

Microbial communities within the human microbiome
Microbial communities associated with the human body are complex ecological systems governed by many of the same processes that shape plant and animal communities. Because the composition of the human-associated microbiome is critical for normal immune development and health [1, 2], there is a pressing need to develop robust models of microbiome structure and function grounded in ecological theory [3, 4]. Therapies such as fecal transplants have tremendous potential to alter the microbiome to improve health [5], but these are currently blunt instruments that modify community structure in an untargeted manner. In order to understand and modulate the structure of the human microbiome – and of other microbial communities – we must better characterize the fundamental ecological processes that underlie microbiome assembly. That is, what properties (organisms, metabolic functions, regulatory circuits, etc.) must be present in ‘normal’ host-associated microbial communities, and how are they established and maintained?
This question was one of the early motivating factors for studies such as the Human Microbiome Project (HMP) [6], which hypothesized that essential microbial species might be distributed ‘universally’ among healthy human beings [7, 8]. Such a ‘core’ microbiome, accompanied by some level of inter-individual variation, would be analogous to the core and pan genomes present in many microbial species [9]. A (near-)universally present, minimal set of microbial species in healthy individuals would have provided a very clear target for microbiological characterization and identification of biomarkers to distinguish between healthy and dysbiotic individuals. However, early results showed that inter-individual variability even among closely related individuals precluded the existence of a meaningful core microbiome in terms of microbial species [7, 10]. Although the same few phyla are typically present within each body site niche of the human microbiome, their relative abundances can vary by 10-fold or more among individuals, and it is not uncommon for even closely matched individuals to share 50% or less of their microbial genera [7]. This result suggested that if a core microbiome exists, it is not defined by the presence of specific microbial taxa.
An alternate hypothesis is that the core human microbiome is defined by a balance of metabolic and other functional capabilities within each body site or microhabitat. That is, the core microbiome maintained in healthy humans may consist of common gene families and pathways rather than common organisms. Theoretical work in metazoan ecology suggests that if selection within each niche (see Glossary) acts primarily on functions that are not phylogenetically unique, the result over evolutionary time is the accumulation of sets of functionally equivalent species [11]. Selection on function rather than phylogenetic identity within niches is plausible in microbial populations given high rates of lateral gene transfer, which means that not all functional traits are stably associated with particular phylogenetic markers [12]. If this was the case, a ‘functional’ core microbiome would act at the gene and pathway level somewhat like the human genome does for small polymorphisms, with less abundant (but highly phenotypically significant) variation occurring outside of the core and basic, common processes maintained within it. Identifying a functional core for the microbiome and other microbial communities has the potential to identify metabolic pathways critical for each environment that are maintained despite stochastic variation in the presence or absence of particular taxa. These core pathways may provide new potential targets for molecular therapies and community engineering.
Sequencing-based techniques for investigating microbial communities are currently quite cost-effective, and consist at a high level of (i) targeted amplicon-based techniques (e.g. 16S rRNA gene sequencing) and (ii) shotgun metagenomic or metatranscriptomic approaches, both reviewed in depth elsewhere [13–15]. Both can be applied to the human microbiome and other communities to study different microbial features (i.e. phylogenetic vs. genomic composition) with various tradeoffs with respect to cost, efficiency, and data quality and biases. For example, primers targeting conserved regions of 16S rRNA can lead to underrepresentation of clades with variation at the primer site. For example, the commonly used 967F primer can underrepresent clades such as the Bacteroidetes [16], 357F/926R (included in HMP protocols [17]) will deplete Bifidobacterium [18], and 515F/806R can poorly amplify Propionibacterium, one of the most common skin microorganisms [13]. Conversely shotgun metagenomics can be heavily influenced by human genomic ‘contamination’ in environments with a high eukaryotic cell fraction, e.g. saliva [19]. However, used in tandem, current technologies can provide a rich picture of microbial community phylogenetic and functional structure in order to determine how these two features interact to assemble a core human microbiome. In this review, we discuss the functional organization of human-associated microbial communities and the underlying ecological processes that affect it to drive microbiome assembly.
Assembly of habitat-specific microbial communities
To understand the processes involved in the generation and maintenance of a core human microbiome, it is important to identify the key ecological processes underlying patterns in microbial distributions. Microbial communities are shaped by a combination of selective and stochastic forces qualitatively similar to, but quantitatively differing from, those in metazoan systems: niche-based selection, dispersal, ecological drift, and mutation [11]. The balance between these forces varies over space and time and can be difficult to determine based on species abundance distributions alone, particularly when diversity is high [20]. Indeed, most studies on the roles of selection and stochasticity on microbial community assembly based on such observations suggest that both factors play significant roles in structuring communities [21, 22]. This is due in part to the large differences in temporal and spatial scales relative to metazoan communities, and it requires microbial ecology to account, for example, for more stochasticity than may be apparent in larger-scale systems.
The neutral theory of community assembly has thus been particularly successful in explaining microbial ecologies [23], although interestingly it was originally developed to explain tropical trees sharing niches (slow-growing shade-tolerant and fast-growing sun-tolerant species) [24]. In its classical form it posits that that steady-state species abundance distributions are based only on ecological drift, dispersal, and the diversity of organisms available for colonization (referred to as the metacommunity). Since it assumes that species are effectively functionally equivalent, it can act as a null model in contrast to selective pressure or niche specialization. However, when combined with a model of specialization and extended to evolutionary time, neutral theory suggests that sets of functionally equivalent species can evolve within the niches that are most prevalent [25]. That is, the dynamics within each niche remain neutral, but selection occurs to set the boundaries of each species set. The necessary condition for this process to occur is the absence of factors that promote competitive exclusion between functionally similar species. This pattern of sets of functionally similar species (‘emergent neutrality’) can appear via several different possible pathways [26, 27] and is more likely to occur in species-rich communities [28]. Finally, these models typically only describe communities at steady-state, such that species sets can also follow each other in successional processes; for example, any of a set of sun-tolerant, fast-growing trees could colonize a vacant spot in a forest followed by any of a set of slow-growing, shade tolerant trees.
Microbial communities, particularly those associated with the dynamic environment of a host, are typically a good fit to such a model. They are often quite diverse, with a relatively small number of high-abundance members coupled with a long tail of low abundance members [16]. High rates of genetic exchange between spatially and phylogenetically associated microbes provide a mechanism by which niche-determining genes can be decoupled from phylogenetic lineages, decreasing the stability of competition between related microbes. Even dispersal is thought to be particularly widespread for microbial populations [29], allowing (among other consequences) inferior competitors to escape from direct competition. Combined, these factors help to explain why a number of empirical studies have found that microbial communities with different taxonomic compositions appear to be functionally similar, ranging from different wastewater treatment reactors [30] to different individual human hosts [6].
Some factors contributing to this behavior in host-associated communities operate primarily through colonization. The microbiome of each individual host is effectively a local community connected to other, similar communities through dispersal. The collective set of microbes shared in this fashion is called a metacommunity, and there is a rich ecological theory describing its dynamics [4]. Metacommunity theory describes the linked dynamics of transitory or more permanent habitat patches that are connected through dispersal of individuals from multiple species [31]. Patch composition can be governed by local selection, dispersal rates, ecological drift, and neutral processes [11]. Each individual host can be viewed as a patch, or discrete area of habitat suitable for microbial colonization. The rate of dispersal between habitats can have a strong influence on community assembly. If dispersal rates are low, then chance colonization by particular microbes (random sampling) can play a large role in initial assembly. Following colonization, dynamics may be determined primarily by environmental selection (stochastic niche theory [32]) or by neutral processes involving immigration of new microbes from both the reservoir and other host individuals.
Other factors in host-associated community assembly operate dynamically after community assembly, although their properties are in turn dictated by long-term co-evolution. This contrasts community kinetics, or the effects of any recent perturbations on community structure, with thermodynamics, or community steady state as dictated by the frequency, extent, and type of perturbations that have occurred over its evolutionary history. Major disruptions in metazoan ecologies are rare and occur only over time scales that are long relative to reproduction, e.g. forest fires or climate change [33, 34]. Similarly, free-living microbial ecologies are often relatively protected from perturbation, whereas differences in assembly strategies for host-associated microbial communities may be explained in part by frequent perturbations such as nutrient availability (diet), washing or antibiotics [35–42]. Thus host-associated communities may be more likely to be away from steady state when observed, and they have co-evolved a robustness to both short- and long-term perturbations in host environment resulting in assembly strategies distinct from those typical for metazoan systems [43].
The combination of deterministic, stochastic, and temporal drivers of ecological assembly shapes any microbial community composition, although how these selection pressures affect host-associated communities in particular (i.e., what specific features of a microbial community result from which specific selection pressures) is still unknown. Functional equivalence between microbes within particular environment could plausibly arise from a combination of both dispersal and selection, if sets of species have adapted to fill particular niches or microhabitats within the host body that have been common over evolutionary time. Overall, both ecological theory and observations in diverse microbial systems [6, 7, 44, 45] support the concept that different organisms can fill comparable functional niches in the microbiome, and it remains to be determined how niches are defined and the kinds of selection pressures operating within them.
Community assembly by functional traits in the human microbiome
The human microbiome is a host-associated microbial ecology of particular interest and consists broadly of the microbial communities that reside in and on the human body: primarily in the gut, but also in the oral cavity, skin, urogenital tract, and almost every other body region. Understanding the principles underlying its assembly is thus especially important, since the role these resident microbiota in human health is vast. Intestinal microbiota salvage energy from carbohydrates and other sources, synthesize vitamins B and K, metabolize bile acids, other sterols and xenobiotics, aid in the development of mucosal immunity, and prevent the overgrowth of harmful bacteria [2, 46]. Across other body sites, vaginal microbes are key in hydrogen peroxide production and pH maintenance to maintain a homeostatic community [47], and on the skin commensal microbes can prevent colonization by potentially pathogenic species [48]. While the culture-independent techniques discussed above have provided extensive ‘parts lists’ of typical microbial residents and metagenomic contents found within and among individuals, our understanding of why and how particular human-associated microbial assemblages come to be remains limited.
One of the early suggestions that the core human microbiome might consist of functional traits arose from the first metagenomic surveys of the gut microbiome, which identified genes in the microbial species associated with the metabolic functions in the gut, paving the way for further functional attributes to be explored in other body sites [49]. The hypothesis was prominently highlighted within the cohort of Turnbaugh et al. in 2009 [7] and finally confirmed on a large scale by the HMP [6], an analysis of the microbial communities of 18 body sites of 300 healthy individuals. By the time of the HMP, both culture-based [50–52] and culture-independent [53–55] studies had shown great diversity of microbial residents across individuals at essentially every body site. The HMP’s large population confirmed that no easily-identifiable phylogenetic core existed across individuals, in the sense of universally carried microbial taxa present across individuals or body sites, although each body site habitat does of course comprise a highly phylogenetically distinct microbial repertoire enriched for characteristic taxa [6, 53, 56]. This result has been echoed by other large projects such as MetaHIT [57], and it is of note that in each such study of the human-associated microbial metagenome, at least two-thirds of the surveyed gene families have had no or minimal assignable molecular function [6, 57].
The characterized components of the functional core observed to date in the human microbiome can be decomposed into at least three components: processes universal to microbial life that are not unique to the human microbiome, pathways that are present across many or all body sites, and core functions within each body site habitat (Figure 1). The first is well-understood from decades of microbiology and includes housekeeping processes such as translation, the cell cycle, and structural components of the cell [58]. Human-associated microbial functions enriched across body sites include biosynthesis of compounds used by a human host, such as essential amino acids (histidine, tyrosine, phenylalanine, methionine, arginine, and lysine); vitamins such as biotin and thiamine; and other families of bioactives such as isoprenoids. Additionally, microbial uptake of several compounds was enriched in the microbiome, including metals (nickel and cobalt), polyamines (spermidine and putrescine) and production of lipopolysaccharide (LPS). Finally, each body site investigated by the HMP also possessed its own enriched microbe-associated functional cores, such as (i) degradation of glycosaminoglycans and uronic acid metabolism in the gut, (ii) fungal transcription and translation on the skin, (iii) arginine transport and methionine biosynthesis in the oral cavity, and (iv) transport of phosphate, glutamate and aspartate in the vagina [59]. Within each of these environments, groups of functionally similar microbes associate with enriched metabolic pathways, sometimes within but often spanning phylogenetic groups (Figure 2). As our understanding of uncharacterized microbial genes and pathways in the human microbiome improves, the lists of typical microbial processes within each habitat will undoubtedly expand as well.
Figure 1. Core microbial processes within and across body sites of the human microbiome.
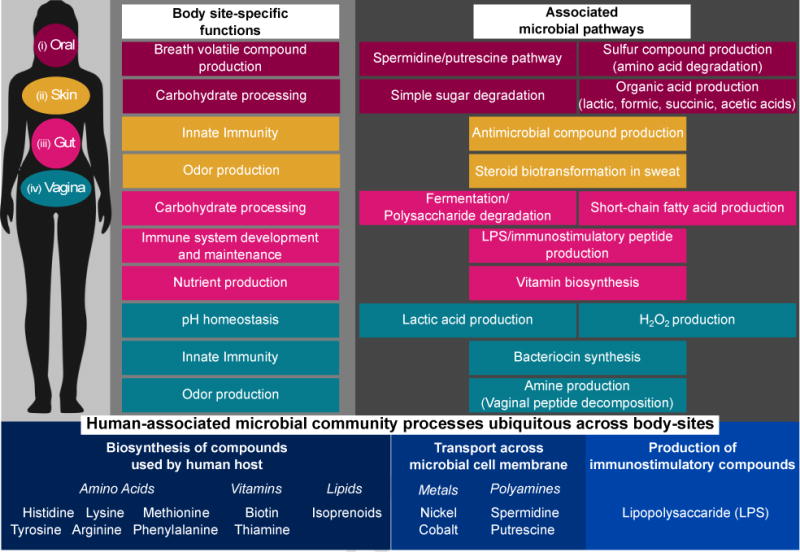
Host-associated microbial communities, particularly those of the human microbiome, often appear to assemble based on collections of functional traits unique to particular niches. Examples shown here include (i) the oral cavity: breath volatile compound production through spermidine/putrescine [59] and sulfur compound pathways (e.g. hydrogen disulfide and methyl mercaptan) [62, 63], as well as energy harvest by amino acid and simple sugar degradation [64]; (ii) skin: release of antimicrobial compounds [65] and odor production by transforming steroids in the sweat [62]; (iii) gut: fermentation and degradation of polysaccharides, producing short-chain fatty acids [6, 57], nutrient (e.g. vitamin) production [6, 57], and immunostimulation [66, 67]; and (iv) vagina: pH maintenance through lactic acid production from glycogen degradation and hydrogen peroxide production [62, 68], bacteriocin synthesis, and odiferous amine production [47, 62]. In addition to niche-specific microbial activities, other host-associated microbial pathways are constituently present across body sites and not typical of non-host-associated microbial (meta)genomes [59], including biosynthesis of host-essential amino acids, cofactors and lipids, transport of metals and polyamines, and production of immunostimulatory compounds [6].
Figure 2. Associations between groups of microbial species and enriched metabolic pathways in the human microbiome.

Significant Spearman correlations between microbial species in three representative body habitats and the abundances of metabolic pathways in their metagenomes. Data shown are from the Human Microbiome Project [6] for the (A) posterior fornix within the vagina, (B) tongue dorsum, and (C) gut. In each case one or more microbial species, often phylogenetically diverse, associate with the metabolic pathways enriched in each body site.
Moving ahead: limitations and future research paradigms
The extent to which the phylogeny of human-associated microbes is linked to their functional roles remains to be determined (Box 1). Regardless of its degree, however, any functional core of the human microbial communities will include molecular processes key to microbial survival in the unique environment of our bodies, to interactions with innate and adaptive immunity, and to our own health. While the core- and pan-genomes across microbial isolates have begun to be well-characterized, the core- and pan-metagenomes found across humans (or other environments) have not. Defining a core microbe-derived metagenome for human health is of particular importance for translational applications, as this will provide a new pool of potentially targetable gene products for therapeutics. Conversely, individual variation in the human pan-metagenome will figure prominently in personalized medicine, as has already been observed for microbially-linked pharmacokinetics [60, 61]. Finally, newer techniques such as metabolomics will enable deeper characterization of the molecular mechanisms and pathways underlying these ecological assembly strategies and their associated host and microbial phenotypes. Just as molecular systems biology has provided new insights into individual organisms’ function over the past decade, a comparable ‘molecular systems ecology’ has the potential to do the same for microbial communities over the coming years.
Box 1. Outstanding questions.
What are the key functional genes and pathways found in the core and pan-metagenomes associated with the human body?
Do these change according to environmental or phenotypic conditions such as location, early life exposures, disease, or diet?
How important is genetic variation between closely related microbes to microbiome community function?
Conversely, how important are taxonomic delineations such as species or genera in defining human-associated microbial function?
Under what conditions are different functional units useful to study in human-associated and other microbial communities, e.g. genes, pathways, strains, taxonomic clades, or phylogenetic clusters?
What is the extent to which the phylogeny of human-associated microbes is linked to their functional roles?
What is the role of horizontal gene transfer in function of the human microbiome?
What is the contribution of low-abundance taxa (the ‘rare biosphere’) to microbiome function?
To what degree are different molecular mechanisms used to implement the same ecological strategies or produce similar phenotypes in microbial communities?
What are the molecular mechanisms and pathways that underlie microbial ecological assembly strategies and their associated host and microbial phenotypes?
Ecological processes influence microbial assembly in human-associated communities.
Microbial functions are core across the human microbiome and specific to each niche.
Technologies/analyses are needed to better define functional traits in microbial communities.
Acknowledgments
We thank Xochitl Morgan, Timothy Tickle and Eric Franzosa for providing helpful feedback and discussion regarding the content of the review. This work was supported in part by NIH R01HG005969, NSF DBI-1053486, and ARO W911NF-11-1-0473RKW to CH.
Glossary
- Ecological drift or stochastic forces
change in population structure over time due to random factors, i.e. neutral evolution not due to selective pressure
- Functional ecology
the study of organisms within one or more habitats based on their functional traits (biochemical capabilities or larger-scale phenotypes) and the effects these have on selective pressures and community steady states
- Metacommunitiy
collection of populations that contribute to a particular ecology, typically connected by genetic or organismal flow of varying rates. For example, the metacommunity of a particular individual’s skin microbiome might include other individuals’ skin (from which organismal flow might vary depending on contact) and other body sites within that host (from which organismal flow might vary depending on similarity of the habitat)
- Niche-based selection
selective pressure based specifically on occupation of a particular habitat, such that organisms with appropriate functional roles ultimately occupy that niche over time
- Selective pressure
processes that result in natural selection operating on a population, changing the composition of genetic material and/or of clades in the community. Examples include nutrient availability, diet, or climate
- Steady-state
composition of features (organisms, genetic material, or compounds) within a population that does not change (or changes only slightly) over time
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol. 2013;14:676–684. doi: 10.1038/ni.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kamada N, et al. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013;14:685–690. doi: 10.1038/ni.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dethlefsen L, et al. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449:811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costello EK, et al. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336:1255–1262. doi: 10.1126/science.1224203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borody TJ, Khoruts A. Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol. 2012;9:88–96. doi: 10.1038/nrgastro.2011.244. [DOI] [PubMed] [Google Scholar]
- 6.Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turnbaugh PJ, et al. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medini D, et al. The microbial pan-genome. Curr Opin Genet Dev. 2005;15:589–594. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Turnbaugh PJ, et al. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci U S A. 2010;107:7503–7508. doi: 10.1073/pnas.1002355107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vellend M. Conceptual synthesis in community ecology. Q Rev Biol. 2010;85:183–206. doi: 10.1086/652373. [DOI] [PubMed] [Google Scholar]
- 12.Wilmes P, et al. The dynamic genetic repertoire of microbial communities. FEMS Microbiol Rev. 2009;33:109–132. doi: 10.1111/j.1574-6976.2008.00144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuczynski J, et al. Experimental and analytical tools for studying the human microbiome. Nat Rev Genet. 2012;13:47–58. doi: 10.1038/nrg3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segata N, et al. Computational meta’omics for microbial community studies. Mol Syst Biol. 2013;9:666. doi: 10.1038/msb.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bornigen D, et al. Functional profiling of the gut microbiome in disease-associated inflammation. Genome Med. 2013;5:65. doi: 10.1186/gm469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sogin ML, et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci U S A. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peterson J, et al. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sim K, et al. Improved detection of bifidobacteria with optimised 16S rRNA-gene based pyrosequencing. PLoS One. 2012;7:e32543. doi: 10.1371/journal.pone.0032543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chisholm RA, Pacala SW. Niche and neutral models predict asymptotically equivalent species abundance distributions in high-diversity ecological communities. Proc Natl Acad Sci U S A. 2010;107:15821–15825. doi: 10.1073/pnas.1009387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dumbrell AJ, et al. Relative roles of niche and neutral processes in structuring a soil microbial community. Isme J. 2010;4:337–345. doi: 10.1038/ismej.2009.122. [DOI] [PubMed] [Google Scholar]
- 22.Jeraldo P, et al. Quantification of the relative roles of niche and neutral processes in structuring gastrointestinal microbiomes. Proc Natl Acad Sci U S A. 2012;109:9692–9698. doi: 10.1073/pnas.1206721109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sloan WT, et al. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ Microbiol. 2006;8:732–740. doi: 10.1111/j.1462-2920.2005.00956.x. [DOI] [PubMed] [Google Scholar]
- 24.P HS. The Unified Neutral Theory of Biodiversity and Biogeography. Princeton Press; 2001. [Google Scholar]
- 25.Hubbell SP. Neutral theory in community ecology and the hypothesis of functional equivalence. Functional Ecology. 2005;19:166–172. [Google Scholar]
- 26.Scheffer M, van Nes EH. Self-organized similarity, the evolutionary emergence of groups of similar species. Proc Natl Acad Sci U S A. 2006;103:6230–6235. doi: 10.1073/pnas.0508024103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gravel D, et al. Reconciling niche and neutrality: the continuum hypothesis. Ecol Lett. 2006;9:399–409. doi: 10.1111/j.1461-0248.2006.00884.x. [DOI] [PubMed] [Google Scholar]
- 28.Holt RD. Emergent neutrality. Trends Ecol Evol. 2006;21:531–533. doi: 10.1016/j.tree.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Martiny JB, et al. Drivers of bacterial beta-diversity depend on spatial scale. Proc Natl Acad Sci U S A. 2011;108:7850–7854. doi: 10.1073/pnas.1016308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ofiteru ID, et al. Combined niche and neutral effects in a microbial wastewater treatment community. Proc Natl Acad Sci U S A. 2010;107:15345–15350. doi: 10.1073/pnas.1000604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leibold MA, et al. The metacommunity concept: a framework for multi-scale community ecology. Ecol Lett. 2004;7:601–613. [Google Scholar]
- 32.Tilman D. Niche tradeoffs, neutrality, and community structure: a stochastic theory of resource competition, invasion, and community assembly. Proc Natl Acad Sci U S A. 2004;101:10854–10861. doi: 10.1073/pnas.0403458101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balanya J, et al. Global genetic change tracks global climate warming in Drosophila subobscura. Science. 2006;313:1773–1775. doi: 10.1126/science.1131002. [DOI] [PubMed] [Google Scholar]
- 34.Stenseth NC, et al. Ecological effects of climate fluctuations. Science. 2002;297:1292–1296. doi: 10.1126/science.1071281. [DOI] [PubMed] [Google Scholar]
- 35.Simon GL, Gorbach SL. The human intestinal microflora. Dig Dis Sci. 1986;31:147S–162S. doi: 10.1007/BF01295996. [DOI] [PubMed] [Google Scholar]
- 36.Dethlefsen L, et al. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS biology. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jernberg C, et al. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. Isme J. 2007;1:56–66. doi: 10.1038/ismej.2007.3. [DOI] [PubMed] [Google Scholar]
- 39.Antonopoulos DA, et al. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect Immun. 2009;77:2367–2375. doi: 10.1128/IAI.01520-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koenig JE, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fierer N, et al. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A. 2008;105:17994–17999. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9:244–253. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Relman DA. The human microbiome: ecosystem resilience and health. Nutrition Review. 70:S2–S9. doi: 10.1111/j.1753-4887.2012.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan L, et al. Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc Natl Acad Sci U S A. 2012;109:E1878–1887. doi: 10.1073/pnas.1203287109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burke C, et al. Bacterial community assembly based on functional genes rather than species. Proc Natl Acad Sci U S A. 2011;108:14288–14293. doi: 10.1073/pnas.1101591108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flint HJ, et al. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9:577–589. doi: 10.1038/nrgastro.2012.156. [DOI] [PubMed] [Google Scholar]
- 47.Hickey RJ, et al. Understanding vaginal microbiome complexity from an ecological perspective. Transl Res. 2012;160:267–282. doi: 10.1016/j.trsl.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kong HH. Skin microbiome: genomics-based insights into the diversity and role of skin microbes. Trends Mol Med. 2011;17:320–328. doi: 10.1016/j.molmed.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gill SR, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hardy PH, Jr, et al. Use of a Bacterial Culture Filtrate as an Aid to the Isolation and Growth of Anerobic Spirochetes. J Bacteriol. 1964;87:1521–1525. doi: 10.1128/jb.87.6.1521-1525.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mata LJ, et al. Fecal microflora in health persons in a preindustrial region. Appl Microbiol. 1969;17:596–602. doi: 10.1128/am.17.4.596-602.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sweet RL. Anaerobic infections of the female genital tract. Am J Obstet Gynecol. 1975;122:891–901. doi: 10.1016/0002-9378(75)90736-x. [DOI] [PubMed] [Google Scholar]
- 53.Costello EK, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bik EM. Composition and function of the human-associated microbiota. Nutr Rev. 2009;67(Suppl 2):S164–171. doi: 10.1111/j.1753-4887.2009.00237.x. [DOI] [PubMed] [Google Scholar]
- 55.Paster BJ, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001;183:3770–3783. doi: 10.1128/JB.183.12.3770-3783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grice EA. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ciccarelli FD, et al. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- 59.Abubucker S, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS computational biology. 2012;8:e1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haiser HJ, et al. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science. 2013;341:295–298. doi: 10.1126/science.1235872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clayton TA, et al. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci U S A. 2009;106:14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thorn RM, Greenman J. Microbial volatile compounds in health and disease conditions. J Breath Res. 2012;6:024001. doi: 10.1088/1752-7155/6/2/024001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Washio J, et al. Hydrogen sulfide-producing bacteria in tongue biofilm and their relationship with oral malodour. J Med Microbiol. 2005;54:889–895. doi: 10.1099/jmm.0.46118-0. [DOI] [PubMed] [Google Scholar]
- 64.Shah HN, Gharbia SE. Ecological events in subgingival dental plaque with reference to Bacteroides and Fusobacterium species. Infection. 1989;17:264–268. doi: 10.1007/BF01639537. [DOI] [PubMed] [Google Scholar]
- 65.Grice EA, Segre JA. Interaction of the microbiome with the innate immune response in chronic wounds. Adv Exp Med Biol. 2012;946:55–68. doi: 10.1007/978-1-4614-0106-3_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kau AL, et al. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cebra JJ. Influences of microbiota on intestinal immune system development. Am J Clin Nutr. 1999;69:1046S–1051S. doi: 10.1093/ajcn/69.5.1046s. [DOI] [PubMed] [Google Scholar]
- 68.Aroutcheva A, et al. Defense factors of vaginal lactobacilli. Am J Obstet Gynecol. 2001;185:375–379. doi: 10.1067/mob.2001.115867. [DOI] [PubMed] [Google Scholar]