

Abstract
Ubiquitylation is an important mechanism for regulating innate immune responses to viral infections. Attachment of lysine 63 (Lys63)–linked ubiquitin chains to the RNA sensor retinoic acid–inducible gene-I (RIG-I) by the ubiquitin E3 ligase tripartite motif protein 25 (TRIM25) leads to the activation of RIG-I and stimulates production of the antiviral cytokines interferon-α (IFN-α) and IFN-β. Conversely, Lys48-linked ubiquitylation of TRIM25 by the linear ubiquitin assembly complex (LUBAC) stimulates the proteasomal degradation of TRIM25, thereby inhibiting the RIG-I signaling pathway. Here, we report that ubiquitin-specific protease 15 (USP15) deubiquitylates TRIM25, preventing the LUBAC-dependent degradation of TRIM25. Through protein purification and mass spectrometry analysis, we identified USP15 as an interaction partner of TRIM25 in human cells. Knockdown of endogenous USP15 by specific small interfering RNA markedly enhanced the ubiquitylation of TRIM25. In contrast, expression of wild-type USP15, but not its catalytically inactive mutant, reduced the Lys48-linked ubiquitylation of TRIM25, leading to its stabilization. Furthermore, ectopic expression of USP15 enhanced the TRIM25- and RIG-I–dependent production of type I IFN and suppressed RNA virus replication. In contrast, depletion of USP15 resulted in decreased IFN production and markedly enhanced viral replication. Together, these data identify USP15 as a critical regulator of the TRIM25- and RIG-I–mediated antiviral immune response, thereby highlighting the intricate regulation of innate immune signaling.
INTRODUCTION
In virus-infected cells, viral RNA is recognized by various Toll-like receptors (TLRs) or the retinoic acid–inducible gene-I (RIG-I)–like receptors (RLRs), RIG-I, and melanoma differentiation-associated gene 5 (MDA5). Whereas TLRs detect extracellular viral RNA that has reached the endosomes or phagosomes of immune cells, RLRs sense RNA replication intermediates in the cytosol of infected nonimmune cells, such as epithelial cells and fibroblasts (1, 2). Specifically, RIG-I binds to the 5′-triphosphate–containing short double-stranded RNA (dsRNA) structures from various negative-sense RNA viruses, including influenza virus, paramyxoviruses, and the rhabdovirus vesicular stomatitis virus (VSV) (3–6). In addition, RIG-I also senses the RNA of hepatitis C virus, a positive-sense, single-stranded RNA virus belonging to the Flaviviridae family (7). In contrast, MDA5 binds to long dsRNA or high–molecular weight RNA aggregates generated during picornavirus replication (6, 8). Furthermore, both RIG-I and MDA5 contribute to the detection of dengue virus, West Nile virus, and reovirus (9). Upon binding to viral RNA through their C-terminal domains (CTDs) and central DExD/H-box helicases, RIG-I and MDA5 use their N-terminal caspase recruitment domains (CARDs) to interact with the mitochondrial adapter protein MAVS (also known as IPS-1, VISA, or Cardif) (10–13). MAVS then initiates signaling cascades that lead to the activation of the transcription factors interferon regulatory factor 3 (IRF3) and IRF7, as well as nuclear factor κB (NF-κB), which results in expression of the genes encoding the type I interferons (IFNs), IFN-α and IFN-β (2, 14).
Modifications by mono- or polyubiquitin, as well as the binding of unanchored ubiquitin chains, play major roles in the regulation of the signaling pathways leading to the production of IFN-α and IFN-β (15). To signal, RIG-I must undergo covalent Lys63-linked ubiquitylation that is mediated by the RING (really interesting new gene)–containing ubiquitin E3 ligase TRIM25 (tripartite motif protein 25) (16). In addition, TRIM25 mediated the noncovalent binding of Lys63-linked polyubiquitin chains to the RIG-I CARDs in a cell-free system (17). Upon viral infection, TRIM25 binds to the first CARD of RIG-I and then delivers Lys63-linked polyubiquitin chains to Lys172 in the second CARD (16, 18). Ubiquitylation of its CARDs enables RIG-I to oligomerize and efficiently interact with MAVS, thereby stimulating downstream signaling (16, 17). That the ubiquitylation of RIG-I by TRIM25 is essential for its antiviral signaling was established by the identification of a splice variant of RIG-I that carries a short deletion (of amino acid residues 36 to 80) within the first CARD and that thereby fails to bind to TRIM25 and stimulate antiviral signaling (18). Furthermore, through their nonstructural protein 1 (NS1), influenza Aviruses specifically target TRIM25 to inhibit the ubiquitylation-dependent activation of RIG-I, further strengthening the vital role of TRIM25 for RIG-I signaling (19). In addition, another ubiquitin E3 ligase, Riplet (also known as RNF135 or REUL), ubiquitylates the CTD of RIG-I, which is also necessary for RIG-I activation (20).
TRIM25 itself is inhibited by ubiquitylation that is mediated by the ubiquitin E3 ligases heme-oxidized IRP2 ubiquitin ligase 1 long (HOIL-1L) and HOIL-1L–interacting protein (HOIP) (21). HOIL-1L and HOIP proteins are increased in abundance in response to type I IFNs, and they act together as the linear ubiquitin assembly complex (LUBAC) to induce the Lys48-linked ubiquitylation of the SPRY domain of TRIM25, which then triggers the proteasomal degradation of TRIM25. In addition, LUBAC competes with TRIM25 for binding to RIG-I. Together, both actions of LUBAC suppress ubiquitylation of the RIG-I CARDs by TRIM25, thereby providing a negative feedback mechanism that regulates the RIG-I signaling pathway (21).
The process of ubiquitylation is reversible because ubiquitin moieties are removed by the enzymatic action of deubiquitylating enzymes (DUBs). Whereas our understanding of the mechanisms of protein ubiquitylation by E3 ligases has improved rapidly over the past 15 years, our knowledge of protein deubiquitylation and its role in regulating innate immune signal transduction is still rudimentary. The human genome encodes more than 90 DUBs that cleave conjugated ubiquitin or ubiquitin-like proteins from substrate proteins, with the ubiquitin-specific proteases (USPs) representing the largest subclass of this protein family (22, 23). Here, we identified USP15 as the DUB responsible for removing Lys48-linked polyubiquitin from TRIM25, thereby leading to stabilization of TRIM25 protein, as well as a sustained antiviral immune response. These results not only show that a dynamic balance between ubiquitylation and deubiquitylation tightly controls the function of TRIM25 but also identify USP15 as a critical regulator of antiviral innate immune responses.
RESULTS
USP15 physically interacts with TRIM25
We previously identified TRIM25 as an essential activator of the RIG-I–mediated production of type I IFN (16, 18). We thus postulated that the activity of TRIM25 or its abundance must be tightly controlled to elicit an effective immune response. To identify previously uncharacterized interaction partners of TRIM25 that might regulate its function, we performed protein purification of defined domains of TRIM25 fused to mammalian glutathione S-transferase (GST). Purification of GST-fused TRIM25–RING–B-box (GST-RING-BB) protein and mass spectrometric analysis revealed that USP15 was specifically present in a complex with GST-RING-BB, but not with GST alone (table S1).
We first confirmed the physical interaction between TRIM25 and USP15 with coimmunoprecipitation experiments. We found that ectopically expressed GST-RING-BB physically interacted with V5-tagged USP15 in human embryonic kidney (HEK) 293T cells (Fig. 1A). Furthermore, there was minimal binding of endogenous USP15 to TRIM25 in HEK 293T cells under normal conditions and at early time points during infection with Sendai virus (SeV), a paramyxovirus that is sensed by RIG-I (Fig. 1B and fig. S1A). In contrast, USP15 substantially interacted with TRIM25 at later time points during SeV infection. Consistently, there was increased binding of USP15 to TRIM25 in infected primary normal human lung fibroblasts (NHLFs), whereas the interaction was minimal in uninfected cells (fig. S1B). In support of this, confocal microscopic analysis showed that endogenous USP15 and TRIM25 substantially colocalized in the cytoplasm of SeV-infected, but not uninfected, HeLa cells (fig. S1C). Moreover, USP15 protein abundance was not detectably increased in response to SeV infection or stimulation with IFN-β (fig. S2, A and B). In contrast, the abundances of RIG-I and TRIM25, which are encoded by IFN-inducible genes (ISGs), were increased in response to SeV infection or IFN (fig. S2, A and B). Together, these results suggest that USP15 is not induced by IFN or viral infection, but instead is recruited to TRIM25 upon viral infection.
Fig. 1. USP15 interacts with TRIM25.

(A) HEK 293T cells were transfected with plasmids encoding GST or GST-TRIM25-RING-BB together with plasmid encoding USP15-V5. Forty-eight hours later, cells were lysed and whole-cell lysates (WCLs) were subjected to GST pull-down (GST-PD) and Western blotting (IB) analysis with anti-V5 antibody (α-V5) and anti-GST antibody (α-GST). Molecular mass markers (kD) are indicated to the left of the blots. (B) HEK 293T cells were left uninfected or were infected with SeV (50 HA U/ml). WCLs were prepared and subjected to immunoprecipitation (IP) with anti-USP15 antibody (α-USP15), and samples were analyzed by Western blotting with anti-TRIM25 antibody (α-TRIM25) and α-USP15 antibody. (C) Domain structures of TRIM25 and USP15, as well as a schematic representation of the GST-fused or Myc-tagged truncation mutants of TRIM25 and USP15, respectively. Numbers indicate amino acid residues. (D) HEK 293T cells were transfected with plasmids encoding the Myc-tagged DUSP, UBL, or His-Box domains of USP15 together with plasmids encoding GST or GST-TRIM25-RING-BB. Forty-eight hours later, WCLs were prepared and subjected to GST pull-down and Western blotting analysis with anti-Myc antibody (α-Myc) and α-GST antibody. WCLs were also analyzed by Western blotting with α-Myc. The upper part of this panel was exposed for a longer time than was used for the lower part. (E) HEK 293T cells were transfected with plasmids encoding GST or the indicated GST fusion constructs together with plasmid encoding Myc-tagged USP15 His-Box. Forty-eight hours later, WCLs were prepared and subjected to GST pull-down analysis, which was followed by Western blotting analysis with α-Myc and α-GST antibodies. Data in all panels are representative of three independent experiments.
TRIM25 and USP15 are both characterized by modular domain structures (Fig. 1C). TRIM25 consists of an N-terminal RING domain, two B-boxes, a central coiled-coil domain (CCD), and a C-terminal SPRY domain. USP15 belongs to a group of USP family members that are characterized by the presence of an N-terminal DUSP (domain present in USPs), a tandem ubiquitin-like (UBL) domain containing the Cys-Box of the catalytic triad, and a C-terminal region in which the His-Box of the catalytic triad is embedded (Fig. 1C). To characterize the TRIM25-binding region of USP15, we generated plasmids encoding specific domains of USP15 and assessed the extent of binding of these domains to GST-TRIM25-RING-BB in coimmunoprecipitation experiments (Fig. 1D). We found that, specifically, the C-terminal His-Box of USP15 bound to TRIM25-RING-BB, whereas there was no interaction between TRIM25-RING-BB and either the DUSP or the UBL domains of USP15 (Fig. 1D). To further characterize the region in TRIM25 required for its binding to USP15, we tested the interaction of the USP15 His-Box with the GST-fused TRIM25-RING domain (GST-RING) and the TRIM25–B-boxes (GST-BB) (Fig. 1E). GST-RING-BB served as a positive control for binding. Coimmunoprecipitations showed that the USP15 His-Box efficiently bound to GST-fused TRIM25-RING-BB and TRIM25-BB, but not the TRIM25-RING domain (Fig. 1E). Collectively, these results indicate that USP15 binds through its C-terminal His-Box to the B-boxes of TRIM25.
USP15 deubiquitylates TRIM25
TRIM25 undergoes Lys48-linked polyubiquitylation at its C-terminal SPRY domain, as well as monoubiquitylation at Lys117, Lys264, Lys320, and Lys416 (21). We next asked whether USP15 functioned as a DUB by removing monoubiquitin, polyubiquitin, or both from TRIM25. Exogenous expression of wild-type USP15 markedly decreased the polyubiquitylation and monoubiquitylation of TRIM25 compared to that in HEK 293T cells expressing empty plasmid (vector) (Fig. 2A). In contrast, expression of a catalytically inactive mutant of USP15, which contains a point mutation in one of the four cysteine residues that form a zinc finger motif that is essential for polyubiquitin cleavage (USP15 C783A) (24), did not have any effect on the ubiquitylation of TRIM25 (Fig. 2A).
Fig. 2. USP15 deubiquitylates TRIM25.

(A) HEK 293T cells were transfected with empty plasmid or with plasmid encoding TRIM25-FLAG together with plasmids encoding V5-USP15 wild type (WT) or the V5-USP15 C783A mutant. Forty-eight hours later, WCLs were prepared and subjected to immunoprecipitation with α-FLAG antibody and Western blotting analysis with anti-ubiquitin (α-Ub) and α-FLAG antibodies. WCLs were further used for Western blotting with α-V5 and anti-actin (α-actin) antibodies. (B) HEK 293T cells were transfected with control plasmid or with plasmid encoding USP15-Myc. Thirty hours later, cells were mock-treated or were infected with SeV (50 HA U/ml) for 16 hours. Ubiquitylation of endogenous TRIM25 was determined by immunoprecipitation of samples with α-TRIM25 antibody followed by Western blotting analysis with α-Ub antibody. (C and D) HEK 293T cells were transfected with empty plasmid or plasmid encoding TRIM25-FLAG together with (C and D) nontargeting control siRNA (si.C), (C) USP15-specific siRNA (si.USP15), or (D) USP11-specific siRNA (si.USP11). Forty-eight hours later, WCLs were subjected to immunoprecipitation with α-FLAG and Western blotting analysis with α-Ub and α-FLAG. The extent of knockdown of USP15 or USP11 was determined by Western blotting analysis of WCLs with the appropriate antibodies. (E) An in vitro deubiquitylation assay was performed with TRIM25-FLAG and the indicated amounts of purified USP15 protein. Nonubiquitylated and monoubiquitylated forms of TRIM25 were detected by Western blotting analysis with α-FLAG. The input amounts of USP15 protein were determined by Western blotting analysis with α-USP15. Data in all panels are representative of three independent experiments.
We next addressed whether USP15 deubiquitylated endogenous TRIM25 in the context of viral infection. Consistent with a previous study (21), we found that infection of HEK 293T cells with SeV enhanced the ubiquitylation of endogenous TRIM25 compared to that in uninfected cells (Fig. 2B). Furthermore, ectopic expression of USP15 substantially decreased the extent of ubiquitylation of endogenous TRIM25 in virus-infected cells (Fig. 2B). To complement these gain-of-function experiments, we knocked down endogenous USP15 with specific small interfering RNA (siRNA) and examined the effect on TRIM25 ubiquitylation. Depletion of USP15 in HEK 293T and primary NHLF cells markedly enhanced the mono- and polyubiquitylation of exogenous and endogenous TRIM25, respectively, and efficient knockdown of USP15 was confirmed by Western blotting analysis (Fig. 2C and fig. S3). In contrast, silencing of endogenous USP11, a DUB closely related to USP15, did not have any effect on the ubiquitylation of TRIM25 (Fig. 2D). Finally, an in vitro deubiquitylation assay showed that purified USP15 protein efficiently deubiquitylated FLAG-tagged TRIM25 (TRIM25-FLAG), demonstrating a direct enzymatic activity of USP15 toward TRIM25 (Fig. 2E).
USP15 counteracts the LUBAC-dependent ubiquitylation of TRIM25 to stabilize the protein
Upon virus infection, LUBAC, the ubiquitin E3 ligase complex containing HOIL-1L and HOIP, mediates the Lys48-linked polyubiquitylation of TRIM25 (21). We thus asked whether USP15 specifically counteracted the LUBAC-mediated ubiquitylation of TRIM25, which would thereby lead to stabilization of TRIM25. Ectopic expression of USP15 efficiently removed Lys48-linked ubiquitin moieties from TRIM25-FLAG (Fig. 3A). Furthermore, wild-type USP15, but not its catalytically inactive C783A mutant, decreased the polyubiquitylation of TRIM25 that was caused by ectopically expressed HOIL-1L and HOIP (Fig. 3B). In addition, we also detected decreased monoubiquitylation of TRIM25-FLAG in the presence of wild-type USP15, but not the catalytically inactive USP15 C783A mutant (Fig. 3B and fig. S4). Whereas monoubiquitylation of TRIM25 does not detectably affect its stabilization or its signaling ability, Lys48-linked poly-ubiquitylation of TRIM25 by LUBAC leads to its degradation, thereby inhibiting the TRIM25–RIG-I signaling pathway (21). Thus, we next tested whether USP15 affected the stability of TRIM25. To this end, we examined the stability of endogenous TRIM25 in cycloheximide-treated HeLa cells that were transfected with empty plasmid or plasmid expressing USP15 (Fig. 3C and fig. S5). We then assessed the abundance of endogenous TRIM25 protein at various time points by Western blotting analysis with an anti-TRIM25 antibody. TRIM25 protein abundance declined in control-transfected cells starting at 4 hours after treatment with cycloheximide (Fig. 3C and fig. S5). In contrast, endogenous TRIM25 protein abundance did not change in USP15-expressing cells, suggesting that USP15 prevented the degradation of TRIM25. In summary, these results suggest that USP15 removes the LUBAC-mediated Lys48-linked polyubiquitin chains from TRIM25, thereby leading to its stabilization.
Fig. 3. USP15 stabilizes TRIM25 by counteracting its ubiquitylation by LUBAC.

(A) HEK 293T cells were transfected with plasmid encoding TRIM25-FLAG together with empty plasmid or plasmid encoding USP15-V5, as well as a plasmid encoding a HA-tagged ubiquitin (Ub) mutant in which all of the lysines except Lys48 (K48) are mutated (HA-K48only-Ub). Thirty-six hours later, cells were treated with 50 μM MG132 for 6 hours. WCLs were subjected to immunoprecipitation with α-FLAG antibody, and samples were then analyzed by Western blotting with α-HA and α-FLAG antibodies. (B) HEK 293T cells were transfected with plasmid encoding TRIM25-FLAG with or without plasmids encoding HOIP-Myc and HOIL-1L-V5, as well as with empty plasmid or plasmids encoding V5-tagged USP15 WT or the USP15 C783A mutant. Mono- and polyubiquitylation of TRIM25 were determined by immunoprecipitation with α-FLAG antibody and Western blotting analysis with α-Ub antibody. The presence of USP15, HOIL-1L, HOIP, and actin was determined in WCLs by Western blotting analysis with α-V5, α-Myc, and α-actin antibodies. (C) Ectopically expressed USP15 enhances the stability of TRIM25. HeLa cells were transfected with empty plasmid or plasmid encoding Myc-tagged USP15. Forty hours later, cells were treated with 100 μM cycloheximide (CHX) for the indicated times. Total protein amounts in WCLs were determined by bicinchoninic acid (BCA) assay, and equivalent amounts of WCLs were then subjected to SDS–polyacrylamide gel electrophoresis (SDS-PAGE), followed by Western blotting analysis with α-TRIM25, α-Myc, and α-actin antibodies. Data in all panels are representative of at least three independent experiments. Data from the densitometric analysis of three experiments represented by the blots in (B) and (C) are presented in figs. S4 and S5, respectively.
USP15 enhances the TRIM25- and RIG-I–mediated expression of type I IFN
Lys63-linked ubiquitylation of the RIG-I CARDs by TRIM25 is essential for RIG-I signaling and for the production of type I IFNs (16). Thus, we next tested the effect of USP15 on signaling stimulated by RIG-I and TRIM25. Consistent with previous reports (5, 16), we found that ectopic expression of the two N-terminal CARDs of RIG-I fused to mammalian GST [GST–RIG-I(2CARD)] stimulated detectable activation of IFN-β– and NF-κB–dependent promoters, and that this activation was markedly enhanced by exogenous TRIM25 (Fig. 4, A and B). Coexpression of increasing amounts of Myc-tagged USP15 further enhanced the IFN-β and NF-κB promoter activation stimulated by GST–RIG-I(2CARD) and TRIM25 in a dose-dependent manner (Fig. 4, A and B). This effect of USP15 on TRIM25 and RIG-I signaling was largely dependent on the enzymatic activity of USP15 because the catalytically inactive USP15 C783A mutant only marginally enhanced IFN-β promoter activity (Fig. 4C). Moreover, as reported previously (21), exogenous HOIL-1L and HOIP markedly reduced the IFN-β promoter activation stimulated by GST–RIG-I(2CARD) and TRIM25, and their suppressive effect on RIG-I and TRIM25 signaling was partially reversed by USP15 (Fig. 4D).
Fig. 4. USP15 enhances TRIM25- and RIG-I–mediated signaling.

(A and B) HEK 293T cells were transfected with the pGK–β-gal plasmid [which constitutively expresses β-galactosidase (β-gal)] and either (A) IFN-β or (B) NF-κB reporter plasmids, together with plasmids encoding GST–RIG-I(2CARD) and TRIM25-V5 and increasing amounts of plasmid encoding USP15-Myc. Forty-eight hours later, luciferase and β-gal values were determined as described previously (16). Data are means ± SD of duplicate samples and are representative of four independent experiments. (C and D) IFN-β luciferase activity in HEK 293T cells that were transfected with the indicated constructs was determined as described in (A). Data are means ± SD of duplicate samples and are representative of (C) three or (D) two independent experiments. (E) Measurement of IFN-β luciferase activity in HEK 293T cells transfected with plasmid encoding GST–RIG-I 2CARD alone or together with plasmid en-coding TRIM25-FLAG and that stably expressed either nontargeting control shRNA (sh.C) or a USP15-specific shRNA (sh.USP15). Luciferase and β-gal values were determined 48 hours after transfection. Data are means ± SD of duplicate samples and are representative of three independent experiments. (F) Twenty-eight hours after transfection with the indicated siRNAs together with the IFN-β luciferase and pGK–β-gal plasmids, HEK 293T cells were mock-treated or were infected with SeV (5 HA U/ml). Luciferase and β-gal values were determined 20 hours later. Data are means ± SD from three independent experiments. **P < 0.01. (G) HEK 293T cells, stably expressing sh.USP15 or sh.C, were infected with SeV (50 HA U/ml) for 16 hours. The extent of ubiquitylation of endogenous RIG-I was then determined by immunoprecipitation with α–RIG-I antibody and Western blotting analysis with α-Ub antibody. Data are representative of three independent experiments.
We next addressed whether USP15 was required for efficient IFN-β production in response to RIG-I and TRIM25 signaling. To this end, we examined RIG-I(2CARD)– and TRIM25-stimulated IFN-β promoter activation in cells in which endogenous USP15 was stably knocked down with a USP15-specific short hairpin RNA (shRNA, sh.USP15) (Fig. 4E). Cells stably expressing a nontargeting control shRNA (sh.C) served as control. Depletion of endogenous USP15 reduced the extent of IFN-β promoter activation stimulated by GST–RIG-I(2CARD) alone or together with TRIM25 (Fig. 4E). Consistent with this, silencing of USP15 decreased the extent of IFN-β promoter activation triggered by infection with SeV (Fig. 4F). Because TRIM25 activates RIG-I signaling by mediating the Lys63-linked ubiquitylation of the RIG-I CARDs, we examined the abundance of ubiquitylated endogenous RIG-I (Fig. 4G). The extent of ubiquitylation of endogenous RIG-I was markedly reduced in cells in which USP15 was stably knocked down compared to that in cells stably expressing control shRNA (Fig. 4G). Together, these results suggest that USP15 sustains the activation of the TRIM25–RIG-I signaling pathway by stimulating the deubiquitylation of TRIM25 and thereby enhancing its stabilization.
USP15 is required for an effective IFN-mediated antiviral response
USP15 modulates various signal transduction pathways, including the transforming growth factor–β (TGF-β), NF-κB, and Wnt–β-catenin pathways (25–28); however, a role for USP15 in the IFN-mediated antiviral innate immune response has not yet been described. We thus tested the effect of expression of USP15 on the abundances of interferon beta 1 (IFN-β) and interferon-stimulated gene 15 (ISG15) mRNAs in the context of viral infection (Fig. 5A). Infection of HEK 293T cells with SeV, but not mock infection, led to the generation of detectable amounts of IFN-β and ISG15 mRNAs, and exogenously expressed USP15 further increased the amounts of these transcripts (Fig. 5A). Conversely, siRNA-mediated silencing of endogenous USP15 in primary NHLF cells markedly reduced IFN-β protein abundance in response to SeV infection compared to that in infected cells transfected with nontargeting control siRNA, and the inhibitory effect of USP15 knockdown on virus-mediated IFN-β production was dependent on the multiplicity of infection (MOI) (Fig. 5, B and C).
Fig. 5. USP15 is required for the RIG-I– and TRIM25-mediated production of IFN and an effective antiviral response.

(A) HEK 293T cells were transfected with empty plasmid or with plasmid encoding USP15-Myc. Thirty-six hours later, the cells were mock-treated or were infected with SeV (25 HA U/ml). Sixteen hours later, the abundances of IFN-β, ISG15, USP15, and actin mRNAs were determined by reverse transcription polymerase chain reaction (RT-PCR) analysis. (B and C) Primary NHLFs were transfected with si.C or si.USP15. Forty-eight hours later, cells were left uninfected or were infected with SeV [5 HA U/ml (left) or 25 HA U/ml (right)]. The amounts of IFN-β protein secreted by the cells were determined by enzyme-linked immunosorbent assay (ELISA) analysis of culture medium ~17 hours after infection. *P < 0.05. N.D. indicates nondetectable. (C) Knockdown of USP15 was confirmed by Western blotting analysis. (D) Forty-eight hours after transfection with the indicated constructs, HEK 293T cells were infected with VSV-eGFP (at an MOI of 0.01). Twenty-seven hours later, eGFP was detected by fluorescence microscopy, and viral titers were determined by plaque assay. PFU, plaque-forming units. Data are representative of four independent experiments. Western blotting analysis was performed to detect the indicated USP15 proteins. (E) HEK 293T cells, stably expressing sh.C or sh.USP15, were infected with VSV-eGFP (MOI, 10−5). Twenty-four hours later, eGFP-positive cells were visualized by fluorescence microscopy and quantified by flow cytometry. Knockdown of endogenous USP15 was confirmed by Western blotting analysis. **P < 0.01. (F) HEK 293T cells transfected with the indicated siRNAs were infected with NDV-GFP (MOI 0.01). Twenty-seven hours later, GFP-positive cells were determined by flow cytometry. Knockdown of USP15 was confirmed by Western blotting analysis. **P < 0.01. Data in bar graphs in (B), (E), and (F) are means ± SD from three independent experiments. Other data are representative of at least three independent experiments.
To determine the role of USP15 in the host antiviral immune response, we first tested the effect of overexpression of USP15 on the replication of VSV, a negative-strand RNA virus of the Rhabdoviridae family that is sensed by RIG-I. Ectopic expression of wild-type USP15 markedly suppressed the replication of a recombinant VSV expressing an enhanced green fluorescent protein (VSV-eGFP) that enables virus replication in infected cells to be monitored. In contrast, the catalytically inactive USP15 C783A mutant only marginally decreased viral titers compared to those in infected cells transfected with control vector (Fig. 5D and fig. S6A). To complement these gain-of-function experiments, we determined the replication of VSV-eGFP in cells in which USP15 was stably knocked down with specific shRNA (Fig. 5E). Cells in which USP15 was knocked down exhibited substantially enhanced VSV-eGFP infection rates compared to those of cells expressing nontargeting control shRNA (Fig. 5E). Consistent with the effects on VSV replication, HEK 293T and NHLF cells in which endogenous USP15 was knocked down exhibited markedly enhanced replication of the paramyxoviruses Newcastle disease virus (NDV) and SeV, respectively, compared to that in cells transfected with control siRNA (Fig. 5F and fig. S6B). In summary, these results indicate that USP15 promotes innate immune signaling by TRIM25 and RIG-I, which leads to efficient IFN production and thus an effective antiviral response.
DISCUSSION
The TRIM25–RIG-I–MAVS axis is recognized as a key signaling pathway in the immediate host response to infection with RNA viruses. Whereas the rapid production of type I IFN by RIG-I and TRIM25 is required to combat virus infection, effective control mechanisms of antiviral signaling are imperative to prevent excessive production of IFNs and pro-inflammatory cytokines that could potentially cause autoimmune disease. Thus, a thorough understanding of the molecular mechanisms that regulate TRIM25 and RIG-I signaling is necessary for the control of infectious diseases and for developing therapeutic targets to treat autoimmune disorders. Here, we identified the DUB USP15 as a critical regulator of TRIM25 activity in human cells. We found that upon binding to TRIM25, USP15 deubiquitylated TRIM25, which stabilized the protein and thereby led to a sustained RIG-I–dependent antiviral response (Fig. 6).
Fig. 6. Model for the activation of TRIM25 and RIG-I signaling by USP15.
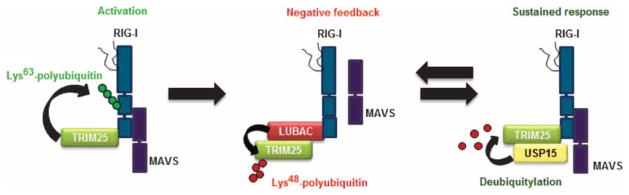
(Left) Upon viral infection, binding of RIG-I to viral RNA induces a conformational change in RIG-I that enables TRIM25 to bind to CARD1 in RIG-I and stimulate the Lys63-linked ubiquitylation of the CARD2, which leads to its subsequent binding to MAVS. (Middle) Later during viral infection, expression of the genes encoding LUBAC is induced. LUBAC then mediates the Lys48-linked ubiquitylation of TRIM25, which leads to its proteasomal degradation. (Right) The action of LUBAC is antagonized by USP15, which removes Lys48-linked polyubiquitin from TRIM25, leading to the stabilization of TRIM25 and a sustained antiviral IFN response.
A series of studies demonstrated an important role of posttranslational modifications in modulating TRIM25–RIG-I signaling, with phosphorylation and ubiquitylation being the most well characterized. In uninfected cells, the RIG-I CARDs are constitutively phosphorylated by protein kinase C–α (PKC-α) and PKC-β, which prevent downstream signaling (29–31). Upon binding of RIG-I to viral RNA, dephosphorylation of the RIG-I CARDs by phosphoprotein phosphatase 1 (PP1) enables the TRIM25-dependent Lys63-linked ubiquitylation of the second CARD of RIG-I, which triggers the binding of RIG-I to MAVS and an effective antiviral response (32). Furthermore, the ubiquitin E3 ligase Riplet mediates the Lys63-linked ubiquitylation of the CTD of RIG-I, which is also important for RIG-I activation (20). Conversely, the ubiquitin E3 ligases RNF125 and LUBAC stimulate Lys48-linked ubiquitylation and subsequent proteasomal degradation of RIG-I and TRIM25, respectively (21, 33). The ubiquitylation of RIG-I and TRIM25 by RNF125 and LUBAC thus provides a negative feedback regulatory mechanism, which is likely required to reduce the abundance of IFN after a successful response to the viral infection.
In contrast to our emerging knowledge about the ubiquitylation of RIG-I and TRIM25, substantially less is known about the role of deubiquitylation for regulating their antiviral activities. Our results support a role for the deubiquitylation of TRIM25 by USP15 for an effective antiviral response. We initially used mass spectrometry analysis to identify USP15 as a binding partner for TRIM25. Through a combination of biochemical and molecular assays, we further found that USP15 removed Lys48-linked ubiquitin moieties from TRIM25, thereby preventing TRIM25 degradation by the proteasome. Moreover, infection studies with viruses of the Paramyxoviridae (SeV and NDV) and Rhabdoviridae families (VSV) demonstrated an antiviral activity of USP15, and this antiviral role was largely dependent on its catalytic activity. Consistent with the suppressive effect of USP15 on viral replication, ectopic expression of USP15 markedly enhanced the TRIM25- and RIG-I–dependent expression of IFN. Crucially, experiments in which USP15 was knocked down demonstrated that USP15 was required for efficient IFN-β production and thus an antiviral response.
Our study also revealed some of the details about how and when during viral infection USP15 modulates TRIM25 activity. Whereas expression of the genes encoding RIG-I and TRIM25 is IFN-inducible, USP15 protein abundance did not change upon virus infection or in response to IFN. Instead, coimmunoprecipitation and confocal microscopy studies indicated that USP15 efficiently bound to TRIM25 in virus-infected cells, whereas, in contrast, a TRIM25-USP15 interaction was barely detectable in un-infected cells. Because USP15 removes ubiquitin moieties from TRIM25, this interaction mode is consistent with previous data showing that TRIM25 is ubiquitylated by LUBAC specifically upon virus infection (21). Together, this suggests a model in which USP15 removes LUBAC-dependent Lys48-linked polyubiquitin chains from the C-terminal SPRY domain of TRIM25 upon virus infection, thereby antagonizing the LUBAC-mediated inhibition of the pathway. In addition, our results showed that USP15 also removed monoubiquitin moieties from TRIM25; however, further analyses are needed to determine the functional role of monoubiquitylation of TRIM25 and its deubiquitylation by USP15.
USP15 belongs to the USP family of DUBs and is structurally closest related to USP4 and USP11, sharing 71 and 60% similarity at the amino acid level, respectively. USP4 removes Lys48-linked ubiquitin chains from RIG-I, leading to its stabilization and thereby to enhanced RIG-I downstream signaling (34). In contrast to USP15, USP4 protein abundance was reduced in HeLa cells and mouse peritoneal macrophages in response to virus infection (34). Furthermore, whereas our results showed that USP15 efficiently interacted with TRIM25 specifically in infected cells, USP4 constitutively interacts with RIG-I (34). Moreover, whereas USP4 binds through its N-terminal DUSP domain to RIG-I, our study showed that the C-terminal His-Box of USP15 interacted with the B-boxes of TRIM25. Although the expression patterns, substrate specificities, and interaction modes of USP15 and USP4 differ, in both cases, their DUB activity promotes RIG-I–mediated innate immune signaling by stabilizing either RIG-I or its upstream regulator TRIM25. Both USP15 and USP4 stimulate TGF-β signaling by deubiquitylating and stabilizing two key signaling molecules in this pathway, TGF-β receptor I (TβRI) and R-SMADs (26, 27), suggesting that these two closely related DUBs act in concert to modulate central signaling processes that are involved in oncogenesis and innate immunity. With respect to the roles for USP15 and USP4 in antiviral signaling, however, much remains to be learned about the kinetics of activation of both DUBs during the course of virus infection, and whether their DUB activity, protein stability, and subcellular localization are subject to additional regulatory mechanisms to efficiently coordinate the antiviral IFN response.
Our study thus adds USP15 to the growing list of DUBs that regulate the innate immune response by RIG-I. Removal of Lys63-linked polyubiquitin chains from RIG-I and TRAF3 by cylindromatosis (CYLD) and deubiquitinating enzyme A (DUBA), respectively, inhibits the RIG-I–dependent activation of IRF3 (35, 36). Furthermore, some viruses encode their own DUBs to evade their detection by RIG-I (37). For example, the Kaposi’s sarcoma–associated herpes virus (KSHV) protein ORF64 inhibits RIG-I signaling by specifically counteracting the TRIM25-dependent Lys63-linked ubiquitylation of RIG-I (38). Furthermore, the arteriviruses and nairo-viruses use their ovarian tumor domain-containing (OTU)–like DUB to inactivate cellular proteins that are involved in RLR-mediated immune signaling (39, 40). In contrast to these host- or virus-encoded DUBs that inhibit RIG-I signaling, our study identifies USP15 as a positive regulator of the TRIM25–RIG-I signaling pathway, which is required for a sustained, IFN-mediated antiviral response. Together, our results unveil a potentially important role for USP15 in fine-tuning TRIM25-dependent antiviral signaling, thus opening new avenues for therapeutic intervention in viral diseases.
MATERIALS AND METHODS
Cell culture and transfection
HEK 293T, HeLa, and NHLF cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 1% penicillin/streptomycin (Gibco-BRL). Transient transfections were performed with Lipofectamine and the Plus reagent (Invitrogen) or with calcium phosphate (Clontech) according to the manufacturers’ instructions. To obtain stable cell lines, HEK 293T cells were transfected with the pEF-IRES-Puro empty plasmid or with pEF-IRES-Puro-USP15-Myc with Lipofectamine and the Plus reagent. Cells were selected by culturing in medium containing puromycin (2 μg/ml).
Plasmids
All constructs for transient and stable expression in mammalian cells were derived from the pEBG-GST fusion plasmid or the pEF-IRES-Puro expression plasmid. Full-length, V5-tagged TRIM25 was described previously (16). The plasmid pEF-TRIM25-FLAG was provided by J. Jung (University of Southern California). The complementary DNAs (cDNAs) encoding GST-TRIM25-RING-BB, GST-TRIM25-RING, and GST-TRIM25-BB were generated by PCR assay from pEF-IRES-TRIM25-V5 as a template, and were subcloned into the pEBG plasmid at the Bam HI and Cla I sites. The cDNA encoding human USP15 (NP_006304.1) was subcloned into the pEF-IRES-Puro plasmid (which encodes a C-terminal V5 or Myc tag) at the Mlu I and Xba I sites. The catalytically inactive mutant USP15 C783A was generated by site-directed mutagenesis from the templates pEF-IRES-USP15-V5 and pEF-IRES-USP15-Myc. Truncation mutants of USP15 (amino acid residues 1 to 130, 125 to 540, and 535 to 952) were generated by PCR from pEF-IRES-USP15-Myc as a template and were subcloned into the pEF-IRES-Myc plasmid between the Mlu I and Xba I sites. All constructs were sequenced to verify that the original sequences were intact. The plasmids pEF-IRES-HOIL-1L-V5 and pcDNA3.1-Myc-HOIP were gifts from J. Jung (University of Southern California) and were described previously (21, 41). The plasmid encoding the hemagglutinin (HA)–tagged Lys48only-ubiquitin mutant was provided by Z. Chen (University of Texas Southwestern Medical Center).
GST pull-downs and mass spectrometry
Procedures for large-scale GST pull-down assays and mass spectrometric analysis were described previously (16, 29). Briefly, 48 hours after transfection with pEBG plasmid or with plasmid encoding GST-TRIM25-RING-BB, HEK 293T cells were collected and lysed with NP-40 buffer [50 mM Hepes (pH 7.4), 150 mM NaCl, 1% (v/v) NP-40, and protease inhibitor cocktail (Sigma)]. After centrifugation, lysates were precleared with Sepharose beads for 1 hour. Lysates were then mixed with a 50% slurry of glutathione-conjugated Sepharose beads (Amersham Biosciences), and the binding reaction was incubated for 4 hours at 4°C. Precipitates were washed extensively with lysis buffer. Proteins bound to glutathione beads were eluted and separated on a NuPAGE 4 to 12% bis-tris gradient gel (Invitrogen). After silver staining, protein bands present specifically in the GST-TRIM25-RING-BB complex, but not with GST alone, were excised and analyzed by ion trap mass spectrometry at the Harvard Taplin Biological Mass Spectrometry Facility.
Knockdown of USP15 with siRNA and shRNA
For transient knockdown of endogenous USP15 in HEK 293T or primary NHLF cells, cells were seeded into 12- or 6-well plates. The next day, cells were transfected with the ON-TARGETplus SMARTpool siRNA specific for USP15 (Thermo Scientific; catalog no. L-006066) with Lipofectamine and the Plus reagent (Invitrogen), according to the manufacturer’s instructions. As a control, cells were transfected with nontargeting control siRNA (Thermo Scientific; catalog no. D-001810-03). A final concentration of 300 nM siRNA per well was used. To generate HEK 293T cells stably depleted of endogenous USP15, cells were transfected with human pSM2 USP15-specific shRNAmir (Open Biosystems) with the Arrest-In Transfection system (Open Biosystems) according to the manufacturer’s instructions. As a control, cells were transfected with pSM2 encoding nontargeting control shRNAmir (Open Biosystems). Transfected cells were then selected in culture medium containing puromycin (2 μg/ml). The efficiency of knockdown of USP15 was determined by Western blotting analysis with an anti-USP15 antibody (1C10; Abnova). Transient knockdown of USP11 was achieved with the ON-TARGETplus SMARTpool siRNA specific for USP11 (Thermo Scientific; catalog no. L-006063).
Coimmunoprecipitations and Western blotting analysis
HEK 293T cells were lysed in NP-40 buffer and then were centrifuged at 15,890g for 20 min. To detect the polyubiquitylation of TRIM25, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer [50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1% (v/v) NP-40, 0.5% deoxycholate, 0.1% SDS, and protease inhibitor cocktail]. For coimmunoprecipitations, 1 ml of postcentrifuged lysates was incubated with 0.5 to 2.5 μg of antibody at 4°C overnight, followed by incubation with a 50% slurry of protein A/G agarose (Amersham) for 2 hours at 4°C. Immunoprecipitated proteins were extensively washed with lysis buffer and were eluted with SDS–Laemmli buffer by boiling for 5 min. For Western blotting analysis, proteins were resolved by SDS-PAGE and were transferred onto polyvinylidene difluoride membranes. The following primary antibodies were used at 1:2000 dilution: anti-V5 (Invitrogen), anti-FLAG (M2; Sigma), anti-Myc (Covance), anti-GST (Sigma), anti-HA (HA-7; Sigma), monoclonal anti-TRIM25 (BD Biosciences), and anti–β-actin (Abcam). The following antibodies were used at 1:500 dilution: anti-USP15 (1C10; Abnova), anti-USP11 [GeneTex (clone EPR4346)], and anti-ubiquitin (P4D1; Santa Cruz Biotechnology). Protein bands were visualized with the enhanced chemiluminescence reagent (Pierce) and were detected by a luminescent imaging system (Fuji LAS-4000).
In vitro deubiquitylation assay
HEK 293T cells were transfected with pEF-TRIM25-FLAG. Forty-eight hours later, TRIM25-FLAG protein was purified from cell lysates with anti-FLAG (M2) agarose (Sigma). Immunoprecipitates were washed extensively with RIPA buffer, which was followed by washing once with NP-40 buffer and phosphate-buffered saline (PBS). An in vitro TRIM25 deubiquitylation reaction was performed at 37°C for 1 hour in deubiquitinase buffer [25 mM tris-HCl (pH 7.5), 150 mM NaCl, 5 mM dithiothreitol] with 0.5 to 5 μg of USP15 protein purified from insect cells (Abcam). The reaction was stopped by adding SDS–Laemmli buffer. Samples were then subjected to SDS-PAGE and Western blotting analysis with anti-FLAG and anti-USP15 antibodies.
Cycloheximide treatment
To determine the stability of endogenous TRIM25 protein, HeLa cells were transfected with either empty plasmid or the pEF-IRES-USP15-Myc plasmid. Forty hours later, cells were treated with 100 μM cycloheximide for the times indicated in the figure legends. Total protein amounts of cell lysates were determined by BCA assay. Lysates with equivalent protein amounts were resolved by SDS-PAGE and analyzed by Western blotting to determine the abundance of endogenous TRIM25 protein.
Luciferase reporter assays
HEK 293T cells seeded into 12-well plates were transfected with 200 ng of either the IFN-β or the NF-κB luciferase construct and 300 ng of the β-gal–expressing pGK–β-gal. In addition, the following amounts of effector plasmids were cotransfected: 1 ng of plasmid encoding GST or GST–RIG-I(2CARD), 200 ng of pEF-IRES-TRIM25-V5 or pEF-TRIM25-FLAG, 15 to 125 ng of pEF-IRES-USP15-Myc or pEF-IRES-USP15-V5, 125 ng of pEF-IRES-USP15 C783A-Myc, 12.5 ng of HOIP-Myc, and 5 ng of HOIL-1L-V5. Forty to 48 hours after transfection, cells were lysed and subjected to a luciferase assay (Promega) according to the manufacturer’s instructions. Luminescence and absorbance were measured, and luciferase values were normalized to β-gal activity to measure transfection efficiency.
Viruses
SeV (Cantell strain) was obtained from Charles River Laboratories. VSV-eGFP and NDV-GFP were provided by S. Whelan (Harvard) and A. García-Sastre (Mount Sinai), respectively.
Measurement of IFN-β by ELISA
NHLFs were seeded into 12-well plates and transfected with 300 nM of either nontargeting control siRNA or USP15-specific siRNA. About 48 hours later, cells were left uninfected or were infected with SeV (5 or 25 HAU/ml) for the times indicated in the figure legends. The supernatants were collected and analyzed for IFN-β production by ELISA (PBL Biomedical Laboratories).
RT-PCR analysis
HEK 293T cells, transfected with the indicated plasmids, were mock-treated or infected with SeV (25 HA U/ml). Sixteen hours after infection, total cellular RNA was extracted with the RNeasy Plus Mini kit (Qiagen). Five micrograms of RNA was used for cDNA synthesis with the SuperScript III First-Strand kit (Invitrogen), which was followed by a PCR assay with primers specific for the genes encoding IFN-β, ISG15, USP15, or actin. Relative amounts of PCR product were determined by agarose gel electrophoresis. To measure SeV RNA in primary NHLF cells, 700 ng of RNA was used for cDNA synthesis, which was followed by a PCR assay with SeV-specific primers [SeV-1068F (5′-GACGCGAGTTATGTGTTTGC-3′) and SeV-1226R (5′-TTCCACGCTCTCTTGGATCT-3′)], which amplify a 159–base pair region in the viral genomic RNA.
Confocal microscopy
HeLa cells, grown on chamber slides, were mock-treated or infected with SeV (80 HA U/ml). Sixteen hours after infection, cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% (v/v) Triton X-100, followed by incubation with 10% goat serum in PBS for 1 hour. For immunostaining USP15, a monoclonal anti-USP15 antibody (Abcam) and an Alexa Fluor 488–conjugated anti-mouse immunoglobulin G (IgG) (Invitrogen) were used. Endogenous TRIM25 was stained with a rabbit anti-TRIM25 antibody (Santa Cruz Biotechnology) and Alexa Fluor 594–conjugated anti-rabbit IgG (Abcam). Laser scanning images were taken on an Olympus IX8I confocal microscope.
VSV replication assays
HEK 293T cells stably transfected with pSM2 encoding nontargeting control shRNAmir or USP15-specific shRNAmir were seeded into 12-well plates. The next day, cells were infected with VSV-eGFP at the MOI indicated in the figure legends. Twenty-four hours later, eGFP was visualized by fluorescence microscopy, and the relative percentages of eGFP-positive cells were determined by flow cytometric analysis. Furthermore, HEK 293T cells were transiently transfected with the indicated plasmids. Forty-eight hours later, the cells were infected with VSV-eGFP at the MOI indicated in the figure legends. Twenty-seven hours after infection, culture media were harvested, and viral titers were determined by standard plaque assay on Vero cells, as described previously (16).
NDV replication assays
HEK 293T cells, seeded into 24-well plates, were transfected with 300 nM of either USP15-specific siRNA or control nontargeting siRNA. Twenty-eight hours after transfection, cells were infected with NDV-GFP at an MOI of 0.01. Twenty-seven hours later, the relative percentages of GFP-positive cells were determined by flow cytometric analysis.
Statistical analysis
Statistical analyses were performed by two-tailed Student’s t test. P < 0.05 was considered statistically significantly different.
Supplementary Material
Fig. S1. Viral infection increases the binding of USP15 to TRIM25.
Fig. S2. USP15 protein abundance is not increased in response to type I IFN or viral infection.
Fig. S3. Depletion of USP15 enhances the ubiquitylation of endogenous TRIM25 in infected primary human lung fibroblasts.
Fig. S4. Quantification of HOIP- and HOIL-1L–mediated monoubiquitylation of TRIM25 in the presence of wild-type USP15 or a catalytically inactive mutant USP15.
Fig. S5. Quantification of TRIM25 protein abundance in the absence or presence of ectopically expressed USP15.
Fig. S6. Quantification of VSV-eGFP replication in cells overexpressing USP15 and of SeV replication in NHLFs in which USP15 is knocked down.
Table S1. Peptides identified in affinity-purified TRIM25-RING-BB complexes analyzed by mass spectrometry.
Acknowledgments
We thank J. Jung, S. Whelan, and A. García-Sastre for providing reagents and R. Tomaino (Taplin Mass Spectrometry Facility of Harvard) for mass spectrometry experiments.
Funding: This work was supported by NIH grants R01 AI087846 and RR000168 to M.U.G. and by a fellowship from the German Research Foundation to E.-K.P. (PA2090/1-1).
Footnotes
Author contributions: E.-K.P. and M.U.G. planned the experiments; E.-K.P., Y.K.C., M.E.D., S.G., M.K.W., and K.F.F. performed the experiments; and E.-K.P. and M.U.G. wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 2.Nakhaei P, Genin P, Civas A, Hiscott J. RIG-I-like receptors: Sensing and responding to RNA virus infection. Semin Immunol. 2009;21:215–222. doi: 10.1016/j.smim.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Hornung V, Ellegast J, Kim S, Brzoóka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 4.Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 5.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 6.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 7.Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol. 2009;83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, García-Sastre A, Katze MG, Gale M., Jr Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 11.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 13.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Paz S, Sun Q, Nakhaei P, Romieu-Mourez R, Goubau D, Julkunen I, Lin R, Hiscott J. Induction of IRF-3 and IRF-7 phosphorylation following activation of the RIG-I pathway. Cell Mol Biol. 2006;52:17–28. [PubMed] [Google Scholar]
- 15.Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458:430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- 16.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 17.Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, Myong S, Ha T, Hopfner KP, Jung JU. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci USA. 2008;105:16743–16748. doi: 10.1073/pnas.0804947105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, García-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-β induction during the early phase of viral infection. J Biol Chem. 2009;284:807–817. doi: 10.1074/jbc.M804259200. [DOI] [PubMed] [Google Scholar]
- 21.Inn KS, Gack MU, Tokunaga F, Shi M, Wong LY, Iwai K, Jung JU. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol Cell. 2011;41:354–365. doi: 10.1016/j.molcel.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baek KH. Conjugation and deconjugation of ubiquitin regulating the destiny of proteins. Exp Mol Med. 2003;35:1–7. doi: 10.1038/emm.2003.1. [DOI] [PubMed] [Google Scholar]
- 23.Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hetfeld BK, Helfrich A, Kapelari B, Scheel H, Hofmann K, Guterman A, Glickman M, Schade R, Kloetzel PM, Dubiel W. The zinc finger of the CSN-associated deubiquitinating enzyme USP15 is essential to rescue the E3 ligase Rbx1. Curr Biol. 2005;15:1217–1221. doi: 10.1016/j.cub.2005.05.059. [DOI] [PubMed] [Google Scholar]
- 25.Schweitzer K, Bozko PM, Dubiel W, Naumann M. CSN controls NF-κB by deubiquitinylation of IκBα. EMBO J. 2007;26:1532–1541. doi: 10.1038/sj.emboj.7601600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eichhorn PJ, Rodón L, Gonzàlez-Juncà A, Dirac A, Gili M, Martínez-Sáez E, Aura C, Barba I, Peg V, Prat A, Cuartas I, Jimenez J, García-Dorado D, Sahuquillo J, Bernards R, Baselga J, Seoane J. USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nat Med. 2012;18:429–435. doi: 10.1038/nm.2619. [DOI] [PubMed] [Google Scholar]
- 27.Inui M, Manfrin A, Mamidi A, Martello G, Morsut L, Soligo S, Enzo E, Moro S, Polo S, Dupont S, Cordenonsi M, Piccolo S. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat Cell Biol. 2011;13:1368–1375. doi: 10.1038/ncb2346. [DOI] [PubMed] [Google Scholar]
- 28.Huang X, Langelotz C, Hetfeld-Pechoc BK, Schwenk W, Dubiel W. The COP9 signalosome mediates β-catenin degradation by deneddylation and blocks adenomatous polyposis coli destruction via USP15. J Mol Biol. 2009;391:691–702. doi: 10.1016/j.jmb.2009.06.066. [DOI] [PubMed] [Google Scholar]
- 29.Gack MU, Nistal-Villán E, Inn KS, García-Sastre A, Jung JU. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J Virol. 2010;84:3220–3229. doi: 10.1128/JVI.02241-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nistal-Villán E, Gack MU, Martínez-Delgado G, Maharaj NP, Inn KS, Yang H, Wang R, Aggarwal AK, Jung JU, García-Sastre A. Negative role of RIG-I serine 8 phosphorylation in the regulation of interferon-β production. J Biol Chem. 2010;285:20252–20261. doi: 10.1074/jbc.M109.089912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maharaj NP, Wies E, Stoll A, Gack MU. Conventional protein kinase C-α (PKC-α) and PKC-β negatively regulate RIG-I antiviral signal transduction. J Virol. 2012;86:1358–1371. doi: 10.1128/JVI.06543-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, Gack MU. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity. 2013;38:437–449. doi: 10.1016/j.immuni.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA. 2007;104:7500–7505. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Zhao W, Zhang M, Wang P, Zhao K, Zhao X, Yang S, Gao C. USP4 positively regulates RIG-I-mediated antiviral response through deubiquitination and stabilization of RIG-I. J Virol. 2013;87:4507–4515. doi: 10.1128/JVI.00031-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friedman CS, O’Donnell MA, Legarda-Addison D, Ng A, Cárdenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, Xavier R, Ting AT. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008;9:930–936. doi: 10.1038/embor.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kayagaki N, Phung Q, Chan S, Chaudhari R, Quan C, O’Rourke KM, Eby M, Pietras E, Cheng G, Bazan JF, Zhang Z, Arnott D, Dixit VM. DUBA: A deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 37.Randow F, Lehner PJ. Viral avoidance and exploitation of the ubiquitin system. Nat Cell Biol. 2009;11:527–534. doi: 10.1038/ncb0509-527. [DOI] [PubMed] [Google Scholar]
- 38.Inn KS, Lee SH, Rathbun JY, Wong LY, Toth Z, Machida K, Ou JH, Jung JU. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J Virol. 2011;85:10899–10904. doi: 10.1128/JVI.00690-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frias-Staheli N, Giannakopoulos NV, Kikkert M, Taylor SL, Bridgen A, Paragas J, Richt JA, Rowland RR, Schmaljohn CS, Lenschow DJ, Snijder EJ, García-Sastre A, Virgin HW., IV Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe. 2007;2:404–416. doi: 10.1016/j.chom.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Kasteren PB, Beugeling C, Ninaber DK, Frias-Staheli N, van Boheemen S, García-Sastre A, Snijder EJ, Kikkert M. Arterivirus and nairovirus ovarian tumor domain-containing deubiquitinases target activated RIG-I to control innate immune signaling. J Virol. 2012;86:773–785. doi: 10.1128/JVI.06277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, Sano S, Tokunaga F, Tanaka K, Iwai K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006;25:4877–4887. doi: 10.1038/sj.emboj.7601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Viral infection increases the binding of USP15 to TRIM25.
Fig. S2. USP15 protein abundance is not increased in response to type I IFN or viral infection.
Fig. S3. Depletion of USP15 enhances the ubiquitylation of endogenous TRIM25 in infected primary human lung fibroblasts.
Fig. S4. Quantification of HOIP- and HOIL-1L–mediated monoubiquitylation of TRIM25 in the presence of wild-type USP15 or a catalytically inactive mutant USP15.
Fig. S5. Quantification of TRIM25 protein abundance in the absence or presence of ectopically expressed USP15.
Fig. S6. Quantification of VSV-eGFP replication in cells overexpressing USP15 and of SeV replication in NHLFs in which USP15 is knocked down.
Table S1. Peptides identified in affinity-purified TRIM25-RING-BB complexes analyzed by mass spectrometry.