport into liver tissue which we checked in parallel.1
A more detailed look at the subcellular localization of the penetratin-scFv revealed that unexpectedly in brain cell, the engineered antibody fragment was found in nuclei, whereas it was located in the cytoplasm of liver cells.1 Although the penetratin sequence in the context of scFv does resemble the nuclear entry signal, it was interesting to see that the same sequence resulted in nuclear location only in brain nerve cells but not in hepatocytes. More work is required to unravel the properties of localization signals that are cell- or tissue-specific, possibly due to a different set of transport and transport-associated proteins.
Prospects
Although construction of the new type of penetratin-equipped scFv and demonstration of its functionality was rather preliminary, it certainly opens new ways toward bioengineered antibody fragments. Generally used linkers that are foreign sequences to the immune system can be efficiently replaced by the Trojan horse peptide sequence with essentially equal linking properties but with the additional benefit of directing scFvs across the BBB. The next step would certainly be applying this same approach on the humanized version of the antibody.44 This could as well be done with any other antibody fragments with targets in the brain. Bioengineered antibodies have thus again shown its great potential and the last word in their optimization has not yet been said.
Acknowledgments
We would like to thank Prof. Vladka Čurin Šerbec from the National Blood Transfusion Centre and to her colleagues for a fruitful cooperation on the prion antibody topic, as well as to Drs. Gorazd Drevenšek, Rok Romih, and Samo Hudoklin from the University of Ljubljana Medical Faculty for their contribution in performing experiments on mice and on immunohistology analyses. Nives Škrlj was supported by the Young Investigator grant from the Slovenian Research Agency. Marko Dolinar was funded in part by the Slovenian Research Agency grant L3–0206 and research program P1–0048.
Glossary
Abbreviations:
- BBB
blood-brain barrier
- BSE
bovine spongiform encephalopathy
- CDR
complementarity determining region
- CJD
Creutzfeldt-Jakob disease
- CPP
cell-penetrating peptide
- FDA
Food and Drug Administration
- PrP
prion protein
- scFv
single-chain antibody variable fragment
- SDR
specificity determining residues
- TSE
transmissible spongiform encephalopathy
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/bioe/article/26069
References
- 1.Škrlj N, Drevenšek G, Hudoklin S, Romih R, Čurin Šerbec V, Dolinar M. Recombinant single-chain antibody with the Trojan peptide penetratin positioned in the linker region enables cargo transfer across the blood-brain barrier. Appl Biochem Biotechnol. 2013;169:159–69. doi: 10.1007/s12010-012-9962-7. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 3.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sikorska B, Liberski PP. Human prion diseases: from kuru to variant Creutzfeldt-Jakob disease. Subcell Biochem. 2012;65:457–96. doi: 10.1007/978-94-007-5416-4_17. [DOI] [PubMed] [Google Scholar]
- 5.Creutzfeldt HG. Über eine eigenartige herdförmige Erkrankung des Zentralnervensystems. Z Gesamte Neurol Psychiatr. 1920;57:1–18. doi: 10.1007/BF02866081. [DOI] [Google Scholar]
- 6.Bradley R, Collee JG, Liberski PP. Variant CJD (vCJD) and bovine spongiform encephalopathy (BSE): 10 and 20 years on: part 1. Folia Neuropathol. 2006;44:93–101. [PubMed] [Google Scholar]
- 7.Ludewigs H, Zuber C, Vana K, Nikles D, Zerr I, Weiss S. Therapeutic approaches for prion disorders. Expert Rev Anti Infect Ther. 2007;5:613–30. doi: 10.1586/14787210.5.4.613. [DOI] [PubMed] [Google Scholar]
- 8.Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M, Burton D, Yang SL, DeArmond SJ. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci U S A. 1993;90:10608–12. doi: 10.1073/pnas.90.22.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A. 2001;98:9295–9. doi: 10.1073/pnas.151242598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412:739–43. doi: 10.1038/35089090. [DOI] [PubMed] [Google Scholar]
- 11.Beringue V, Vilette D, Mallinson G, Archer F, Kaisar M, Tayebi M, Jackson GS, Clarke AR, Laude H, Collinge J, et al. PrPSc binding antibodies are potent inhibitors of prion replication in cell lines. J Biol Chem. 2004;279:39671–6. doi: 10.1074/jbc.M402270200. [DOI] [PubMed] [Google Scholar]
- 12.Féraudet C, Morel N, Simon S, Volland H, Frobert Y, Créminon C, Vilette D, Lehmann S, Grassi J. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem. 2005;280:11247–58. doi: 10.1074/jbc.M407006200. [DOI] [PubMed] [Google Scholar]
- 13.Sigurdsson EM, Sy MS, Li R, Scholtzova H, Kascsak RJ, Kascsak R, Carp R, Meeker HC, Frangione B, Wisniewski T. Anti-prion antibodies for prophylaxis following prion exposure in mice. Neurosci Lett. 2003;336:185–7. doi: 10.1016/S0304-3940(02)01192-8. [DOI] [PubMed] [Google Scholar]
- 14.White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422:80–3. doi: 10.1038/nature01457. [DOI] [PubMed] [Google Scholar]
- 15.Sadowski MJ, Pankiewicz J, Prelli F, Scholtzova H, Spinner DS, Kascsak RB, Kascsak RJ, Wisniewski T. Anti-PrP Mab 6D11 suppresses PrP(Sc) replication in prion infected myeloid precursor line FDC-P1/22L and in the lymphoreticular system in vivo. Neurobiol Dis. 2009;34:267–78. doi: 10.1016/j.nbd.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Čurin Serbec V, Bresjanac M, Popović M, Pretnar Hartman K, Galvani V, Rupreht R, Cernilec M, Vranac T, Hafner I, Jerala R. Monoclonal antibody against a peptide of human prion protein discriminates between Creutzfeldt-Jacob’s disease-affected and normal brain tissue. J Biol Chem. 2004;279:3694–8. doi: 10.1074/jbc.M310868200. [DOI] [PubMed] [Google Scholar]
- 17.Ahmad ZA, Yeap SK, Ali AM, Ho WY, Alitheen NB, Hamid M. scFv antibody: principles and clinical application. Clin Dev Immunol. 2012;2012:980250. doi: 10.1155/2012/980250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilliland GL, Luo J, Vafa O, Almagro JC. Leveraging SBDD in protein therapeutic development: antibody engineering. Methods Mol Biol. 2012;841:321–49. doi: 10.1007/978-1-61779-520-6_14. [DOI] [PubMed] [Google Scholar]
- 19.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–7. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 20.Mirick GR, Bradt BM, Denardo SJ, Denardo GL. A review of human anti-globulin antibody (HAGA, HAMA, HACA, HAHA) responses to monoclonal antibodies. Not four letter words. Q J Nucl Med Mol Imaging. 2004;48:251–7. [PubMed] [Google Scholar]
- 21.Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci U S A. 1984;81:6851–5. doi: 10.1073/pnas.81.21.6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almagro JC, Fransson J. Humanization of antibodies. Front Biosci. 2008;13:1619–33. doi: 10.2741/2786. [DOI] [PubMed] [Google Scholar]
- 23.Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 1986;321:522–5. doi: 10.1038/321522a0. [DOI] [PubMed] [Google Scholar]
- 24.Padlan EA. A possible procedure for reducing the immunogenicity of antibody variable domains while preserving their ligand-binding properties. Mol Immunol. 1991;28:489–98. doi: 10.1016/0161-5890(91)90163-E. [DOI] [PubMed] [Google Scholar]
- 25.Tamura M, Milenic DE, Iwahashi M, Padlan E, Schlom J, Kashmiri SV. Structural correlates of an anticarcinoma antibody: identification of specificity-determining residues (SDRs) and development of a minimally immunogenic antibody variant by retention of SDRs only. J Immunol. 2000;164:1432–41. doi: 10.4049/jimmunol.164.3.1432. [DOI] [PubMed] [Google Scholar]
- 26.Reichert JM. Marketed therapeutic antibodies compendium. MAbs. 2012;4:413–5. doi: 10.4161/mabs.19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lonberg N. Human antibodies from transgenic animals. Nat Biotechnol. 2005;23:1117–25. doi: 10.1038/nbt1135. [DOI] [PubMed] [Google Scholar]
- 28.Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods. 2005;36:3–10. doi: 10.1016/j.ymeth.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Bender NK, Heilig CE, Dröll B, Wohlgemuth J, Armbruster FP, Heilig B. Immunogenicity, efficacy and adverse events of adalimumab in RA patients. Rheumatol Int. 2007;27:269–74. doi: 10.1007/s00296-006-0183-7. [DOI] [PubMed] [Google Scholar]
- 30.Lu ZJ, Deng SJ, Huang DG, He Y, Lei M, Zhou L, Jin P. Frontier of therapeutic antibody discovery: The challenges and how to face them. World J Biol Chem. 2012;3:187–96. doi: 10.4331/wjbc.v3.i12.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pardridge WM. Drug targeting to the brain. Pharm Res. 2007;24:1733–44. doi: 10.1007/s11095-007-9324-2. [DOI] [PubMed] [Google Scholar]
- 32.Khawli LA, Prabhu S. Drug delivery across the blood-brain barrier. Mol Pharm. 2013;10:1471–2. doi: 10.1021/mp400170b. [DOI] [PubMed] [Google Scholar]
- 33.Misra A, Ganesh S, Shahiwala A, Shah SP. Drug delivery to the central nervous system: a review. J Pharm Pharm Sci. 2003;6:252–73. [PubMed] [Google Scholar]
- 34.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–93. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 35.Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–88. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 36.Joliot AH, Triller A, Volovitch M, Pernelle C, Prochiantz A. alpha-2,8-Polysialic acid is the neuronal surface receptor of antennapedia homeobox peptide. New Biol. 1991;3:1121–34. [PubMed] [Google Scholar]
- 37.Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18:385–93. doi: 10.1016/j.molmed.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 38.Škrlj N, Čurin Šerbec VC, Dolinar M. Single-chain Fv antibody fragments retain binding properties of the monoclonal antibody raised against peptide P1 of the human prion protein. Appl Biochem Biotechnol. 2010;160:1808–21. doi: 10.1007/s12010-009-8699-4. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roy A, Yang J, Zhang Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012;40(Web Server issue):W471-7. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porollo A, Meller J. Versatile annotation and publication quality visualization of protein complexes using POLYVIEW-3D. BMC Bioinformatics. 2007;8:316. doi: 10.1186/1471-2105-8-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Škrlj N, Erčulj N, Dolinar M. A versatile bacterial expression vector based on the synthetic biology plasmid pSB1. Protein Expr Purif. 2009;64:198–204. doi: 10.1016/j.pep.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 44.Škrlj N, Vranac T, Popović M, Curin Šerbec V, Dolinar M. Specific binding of the pathogenic prion isoform: development and characterization of a humanized single-chain variable antibody fragment. PLoS One. 2011;6:e15783. doi: 10.1371/journal.pone.0015783. [DOI] [PMC free article] [PubMed] [Google Scholar]