

Abstract
Glutamate-triggered signal transduction is thought to contribute widely to cancer pathogenesis. In melanoma, over-expression of the metabotropic glutamate receptor GRM1 occurs frequently and its ectopic expression in melanocytes is sufficient for neoplastic transformation. Clinical evaluation of the GRM1 signaling inhibitor riluzole in patients with advanced melanoma has demonstrated tumor regressions that are associated with a suppression of the MAPK and PI3K/AKT pathways. Together, these results prompted us to investigate the downstream consequences of GRM1 signaling and its disruption in more detail. We found that melanoma cells with enhanced GRM1 expression generated larger tumors in vivo marked by more abundant blood vessels. Media conditioned by these cells in vitro contained relatively higher concentrations of IL-8 and VEGF, due to GRM1-mediated activation of the AKT-mTOR-HIF1 pathway. In clinical specimens from patients receiving riluzole, we confirmed an inhibition of MAPK and PI3K/AKT activation in post-treatment as compared to pre-treatment tumor specimens, which exhibited a decreased density of blood vessels. Together, our results demonstrate that GRM1 activation triggers pro-angiogenic signaling in melanoma, offering a mechanistic rationale to design treatment strategies for the most suitable combinatorial use of GRM1 inhibitors in patients.
Keywords: melanoma, angiogenesis, glutamatergic signaling, glutamate, melanomagenesis
Introduction
Melanoma is the most aggressive form of skin cancer and its incidence is on the rise worldwide (1). Although surgically curable at early stages, metastatic melanoma is relatively refractory to current therapies and has a poor prognosis (2). Cytotoxic chemotherapeutic agents such as dacarbazine have response rates of only 7 to 12% with few long-term survivors. Agents, such as vemurafenib, have shown promising early results (3), but the majority of patients who initially respond eventually progress and die (4). Combinations of Raf-inhibitors with other small molecule inhibitors have shown some improvement over single-agent therapy (5) and confirmatory trials are ongoing. Newer immunotherapies, such as anti-CTLA4 or anti-PD1 antibodies, have shown improved response rates and some improvement in survival in patients with advanced melanoma, but the majority of patients still do not respond or recur after initial response (6). Clearly, new therapeutic targets are needed in this patient population.
Activation of the mitogen-activated protein kinase (MAPK) and Phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) signaling pathways are important early steps in melanomagenesis (7). Activating mutations in NRAS or BRAF combined with inactivation of phosphatase and tensin homolog (PTEN) appear to be important driver mutations in melanoma, activating both the MAPK and PI3K/AKT pathways (7). However, this highly regulated process has other potential drivers (8), including the ectopic expression of metabotropic glutamate receptors (GRMs). Our group was the first to discover the expression of these cell surface receptors in melanoma (9–11).
A gain-of-function of murine metabotropic glutamate receptor 1 (mGRM1) in mouse melanocytes is sufficient to induce spontaneous melanoma in vivo with 100% penetrance (10, 12) and expression of mGRM1 in an immortalized (but not transformed) melanocytic cell line (melanA), results in transformation, rendering it capable of producing tumors in immunocompetent syngenic mice (11). Recently, ectopic expression of the other group I metabotropic glutamate receptor, mGRM5, has been shown to also produce melanoma in a second transgenic model (13).
These transgenic mouse studies prompted us to examine human melanoma for expression of the human form of this receptor, GRM1. Of 25 human melanoma cell lines tested, 23 express GRM1 (9). We also found that 60% to 80% of human melanoma specimens express GRM1 mRNA and protein, while normal skin and melanocytes from the same patients did not (9). Activation of GRM1 results in the release of glutamate from neurons and melanoma cells, setting up paracrine feedback loops that enhance GRM1-activation and signal transduction (9, 14). In preclinical studies we found that inhibition of GRM1 signaling in vitro and in vivo results in a G2/M cell cycle arrest and subsequent apoptosis in human melanoma. GRM1 inhibition also results in decreased human melanoma xenograft growth (9).
Riluzole (2-amino-6-trifluoromethoxybenzothiazole) is a potent inhibitor of glutamate release by GRM1-expressing cells and is currently the only FDA-approved agent for amyotrophic lateral sclerosis (ALS) (15). Using riluzole, we have translated our laboratory findings into the clinic through Phase 0 (16) and Phase II (17) trials in patients with advanced melanoma. In our Phase 0 trial administration of oral riluzole resulted in suppression of MAPK and PI3K/AKT signaling, and involution of tumor in 34% of patients, independent of BRAF and NRAS mutational status. We also found an increase in the number of apoptotic cells in post-treatment tumor samples (16). In the Phase II trial of single-agent riluzole, similar evidence of biologic activity was seen that correlated with initial stable disease in 30% of patients (17), consistent with our pre-clinical findings (18).
Logical clinical trial design requires a better understanding of how GRM1 signaling affects melanomagenesis and disease progression. We began by enhancing the expression of GRM1 in three human melanoma cell lines; a low GRM1-expressing subclone derived from UACC903, the metastatic line C8161 that has moderate GRM1 expression, and a related line, C81-61, (derived from the same patient as C8161) that does not express GRM1. We introduced exogenous full-length GRM1 cDNA into these lines to increase the expression of GRM1 and found that this did not increase in vitro proliferation of UACC903 or C8161 cells, though the normally slowly proliferating C81-61 cells did have a moderate increase in proliferation. What we did find was a marked increase in tumor growth and blood vessel formation when enhanced GRM1-expressing cells were used to produce xenografts. We hypothesized that increasing GRM1 signal transduction triggered an increase in the production of pro-angiogenic factors. Examination of the parental and enhanced GRM1-expressing cells confirmed this hypothesis revealing that AKT, mTOR, and HIF-1α participated in regulating the secretion of IL8 and VEGF secondary to enhanced GRM1 expression. We confirmed these findings using pre- and post-treatment tumors from our Phase II clinical trial (17). We have therefore discovered that GRM1 signal transduction promotes angiogenesis in melanoma through activation of the AKT/mTOR/HIF-1α signaling pathway. Results from these studies provide valuable insights that will help in the design of new combinatorial therapies for patients with advanced melanoma.
Materials and Methods
Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of Robert Wood Johnson Medical School (protocol I11-007). Human tumor samples were obtained from patients enrolled on a Phase II trial approved by our Institutional Review Board (IRB). Informed, signed consent for the protocol therapy and the use of the tumor samples was obtained from all patients.
Cell culture and cell proliferation assay
Melanoma lines were maintained in RPMI-1640 (Invitrogen, Carlsbad, CA) containing 10% FBS. SVEC4-10 immortalized endothelial cells were grown in DMEM containing 10% FBS. Primary endothelial cells (HMVECnd) were grown following the manufacture's protocol (Invitrogen). All cell were maintained in a humidified incubator at 37 °C with 5% CO2. Cell viability was ascertained using MTS reagent (Promega) following the manufacturer's protocol.
Constructing melanoma cell lines with increased GRM1 expression
GRM1 cDNA was cloned into the XhoI and EcoRI sites of pcDNA6/V5-HisA. The open reading frame was sequenced and matched the Gene Bank GRM1 sequence (BC111844). Lipofectin (Invitrogen) was used for the transfections following the manufacture's protocol. Stable clones UACC903-G2, UACC903-G4, C8161-G21, C81-61-G6, and C81-61-G7 that express GRM1 at significantly higher levels than the parental lines were propagated under selection with 10ug/ml Blasticidin. Stable clones transfected with empty vector were produced as controls and designated UACC903-V1, C8161-V2, and C81-61-V3.
Tumor induction in nude mice, in vivo drug treatment, and vessel imaging
Cells from each line were grown to 80% confluence, trypsinized, counted and re-suspended in ice-cold PBS. 106 cells in 0.1ml PBS were injected subcutaneous into each flank of nude mice (nu/nu), with each line injected into five mice. Tumors were measured twice a week (19). The mice were treated with either riluzole (20mg/kg), MK-2206 (60mg/kg), rapamycin (20mg/kg), or YC-1 (30mg/kg) by intraperitoneal (IP) injection every other day for 2 weeks starting when the tumors reached ~6-10 mm3. Tumor blood vessels were visualized by injecting 200ul of high molecular weight (average 2,000,000MW) FITC-dextran (Sigma) into the mice 10 minutes prior to sacrifice. Xenografts were removed, fixed in 10% formalin, thinly sliced, and photographed under UV-illumination.
Conditioned Media
Conditioned media were generated by seeding 106 cells from each cell line in 1 ml serum-free RPMI in tissue-culture wells for 20 hours.
Angiogenesis and apoptosis antibody arrays
The human angiogenesis (Panomics, Affymetrix) and the human apoptosis (R&D Systems) antibody array assays were performed using conditioned media from UACC903-G2 and UACC903-V1 cells following the manufacturers’ instructions.
Tumor cell migration assay
Cell migration assays using UACC903-V1, UACC903-G2, and UACC903-G4 cells were performed using 24-well plates and PET membrane inserts (pore size 8uM [BD Falcon]). 5x104 cells in 0.25ml serum-free RPMI were placed in the upper wells and the bottom wells were filled with 0.75ml RPMI with 0.5% FBS. Plates were incubated at 37°C and 5% CO2 overnight and then processed per manufacturer's protocol. Three pictures were taken of each insert and the number of migrated cells quantified using imageJ software (http://rsb.info.nih.gov/ij/).
Endothelial cell migration assay
A similar 24-well/PET membrane inserts set up was used for this assay. 1x 105 HMVECnd endothelial cells in 250ul serum-free RPMI were seeded into the upper wells while the lower wells contained 750ul serum-free RPMI and either 7.5x105 UACC903-V1, UACC903-G2, or UACC903-G4 cells. All other procedures were performed in the same fashion as the tumor cell migration assay. The endothelial cell migration assay was repeated using a transformed endothelial cell line (2x104 SVEC4-10 cells in 0.25ml serum free RPMI medium per upper well).
IL8 and VEGF ELISA
ELISAs (Invitrogen) were used to measure IL8 and VEGF levels in conditioned media. 2x105 cells were seeded in triplicate into a 96-well plate, each well had 200ul serum-free RPMI containing DMSO, MK-2206 (2uM), riluzole (10µM), or aminoflavone (50uM). After 9 hours the media were replaced with fresh serum-free RPMI with matching treatments and the cells were incubated for another 11 hours. Conditioned medium was collected for ELSA, fresh RPMI (100ul with serum) and 20ul of MTS reagent was added to the wells, and the plates were incubated at 37°C for one hour followed by measurement of cell viability.
Inducible HIF1 transcription reporter activity assay
100µl of resuspended UACC903-V1, UACC903-G2, and UACC903-G4 cells (0.5-1X104 cells) were seeded in triplicate in a 96-well plate and transfected with inducible HIF1 transcription reporter lentiviral particles (Qiagen) as per the manufacturers' protocol. Reporter gene expression level was measured using dual-glo luciferase assay system (Promega), using the manufacturer's protocol.
Western blot
Protein lysates from clinical tumor samples and cultured cells were prepared, and immunoblotting was performed, as previously described (18, 20). Cells were incubated with each inhibitor for 18 hours in 24-well plates to assess changes in signal transduction. Inhibitors used include riluzole (10µM), AF (50µM), or MK-2206 (10µM).
Statistical analysis
Heterogeneous variance models (21) were used to account for the different variances among groups. The comparisons of treatment groups with controls were conducted with Dunnett’s adjustment (22) to account for multiplicity in multiple comparisons. Adjusted p-values were provided when multiple comparisons were performed.
Results
1) Enhanced expression of GRM1 in human melanoma cells promotes cell migration and activates AKT and MAPK signaling
The melanoma cell lines that express GRM1 at significantly higher levels than the parental cell lines (UACC903-G2, UACC903-G4, C8161-G21, C81-61-G6, and C81-61-G7) were found to have increased AKT and ERK activation compared to controls (Figures 1A-1C), consistent with our previous findings that GRM1 activates the MAPK and AKT pathways (9, 18). Increased expression of GRM1 in UACC903 and C8161 did not significantly alter in vitro proliferation (Figures 1D and 1E). In contrast, expression of GRM1 in C81-61 (C81-61-G6, and C81-61-G7) cells led to increase proliferation in vitro, compared to the control C81-61-V3 cells (Figure 1F).
Figure 1.

Immunoblotting was performed by loading 10ug of protein lysate from each cell line into the lanes. Membranes were probed with antibodies to GRM1, GAPDH, total AKT, p-AKT473, p-AKT308, total ERK, and p-ERK. A) There is an increase in p-ERK/total ERK and p-AKT/Total AKT in UACC903-G2 (G2) and UACC903-G4 (G4) cells compared to UACC903-V1 (V1) cells, with the quantified relative fold increase shown in the graph. B and C) Similar results were seen when comparing C8161-G21 (G21) to its control (V2) and C81-61-G6 (G6) and C81-61-G7 (G7) to their control (V3). (Total ERK levels were unchanged in G21, G6, and G7 as compared to V2 and V3, not shown). D and E) MTS assay at 48 hours post-cell plating showing control cells V1 and V2 proliferate in vitro at a slightly higher rate than cells with enhanced GRM1 expression (G2, G4, and G21) (p<0.0001). F) C81-61 cells with higher GRM1 expression (G6 and G7) displayed increased cell proliferation compared to their control cells (V3) (p<0.01). All MTS assays were done in triplicate. G) UACC903-G2 and UACC903-G4 cells show more migration in a Boyden-chamber assay compared to the control UACC903-V1. Quantification of migration is average number of migrating cells ± SEM (p<0.05).
We used a Boyden Chamber assay to examine how increased expression of GRM1 affects migration of melanoma cells. We found that UACC903-G2 and UACC903-G4 cells had increased migration compared to UACC903-V1 control cells using 0.5% FBS as the chemo-attractant (Figure 1G). This data supports our previous findings that inhibiting GRM1 signaling with riluzole or a non-competitive inhibitor Bay36-7620 decreased the migration of GRM1-expressing melanoma cells (18).
2) Increased GRM1 expression in melanoma cells leads to larger xenografts with more abundant blood vessels
UACC903-G2, UACC903-G4, and C8161-G21 xenografts grew faster than UACC903-V1 or C8161-V2 xenografts (Figures 2A and 2B). The control C81-61-V3 cells did not produce xenografts though the GRM1-expressing C81-61-G7 (G7) cells did (Figure 2C). It is unlikely that the larger UACC903-G2, UACC903-G4, and C8161-G21 xenografts were produced by faster cell proliferation as these cell lines proliferated slightly slower in vitro than the controls (Figures 1D and 1E). There was also an absence of hemorrhagic necrosis in UACC903-G2, UACC903-G4, and C8161-G21 xenografts while UACC903-V1 and C8161-V2 xenografts showed extensive hemorrhagic necrosis (Figure 2D). These results suggest that enhanced GRM expression increased angiogenesis in vivo. This hypothesis was supported by the appearance of significantly more blood vessels in the UACC903-G2, UACC903-G4, and C8161-G21 xenografts than UACC903-V1 and C8161-V2 xenografts (Figure 2E).
Figure 2.

106 cells (UACC903-V1, UACC903-G2, UACC903-G4, C8161-V2, C8161-G21, C81-61-V3, or C81-61-G7) were injected subcutaneous into each flank of nu-/nu mice. Tumors were measured twice per week with a digital caliper starting at day 18 (UACC903-derived cells) or day 19 (C8161- and C81-61-derived cells). Tumor volumes were calculated in cubic millimeters. A) UACC903-G2 and UACC903-G4 xenografts (G2 and G4) grew more rapidly than control xenografts (V1). B) A similar difference was seen between C8161-G21 xenografts and control xenografts (V2). C) C81-61-G7 cells (G7) form xenografts in nude mice while the control C81-61-V3 cells (V3) do not. Data is the average tumor size ± SEM with p<0.001, n=6, t-test was performed on day 40 (V1, G2, and G4) or day 42 (V2, G21, V3, and G7). D) UACC903-V1 xenografts showed hemorrhagic necrosis while UACC903-G2 xenografts did not. E) Blood vessels were more abundant in UACC903-G2, UACC-903-G4, and C8161-G21 xenografts compared to control xenografts (Fluorescent microscopy magnification is 4x).
3) Increased expression of GRM1 results in increased IL8 and VEGF secretion
We performed an endothelial cell migration assay to determine if conditioned media produced by cells with enhanced GRM1 expression would act as a chemo-attractant for endothelial cells. Significantly more migration of primary human endothelial cells (HMVECnd) (Figures 3A and 3B) and transformed endothelial cells (SVEC4-10; data not shown) (23) was detected using conditioned medium from UACC903-G2 and UACC903-G4 cells as the chemo-attractant, compared to conditioned medium from UACC903-V1, suggesting that higher levels of pro-angiogenic factors were being produced by the cells with enhanced GRM1-expression.
Figure 3.

A) Endothelial cell migration assays were performed in triplicate for each cell line (UACC903-V1, UACC903-G2, and UACC903-G4) and repeated 3 times. HMVECnd migration is increased by the use of conditioned media from UACC903-G4 and UACC903-G3 cells compared to medium from control cells. Pictures taken with a Nikon eclipse TS100 inverted microscope with digital camera (Scale bar equals 100µM). B) Quantification of results shown in 3A (Average number of migrating cells ± SEM) (p<0.01). C) Angiogenic antibody array assay results show increased IL8 and VEGF secretion by cells with enhanced GRM1 expression compared to control cells. Row 1 (top row) is positive control. Row 5 dot 3 and 4 are IL-8, and Dot 7 and 8 are TIMP-1. Row 6 dot 1 and 2 are VEGF and dot 7 and 8 are TIMP-2. Validation of an increase in IL-8 (D, E, and F) and VEGF (G, H, and I) concentration in serum-free conditioned medium from UACC903-G2, UACC903-G4 (D and G), C8161-G21(E and H), C81-61-G6, and C81-61-G7 (F and I) cells compared to corresponding control cells (UACC903-V1, C8161-V2 C81-61-V3) using ELISA. Conditioned medium was diluted 20x for ELISA measurement. Experiments were repeated three times. Data presented are average ± SEM, p<0.001 for Figures 3 D-3I.
To determine which angiogenic factors were modulated by increased expression of GRM1, we performed an angiogenesis antibody array assay using serum-free conditioned media from UACC903-V1 and UACC903-G2 cells. We found increases in IL8 and VEGF in UACC903-G2 medium compared to UACC903-V1 medium (Figure 3C). We confirmed that all of the human melanoma cell lines with enhanced GRM1-expression released more IL8 and VEGF into the medium compared to controls by ELISA (Figures 3D-3I). We conclude that increased expression of GRM1 in melanoma cells results in increased IL8 and VEGF expression.
4) Enhanced expression of GRM1 increases the activation of HIF-1α
We treated UACC903-G2 and UACC903-G4 cells with an inhibitor of glutamate release, riluzole, in an in vitro cell viability assay. UACC903-G2 and UACC903-G4 cells were more sensitive to riluzole compared to control UACC903-V1 cells (Figure 4A). Previously we found that the reduced number of viable cells after riluzole treatment was due to an increase in apoptotic cells (9, 18). We therefore performed an apoptosis antibody array assay using protein lysates from cultured UACC903-V1 and UACC903-G2 cells. We found differences in the protein levels of cIAP-1, Claspin, TRAILR1, TRAILR2, FADD, HIF-1α, HO-2/HMOX2, HSP70, phospho-p53 (S15), phospho-p53 (S392), SMAC/Diablo, and Survivin (Supplemental Figure 1), confirmed by Western Blotting (data not shown).
Figure 4.

A) Cell viability of riluzole-treated UACC903-V1, UACC903-G2, and UACC903–G4 cells at 96 hours post-treatment by MTS assays. Relative viability is normalized to untreated cells. B) HIF-1α, p-mTOR, p70S6K, and Survivin protein levels increased in response to enhanced GRM1 expression. Immunoblots were performed as described in the Materials and Methods section. C) Increased HIF-1α transcriptional activity is seen in UACC903-G2 and UACC903-G4 cells compared to UACC903-V1 cells (p<0.001). D) IL8 levels in conditioned medium from C8161-G21 cells is suppressed by 6 hours of treatment with aminoflavone, (AF, 50µM) an anti-HIF-1α agent, riluzole (10µM), or the anti-AKT agent MK-2206 (2µM) (measured by ELISA, p< 0.01). E) Protein lysates from C8161-21 cells treated as in D were subjected to immunoblotting and show reduced levels of HIF-1α with each treatment.
One of these factors, HIF-1α, is a transcription factor known to regulate VEGF and IL8 expression (24). HIF-1α transcriptional activity can be activated by PI3K/AKT/mTOR signaling (25) and because HIF-1α expression is increased in UACC903-G2 cells compared to UACC903-V1 cells, it may be at least partially responsible for the angiogenesis-enhancing effects of GRM1. Another molecule up-regulated in UACC903-G2 cells was Survivin, an anti-apoptotic molecule whose transcription is also regulated by HIF-1α (26). Immunoblotting protein lysates from UACC903-V1, UACC903-G2, and UACC903-G4 cells showed that HIF-1α, p-mTOR, p-70S6K [a downstream target of mTOR], and Survivin, were all up-regulated in cell lines with increased GRM1 expression (Figure 4C). To confirm that increased expression of HIF-1α resulted in an increase in its transcriptional activity we transiently transfected an inducible HIF-1α luciferase-reporter construct into UACC903-V1, UACC903-G2, and UACC903-G4 cells. We found that HIF-1α transcriptional activity was significantly higher in UACC903-G2 and UACC903-G4 cells as compared to UACC903-V1 cells (Figure 4D). We found a similar increase in HIF-1α expression and transcriptional activity in C8161-G21, C81-61-G6, and C81-61-G7 cells as compared to C8161-V2 and C81-61-V3 cells (data not shown). We next treated C8161-V2, C8161-G21, UACC903-V1, UACC903-G2, and UACC903-G4 cells with inhibitors targeting the GRM1/AKT/mTOR/HIF-1α pathways and looked for alterations in IL8 and VEGF expression. Regardless of the inhibitors used, levels of HIF1α and secreted IL8 and VEGF were all reduced (Figures 4E, 4F, and Supplemental Figure 2). These results demonstrated that up-regulated HIF-1α expression and transcriptional activity in response to an increase in GRM1 expression in human melanoma cells is likely mediated via direct activation of the PI3K/AKT/mTOR pathway.
5). GRM1/AKT/mTOR/HIF-1α signaling promotes xenograft vessel formation
Xenografts generated using UACC903-G4 cells and treated by IP injection every other day with riluzole (20mg/kg), MK-2206 (60mg/kg), rapamycin (20mg/kg), or YC-1 [HIF-1α inhibitor, 30mg/kg] (27) all showed significant inhibition of xenograft growth compared to vehicle-treated controls (Figure 5A). Treated UACC903-G4 xenografts showed a significant decrease in blood vessel growth compared to vehicle-treated xenografts and residual vessels in treated xenografts were decreased in quantity and highly fragmented (Figure 5B). Signaling analysis revealed that HIF-1α expression in UACC903-G4 xenografts was much higher than in UACC903-V1 xenografts and decreased significantly with riluzole, MK-2206, rapamycin, or YC-1 treatment (Figure 5C). We repeated these xenograft experiments using the C8161-G21 cell line and found that these cells formed larger xenografts than the control C8161-V2 cells (Figure 2B) and were exquisitely sensitive to anti-GRM1 (riluzole) or anti-HIF-1α (YC-1) treatment (Figure 5D). Therefore, larger tumors with more abundant blood vessels seen in xenografts produced from melanoma cell lines with enhanced GRM1 expression (Figure 2) are partially due to increased activation of the PI3K/AKT pathway and can be inhibited by agents targeting GRM1, AKT, mTOR, and HIF-1α. These data support our hypothesis that the AKT/mTOR/HIF-1α signaling pathway is downstream of GRM1 and functions to promote blood vessel formation as a consequence of GRM1 signal transduction.
Figure 5.

A) UACC903-G4 xenografts were produced as in Figure 2 and treated with riluzole (20mg/kg), MK-2206 (60mg/kg), rapamycin (20mg/kg), or YC-1 (30mg/kg) by IP injection. Tumor volumes were calculated in cubic millimeters using a standard formula (19). Results represent average tumor size ± SEM. p<0.005, n=6, with t-test performed on day 25. B) Blood vessels in UACC903-G4 xenografts on day 25. C) Signaling response in UACC903-G4 xenografts treated with the four agents show expected suppression of downstream signal transduction. D) A second xenograft experiment using C8161-G21 and control C8161-V2 cells. We again see that enhanced GRM1 expression leads to larger xenografts with xenograft growth suppressed by inhibiting glutamatergic signaling (riluzole) or HIF1 activity (YC-1).
6) Response to riluzole corresponds to a decrease in activation of the AKT/mTOR/HIF-1α pathway and decreased CD31 and Survivin in clinical melanoma samples
We conducted a Phase II trial of single-agent riluzole in patients with advanced melanoma. All 13 patients enrolled on the trial were GRM1-positive (by IHC) and treated with the maximum FDA-approved dose of riluzole (100mg BID). Although no RECIST responses were seen (17), stable disease at first restaging was seen in 46% of previously progressing patients. We collected pre- and post-treatment tumors from 8 patients and 3 of these 8 patients were among those with initial stable disease. Immunoblotting results comparing pre- and post-treatment tumor samples show a reduction in pERK, pAKT, CD31, Survivin, HIF-1α, and p-mTOR levels only in the post-treatment samples from these 3 patients (1, 5 and 11) (Figure 6). CD31 is highly-expressed by endothelial cells and is used as a marker of vessel density (28). Decreased CD31 expression suggests riluzole may have inhibited angiogenesis in these patients. These results demonstrate that signaling molecules whose activity is modulated in response to increased expression of GRM1 in our pre-clinical models are also modulated in melanoma samples from patients treated with an inhibitor of GRM1 signal transduction.
Figure 6.
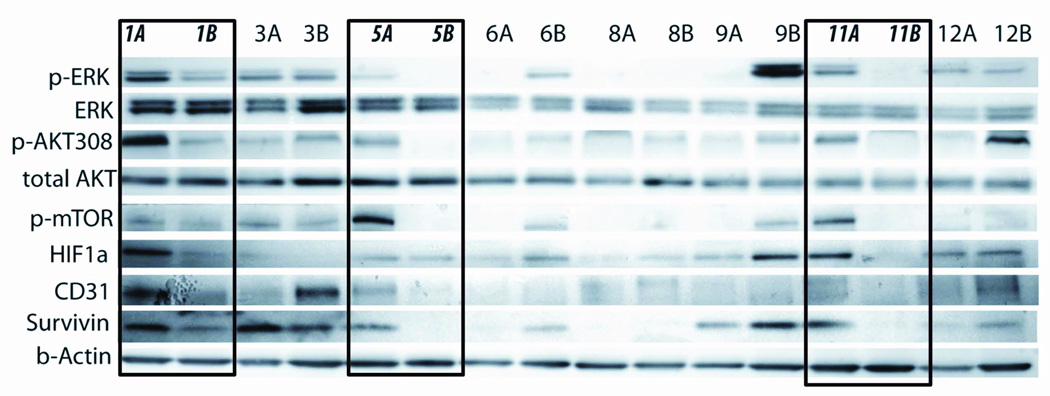
Eight pairs of pre- and post-treatment melanoma samples from patients on a phase II trial of single-agent riluzole were snap-frozen in liquid nitrogen immediately after resection. Protein lysates were prepared and immunoblots performed as described in the Materials and Methods section. Patient number 1, 5, and 11 had mixed clinical responses to riluzole treatment with stable disease on first post-treatment evaluation. These three paired patient samples were the only ones to show post-treatment decreases in pERK, pAKT, p-mTOR, HIF-1α, CD31, and Survivin. A = pre-treatment samples and B = post-treatment samples.
Discussion
Constitutive activation of the MAPK and PI3K/AKT pathways is found in the vast majority of melanomas and agents that target these pathways have been developed (29). However, regulation of signal transduction through these pathways is more complicated than originally thought (30, 31). Driver mutations such as V600E BRAF are likely early events in the development of melanoma but are not necessary for continued progression of the disease as evidenced by its loss in patients who recur after initial response to vemurafenib (30, 31). It appears that after initial transformation subsequent mutations, feedback loops, changes in the tumor microenvironment, and other factors add layers of genotypic and phenotypic complexity to the neoplasm that requires a more complex treatment approach. Ectopic expression of seven transmembrane G-protein coupled receptors (GPCRs) appears to be one of the subsequent changes found in many different cancers (32).
GRM1 is a GPCR belonging to the metabotropic glutamate receptor family that has 8 different isoforms that bind glutamate as their natural ligand (33). In the central nervous system L-glutamate is a major excitatory neurotransmitter and activates both ionotropic and metabotropic glutamate receptors. GRM1 and GRM5 belong to Group I and activate PLCβ, resulting in intracellular Ca2+ release and protein kinase C (PKC) activation (33).
Our discovery that GRM1 is expressed by the majority of human melanomas adds to our understanding of the multiple mechanisms of MAPK and AKT pathway activation in this disease (9, 10, 18). Recently, others have reported that other metabotropic glutamate receptors can also affect melanoma pathogenesis and progression. As mentioned, Choi et al. reported that ectopic expression of GRM5 in a transgenic model results in an aggressive form of melanoma with high penetrance (13), reminiscent of our transgenic models (10). Prickett et al. have recently reported that an activating mutation in a group 2 metabotropic glutamate receptor, GRM3, is common in human melanoma and correlates with a more aggressive form of the disease (34). It is now evident that metabotropic glutamate signal transduction is an important component of the pathogenesis of melanoma.
We have translated our preclinical findings into the clinic with the use of the agent riluzole, an oral inhibitor of GRM1 signal transduction. We observed significant tumor shrinkage and evidence of biologic activity with prolonged administration of riluzole in some patients with advanced melanoma (16, 17). Accumulated evidence demonstrates that riluzole inhibits GRM1 signaling in melanoma, though durable responses were not obtained in these clinical trials. As has been found with RAF inhibitors (5), it now appears likely that combinatorial approaches will be needed to have a significant effect on outcome. We report here the effects of increasing the expression of GRM1 in human melanoma cell lines that allowed us to examine GRM1 function and downstream signaling. First, we found that enhanced expression of GRM1 increases cell migration and activates ERK and AKT, consistent with our previous findings (18). Second, it appears that in cells that express this receptor, GRM1 is essential for survival, with downstream signal transduction likely activating multiple pathways needed to maintain the transformed phenotype and drive tumor progression (35). We now demonstrate that GRM1 signal transduction results in downstream activation of the AKT/mTOR/HIF-1α pathway with increased expression of IL8 and VEGF. We confirmed these preclinical findings using pre- and post-treatment tumor samples from patients with advanced melanoma who received oral riluzole on a Phase II trial. Therefore, one of the consequences of GRM1 expression and activation in melanoma is the production of pro-angiogenic factors and a corresponding increase in tumor vasculature.
Interestingly we are not the first investigators to suggest that suppression of glutamatergic signaling with riluzole decreases the expression of VEGF and inhibits angiogenesis. Yoo et al. reported in two different studies that riluzole can inhibit VEGF-induced endothelial cell proliferation and block vascular retinopathy induced by hyperglycemia (36, 37). Retinopathy resulting from overgrowth of blood vessels is common in diabetic patients and pre-mature infants and is likely caused by an increase in VEGF expression brought on by tissue hypoxia and stimulation of insulin-like growth factor receptors (IGFR) (38). Yoo et al. demonstrated that increased VEGF expression, endothelial cell proliferation, and angiogenesis seen in pre-clinical models of retinopathy could be inhibited by riluzole. They hypothesized that riluzole inhibited PKCβII activation and did not link its effects to the expression of metabotropic glutamate receptors. As part of the CNS, retinal tissue does express GRM1 and other metabotropic glutamate receptors (39) and riluzole likely exerts its effects on angiogenesis in the retina through inhibition of glutamatergic signaling, supporting our findings of a similar effect in melanoma.
IL8 has been reported to function as an autocrine/paracrine factor stimulating cell growth, enhancing metastasis, and promoting angiogenesis in melanoma (40). Up-regulation of IL8 and VEGF is involved in the resistance of melanomas to chemotherapeutic agents such as dacarbazine (41). The roles of VEGF in solid tumors have been well validated and therapies targeting VEGF/VEGFR are approved for clinical use in patients with colon, renal, non-small cell lung, and other types of human cancers (42). Our prior research demonstrated high levels of VEGF, VEGF-R1, VEGF-R2, and VEGF-R3 expression in melanoma (43) and a recently published Phase II study of bevacizumab and temozolomide versus nab-paclitaxel and carboplatin in patients with advanced melanoma showed promising results with the addition of the anti-angiogenic agent, though the tolerability of the regimens was a concern (44). It is also possible that resistance to therapies that target VEGF, such as bevacizumab, in patients with advanced melanoma could be secondary to simultaneous up-regulation of IL8 with continued angiogenesis even as VEGF activity is inhibited (42).
Our finding that activation of the AKT/mTOR/HIF-1α pathways and downstream up-regulation of VEGF and IL8 is consistent with previous reports (24, 25, 28). However, it has also been reported that voltage-gated sodium channels are involved in invasion and metastasis in both prostate cancer and triple negative breast cancer and riluzole can inhibit the effects of these ion channels in cancer cells (45). Therefore, we cannot exclude the possibility that the effects of riluzole in melanoma may be partly due to the inhibition of sodium channels. We are currently conducting studies to test this hypothesis.
Collectively, our findings support the hypothesis that GRM1 plays important roles in melanoma development and progression. These data will be useful in the design of further preclinical studies and future clinical trials combining GRM1-blockade with agents inhibiting components of the MAPK, PI3K/AKT, mTOR/HIF-1α and other cellular pathways.
Supplementary Material
Acknowledgements
We thank Joseph L.-K. Chan for his help with original assay design and the Histopathology Shared Resource of Rutgers Cancer institute of New Jersey for slide processing and immunohistochemical staining. We are grateful to Dr. Marina Chekmareva, Department of Pathology, Rutgers Robert Wood Johnson Medical School for assessing the quality and specificity of the immunohistochemical staining.
Grant Support: This work was supported by National Institutes of Health grants R01CA124975, R01CA149627, and R21CA139473 (PI: Goydos) and a Grant from the Elizabeth and Barets O. Benjamin Foundation.
Footnotes
Conflict of Interest: The authors disclose no potential conflicts of interest.
Literature Cited
- 1.Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62:220–241. doi: 10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- 2.Mouawad R, Sebert M, Michels J. Treatment for metastatic malignant melanoma: old drugs and new strategies. Crit Rev Oncol Hematol. 2010;74:27–39. doi: 10.1016/j.critrevonc.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. New England Journal of Medicine. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N Engl J Med. 2012 doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simeone E, Ascierto PA. Immunomodulating antibodies in the treatment of metastatic melanoma: the experience with anti-CTLA-4, anti-CD137, and anti-PD1. J Immunotoxicol. 2012;9:241–247. doi: 10.3109/1547691X.2012.678021. [DOI] [PubMed] [Google Scholar]
- 7.Yajima I, Kumasaka MY, Thang ND, Goto Y, Takeda K, Yamanoshita O, et al. RAS/RAF/MEK/ERK and PI3K/PTEN/AKT Signaling in Malignant Melanoma Progression and Therapy. Dermatol Res Pract. 2012;2012:354191. doi: 10.1155/2012/354191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett DC. How to make a melanoma: what do we know of the primary clonal events? Pigment Cell Melanoma Res. 2008;21:27–38. doi: 10.1111/j.1755-148X.2007.00433.x. [DOI] [PubMed] [Google Scholar]
- 9.Namkoong J, Shin SS, Lee HJ. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007;67:2298–2305. doi: 10.1158/0008-5472.CAN-06-3665. [DOI] [PubMed] [Google Scholar]
- 10.Pollock PM, Cohen-Solal K, Sood R, Namkoong J, Martino JJ, Koganti A, et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat Genet. 2003;34:108–112. doi: 10.1038/ng1148. [DOI] [PubMed] [Google Scholar]
- 11.Shin S-S, Namkoong J, Wall BA, Gleason R, Lee HJ, Chen S. Oncogenic activities of metabotropic glutamate receptor 1 (Grm1) in melanocyte transformation. Pigment Cell & Melanoma Research. 2008;21:368–378. doi: 10.1111/j.1755-148X.2008.00452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohtani Y, Harada T, Funasaka Y, Nakao K, Takahara C, Abdel-Daim M, et al. Metabotropic glutamate receptor subtype-1 is essential for in vivo growth of melanoma. Oncogene. 2008;27:7162–7170. doi: 10.1038/onc.2008.329. [DOI] [PubMed] [Google Scholar]
- 13.Choi KY, Chang K, Pickel JM, Badger JD, Roche KW. Expression of the metabotropic glutamate receptor 5 (mGluR5) induces melanoma in transgenic mice. Proceedings of the National Academy of Sciences. 2011;108:15219–15224. doi: 10.1073/pnas.1107304108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heasley L. Autocrine and paracrine signaling through neuropeptide receptors in human cancer. Oncogene. 2001;20:1563–1569. doi: 10.1038/sj.onc.1204183. [DOI] [PubMed] [Google Scholar]
- 15.Swash M. New ideas for therapy in ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:3–4. doi: 10.1080/14660820510035379. [DOI] [PubMed] [Google Scholar]
- 16.Yip D, Le MN, Chan JL-K, Lee JH, Mehnert JA, Yudd A, et al. A Phase 0 Trial of Riluzole in Patients with Resectable Stage III and IV Melanoma. Clinical Cancer Research. 2009;15:3896–3902. doi: 10.1158/1078-0432.CCR-08-3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mehnert J, Wen Y, Lee J, Dudek L, Jeong B, Li J, et al. A phase II trial of riluzole, an antagonist of metabotropic glutamate receptor (GRM1) signaling, in advanced melanoma. 2013 doi: 10.1111/pcmr.12694. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le MN, Chan JL, Rosenberg SA, Nabatian AS, Merrigan KT, Cohen-Solal KA, et al. The glutamate release inhibitor Riluzole decreases migration, invasion, and proliferation of melanoma cells. J Invest Dermatol. 2010;130:2240–2249. doi: 10.1038/jid.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feldman J, Goldwasser R, Mark S, Schwartz J, Orion I. A Mathematical Model For Tumor Volume Evaluation Using Two-Dimensions. Journal of Applied Quantitative Methods. 2009;4:455–462. [Google Scholar]
- 20.Yan X, Shen H, Jiang H, Hu D, Wang J, Wu X. External Qi of Yan Xin Qigong Inhibits Activation of Akt, Erk1/2 and NF-ĸB and Induces Cell Cycle Arrest and Apoptosis in Colorectal Cancer Cells. Cellular Physiology and Biochemistry. 2013;31:113–122. doi: 10.1159/000343354. [DOI] [PubMed] [Google Scholar]
- 21.Wolfinger RD. Heterogeneous variance covariance structure for repeated measures. Journal of agricultural, biological, and environmental statistics. 1996;1:205–230. [Google Scholar]
- 22.Dunnett CW. A multiple comparison procedure for comparing several treatments with a control. Journal of the American Statistical Association. 1955;50:1096–1121. [Google Scholar]
- 23.O'Connell K, Edidin M. A mouse lymphoid endothelial cell line immortalized by simian virus 40 binds lymphocytes and retains functional characteristics of normal endothelial cells. The Journal of Immunology. 1990;144:521–525. [PubMed] [Google Scholar]
- 24.Cane G, Ginouvès A, Marchetti S, Buscà R, Pouysségur J, Berra E, et al. HIF-1α mediates the induction of IL-8 and VEGF expression on infection with Afa/Dr diffusely adhering E. coli and promotes EMT-like behaviour. Cellular Microbiology. 2010;12:640–653. doi: 10.1111/j.1462-5822.2009.01422.x. [DOI] [PubMed] [Google Scholar]
- 25.Toschi A, Lee E, Gadir N, Ohh M, Foster DA. Differential Dependence of Hypoxia-inducible Factors 1α and 2α on mTORC1 and mTORC2. Journal of Biological Chemistry. 2008;283:34495–34499. doi: 10.1074/jbc.C800170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Zhao C, Li W. Effect of hypoxia-inducible factor-1alpha on transcription of survivin in non-small cell lung cancer. Journal of Experimental and Clinical Cancer Research. 2009;28:8. doi: 10.1186/1756-9966-28-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeo E-J, Chun Y-S, Cho Y-S, Kim J, Lee J-C, Kim M-S, et al. YC-1: A Potential Anticancer Drug Targeting Hypoxia-Inducible Factor 1. Journal of the National Cancer Institute. 2003;95:516–525. doi: 10.1093/jnci/95.7.516. [DOI] [PubMed] [Google Scholar]
- 28.Argyriou P, Papageorgiou S, Panteleon V, Psyrri A, Bakou V, Pappa V, et al. Hypoxia-inducible factors in mantle cell lymphoma: implication for an activated mTORC1→HIF-1α pathway. Annals of Hematology. 2010:1–8. doi: 10.1007/s00277-010-1070-6. [DOI] [PubMed] [Google Scholar]
- 29.Flaherty KT, Hodi FS, Bastian BC. Mutation-driven drug development in melanoma. Curr Opin Oncol. 2010;22:178–183. doi: 10.1097/cco.0b013e32833888ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yadav V, Zhang X, Liu J, Estrem S, Li S, Gong XQ, et al. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J Biol Chem. 2012;287:28087–28098. doi: 10.1074/jbc.M112.377218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu J, Xie N, Zhao X, Nice EC, Huang C. Dissection of aberrant GPCR signaling in tumorigenesis--a systems biology approach. Cancer Genomics Proteomics. 2012;9:37–50. [PubMed] [Google Scholar]
- 33.Kim CH, Lee J, Lee J-Y, Roche KW. Metabotropic glutamate receptors: Phosphorylation and receptor signaling. Journal of Neuroscience Research. 2008;86:1–10. doi: 10.1002/jnr.21437. [DOI] [PubMed] [Google Scholar]
- 34.Prickett TD, Wei X, Cardenas-Navia I, Teer JK, Lin JC, Walia V, et al. Exon capture analysis of G protein-coupled receptors identifies activating mutations in GRM3 in melanoma. Nat Genet. 2011;43:1119–1126. doi: 10.1038/ng.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wangari-Talbot J, Wall BA, Goydos JS, Chen S. Functional Effects of GRM1 Suppression in Human Melanoma Cells. Molecular Cancer Research. 2012 doi: 10.1158/1541-7786.MCR-12-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoo MH, Yoon YH, Chung H, Cho KS, Koh JY. Insulin increases retinal hemorrhage in mild oxygen-induced retinopathy in the rat: inhibition by riluzole. Invest Ophthalmol Vis Sci. 2007;48:5671–5676. doi: 10.1167/iovs.07-0395. [DOI] [PubMed] [Google Scholar]
- 37.Yoo MH, Hyun HJ, Koh JY, Yoon YH. Riluzole inhibits VEGF-induced endothelial cell proliferation in vitro and hyperoxia-induced abnormal vessel formation in vivo. Invest Ophthalmol Vis Sci. 2005;46:4780–4787. doi: 10.1167/iovs.05-0376. [DOI] [PubMed] [Google Scholar]
- 38.Witmer AN, Vrensen GF, Van Noorden CJ, Schlingemann RO. Vascular endothelial growth factors and angiogenesis in eye disease. Prog Retin Eye Res. 2003;22:1–29. doi: 10.1016/s1350-9462(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 39.Hanna MC, Calkins DJ. Expression and sequences of genes encoding glutamate receptors and transporters in primate retina determined using 3'-end amplification polymerase chain reaction. Mol Vis. 2006;12:961–976. [PubMed] [Google Scholar]
- 40.Huang S, Mills L, Mian B, Tellez C, McCarty M, Yang X, et al. Fully humanized neutralizing antibodies to interleukin-8 (ABX-IL8) inhibit angiogenesis, tumor growth, and metastasis of human melanoma. Am J Pathol. 2002;161:125–134. doi: 10.1016/S0002-9440(10)64164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zigler M, Villares GJ, Lev DC, Melnikova VO, Bar-Eli M. Tumor Immunotherapy in Melanoma: Strategies for Overcoming Mechanisms of Resistance and Escape. American Journal of Clinical Dermatology. 2008;9:307–311. doi: 10.2165/00128071-200809050-00004. [DOI] [PubMed] [Google Scholar]
- 42.Grothey A, Galanis E. Targeting angiogenesis: progress with anti-VEGF treatment with large molecules. Nat Rev Clin Oncol. 2009;6:507–518. doi: 10.1038/nrclinonc.2009.110. [DOI] [PubMed] [Google Scholar]
- 43.Mehnert JM, McCarthy MM, Jilaveanu L, Flaherty KT, Aziz S, Camp RL, et al. Quantitative expression of VEGF, VEGF-R1, VEGF-R2, and VEGF-R3 in melanoma tissue microarrays. Human Pathology. 2010;41:375–384. doi: 10.1016/j.humpath.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kottschade LA, Suman VJ, Perez DG, McWilliams RR, Kaur JS, Amatruda TT, 3rd, et al. A randomized phase 2 study of temozolomide and bevacizumab or nab-paclitaxel, carboplatin, and bevacizumab in patients with unresectable stage IV melanoma : A North Central Cancer Treatment Group study, N0775. Cancer. 2012 doi: 10.1002/cncr.27760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Djamgoz M, Onkal R. Persistent current blockers of voltage-gated sodium channels: a clinical opportunity for controlling metastatic disease. Recent Pat Anticancer Drug Discov. 2013;8:66–84. doi: 10.2174/15748928130107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.