

Abstract
Pancreatic cancer is often diagnosed at an advanced stage and it has a poor prognosis which points to an increased need to develop effective chemoprevention strategies for this disease. We examined the ability of phenethyl isothiocyanate (PEITC), a naturally-occurring isothiocyanate found in cruciferous vegetables, to inhibit the growth of pancreatic cancer cells in vitro and in a MIAPaca2 xenograft animal model. Exposure to PEITC inhibited pancreatic cancer cell growth in a dose-dependent manner, with an IC50 of approximately 7 μmol/L. PEITC treatment induced G2/M phase cell cycle arrest, down-regulated the anti-apoptotic proteins Bcl-2 and Bcl-XL, up-regulated the pro-apoptotic protein Bak, and suppressed Notch 1 and 2 levels. In addition, treatment with PEITC induced cleavage of poly-(ADP-ribose) polymerase and led to increased cytoplasmic histone-associated DNA fragmentation and subdiploid (apoptotic) fraction in pancreatic cancer cells. Oral administration of PEITC suppressed the growth of pancreatic cancer cells in a MIAPaca2 xenograft animal model. Our data show that PEITC exerts its inhibitory effect on pancreatic cancer cells through several mechanisms including G2/M phase cell cycle arrest and induction of apoptosis, and support further investigation of PEITC as a chemopreventive agent for pancreatic cancer.
Keywords: pancreatic cancer, phenethyl isothiocyanate, G2/M arrest, apoptosis
INTRODUCTION
Pancreatic cancer is a rapidly progressing disorder that is often diagnosed at an advanced, non-resectable stage and it is highly resistant to chemotherapy. Pancreatic ductal adenocarcinoma is believed to result from the accumulation of several genetic mutations, including Kras, p16, p53, SMAD4, and BRCA2 (1,2), and may arise from precursor lesions called pancreatic intraepithelial neoplasias (1,3). The low 5-year survival rate of patients with pancreatic cancer of ~6% (4) is related to the challenges in diagnosing a pancreatic adenocarcinoma at an early stage when curative resection is still possible. A recent study analyzed the timing of genetic evolution from the genomic sequencing data of seven pancreatic cancer metastases and primary tumors and concluded that there was at least 15 years from the original initiating mutation to when the primary tumor cancer cells acquire the ability to metastasize (5). Therefore there is clearly a need for pancreatic cancer chemoprevention and there appears to be an adequate window of opportunity to improve on the dismal survival of this disease through the development of effective chemoprevention strategies aimed at prevention of the primary tumor or the development of metastases (6).
Epidemiological studies have shown an inverse association between consumption of cruciferous vegetables and cancer incidence, including pancreatic cancer (7-12). For example, a statistically significant negative trend in risk of pancreatic cancer with cruciferous vegetables consumption was observed in a case-control study in both men and women, with a combined odds ratio (OR) for the highest quartile (i.e., > 4 servings/week) of 0.5 (95% CI = 0.4-0.8, P for trend = 0.0004) (11). Cabbage consumption was associated with a statistically lower risk of developing pancreatic cancer (> 1 serving/week versus never consumption, HR, 0.62; 95% CI, 0.39-0.99) (12). Other inverse associations, although not-statistically significant, between consumption of cruciferous vegetables and pancreatic cancer risk were reported in two other case control studies (7,9) and in a prospective study (12).
Phenethyl isothiocyanate (PEITC) is a naturally occurring isothiocyanate (ITC) found in cruciferous vegetables such as watercress and garden cress. PEITC has been intensively studied as a cancer chemopreventive agent and was shown to inhibit cancer growth of various tissues including lung, esophagus, colorectum, mammary gland, prostate, liver, pancreas, and bladder (13-16). The inhibitory effects of ITCs have been attributed to their ability to regulate multiple molecular mechanisms including inhibition of Phase I drug-metabolizing enzymes (e.g., cytochrome P-450), induction of Phase II detoxifying enzymes (e.g., glutathione-S-transferases), induction of cell cycle arrest and apoptosis, inhibition of histone deacetylases, regulation of androgen and estrogen receptors, generation of reactive oxygen species, induction of autophagy, modulation of immune response, and suppression of angiogenesis (16-20). PEITC is currently in clinical trials for lung cancer and lymphoproliferative disorders (21). Prior preclinical studies showed that PEITC inhibits pancreatic tumors induced by N-nitrosobis(2-oxopropyl)amine in hamsters (22). However, little is known about the chemopreventive potential of PEITC against pancreatic cancer or the underlying mechanism of action of PEITC in pancreatic cancer cells.
We aimed this study to determine the efficacy of PEITC in inhibiting the growth of human pancreatic cancer cells in vitro and in a xenograft animal model. Here we demonstrate that PEITC inhibits the growth of pancreatic cancer cells through multiple mechanisms including induction of G2/M phase cell cycle arrest, apoptosis, and modulation of Notch 1 and 2 expression, and inhibits the growth of MIAPaca2 cells in a xenograft animal model.
MATERIALS AND METHODS
Cell Lines and Reagents
MIAPaca2, PL-45, and BxPC3 pancreatic cancer cells were purchased from ATCC (American Type Culture Collection, Manassas, VA) and grown in a humidified incubator at 37°C under 5% CO2. MIAPaca2 and PL-45 cell lines were authenticated by Research Animal Diagnostic Laboratory (University of Missouri, Columbia, MO). MIAPaca2 cells were grown in DMEM (Invitrogen, Grand Island, NY) supplemented with 10% FBS (Invitrogen) and 2.5% horse serum (Invitrogen). PL-45 cells were grown in DMEM supplemented with 10% FBS. BxPC3 cells were grown in RPMI 1640 (Fisher, Pittsburgh, PA) supplemented with 10% FBS, 10 mM HEPES (Invitrogen) and 1 mM sodium pyruvate (Invitrogen); all media contained 1% PSN antibiotic mixture (Invitrogen). PEITC (≥ 98% purity) was obtained from LKT Laboratories (St. Paul, MN), and DMSO was obtained from Sigma-Aldrich (St. Louis, MO). Bcl-2 antibody was purchased from Dako Cytomation (Carpinteria, CA), and Bcl-XL and Notch 1 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The antibodies against Bak, cleaved poly(ADP) ribose polymerase (PARP), and Notch 2 were purchased from Cell Signaling (Danvers, MA), and the antibody against phospho-histone H3 was obtained from Upstate (Lake Placid, NY).
Cell Viability Assay
PL-45, MIAPaca2, and BxPC3 cells were plated in 12-well plates and allowed to attach by overnight incubation. Cells were treated with DMSO (control) or PEITC (2.5, 5, and 10 μmol/L) for 24 h. The final concentration of DMSO was less than 0.1%. The effect of PEITC on pancreatic cell viability was determined by trypan blue dye exclusion assay. Briefly, cells were collected by trypsinization and incubated with trypan blue for 5 min. Live (unstained) cells were counted and reported as % of DMSO-treated control cells.
Clonogenic Assay
Cells were plated in 100-mm dishes and incubated overnight. The cells were treated with DMSO (control) or PEITC (2.5, 5, and 10 μmol/L) for 24 h. After treatment, the cells were detached using trypsin, and an equal number of cells from each treatment was re-seeded in triplicate in 6-well plates (300 cells/well). The medium was changed every 3 days for 10 days when the colonies were counted after staining with 0.5% crystal violet.
Cell Cycle Assay
Cell cycle distribution was determined by flow cytometry analysis as described (23). Briefly, MIAPaca2 cells were treated with either DMSO (control) or PEITC for 24 h and then collected by trypsinization and fixed with 70% ethanol. The fixed cells were stained with 80 μg/mL RNase A and 50 μg/mL propidium iodide for 30 min before analysis of cell cycle distribution by flow cytometry using a Coulter Epics XL Flow Cytometer (Beckman Coulter, Fullerton, CA).
Apoptosis Assay
Pancreatic cancer cells were plated in 12-well plates, incubated overnight, and treated for 24 h with DMSO (control) or PEITC (2.5, 5 and 10 μmol/L). Apoptosis was determined by quantification of cytoplasmic histone-associated DNA fragmentation using a cell death detection ELISA kit from Roche Applied Science (Indianapolis, IN) according to the manufacturer’s instructions.
Western Blotting
Pancreatic cancer cells were plated in 100-mm dishes and incubated overnight. The cells were either treated with various concentrations of PEITC (0, 2.5, 5, and 10 μmol/L) for 48 h or with 5 μmol/L PEITC for various time intervals (24, 48, and 72 h). After treatment, the cells were washed with PBS and lysed with lysis buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1% SDS, 1% Triton X-100, and protease and phosphatase inhibitors (Sigma-Aldrich). The cells were kept on ice for 30 min and regularly vortexed before centrifugation at 14,000 rpm for 30 min at 4°C. Protein concentration was determined using a BioRad (Hercules, CA) protein assay kit. The proteins were resolved using SDS-polyacrylamide gel electrophoresis and transferred to a PVDF membrane. After blocking for 1 h at room temperature in 5% (w/v) nonfat dry milk, the membrane was incubated overnight with the specific primary antibody and then washed three times in TTBS and incubated with the appropriate secondary antibody for 1 h at room temperature. The membrane was developed using enhanced chemiluminescence (ECL) reagent (Perkin Elmer, Waltham, MA).
Animal Study
Male athymic nude mice (6 weeks old) were obtained from Harlan (Madison, WI) and acclimated for 1 week before the start of the experiment. Mice were housed under environmentally controlled conditions (50% humidity, 12 h light/12 h dark cycle) and allowed access to water and AIN-76A diet (Harlan) ad libitum. To mimic a prevention protocol, mice were treated with PEITC (12 μmol/day, 5 days/week, n=10) or vehicle (control, n=8) for 1 week before injection of cells. Exponentially growing MIAPaca2 cells were collected by trypsinization, resuspended in PBS and mixed with matrigel (BD Biosciences, San Diego, CA) in a 1:1 ratio; 2.5 million cells were injected subcutaneously into one flank of each mouse. Treatment of mice continued for total 7 weeks. Body weights and tumor volumes were monitored weekly. At necropsy, tumor and organ tissues were fixed in 10% phosphate buffered formalin and embedded in paraffin for immunohistochemistry analysis. The experimental protocol was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Immunohistochemistry
Tissue sections were processed for Ki-67, PCNA, and TUNEL assays. Ki-67 and PCNA antibodies were purchased from Dako Cytomation. Sections were deparaffinized and hydrated with deionized water. Heat induced epitope retrieval was performed using citrate buffer pH 6 (Dako). Endogenous peroxidase was quenched using 3% hydrogen peroxide (Fisher). Blocking was performed using either 5% normal rabbit serum followed by avidin/biotin block (Ki-67 staining) or with rodent block (Biocare, Concord, CA) (PCNA staining). Slides were incubated with primary antibody followed by incubation with the appropriate secondary antibody. Slides were labeled with ABC reagent (Vector) (Ki-67 staining) and treated with DAB Chromagen (Dako) followed by counterstaining with Harris Hematoxylin. Terminal deoxynucleotidyl transferase mediated deoxyuridine triphosphate (dUTP) nick-end labeling (TUNEL) assay was performed using the ApopTag® Plus Peroxidase In Situ Apoptosis Kit from Millipore (Billerica, MA) according to the manufacturer’s instructions. A total of five 400× fields were counted from each mouse in the control and treated groups.
Statistical Analysis
Results are shown as mean ± SEM and were analyzed by Student’s t test or one-way ANOVA followed by Dunnett’s test. Difference between groups was considered to be statistically significant at P < 0.05.
RESULTS
PEITC Inhibits Growth and Reduces Colony Forming Ability of Pancreatic Cancer Cells
Effect of PEITC treatment on viability of pancreatic cancer cells was determined by trypan blue-dye exclusion assay using three well characterized pancreatic cancer cell lines (MIAPaca2, PL-45, and BxPC3) which differ with respect to their genetic mutations. Cells were treated with DMSO (control) or PEITC (2.5, 5, and 10 μmol/L) for 24 h. Exposure to PEITC potently decreased survival of pancreatic cancer cells in a dose-dependent manner with an IC50 of approximately 7 μmol/L (Fig. 1A). We further examined the efficacy of PEITC to inhibit the ability of pancreatic cancer cells to form viable colonies after removal of PEITC. As shown in Fig. 1B, 24-h PEITC treatment significantly reduced, in a dose-dependent manner, the number of viable colonies formed by MIAPaca2 cells following removal of PEITC. Similar results were obtained with PL-45 cells (data not shown).
FIGURE 1. PEITC inhibits growth and induces G2/M arrest in pancreatic cancer cells.
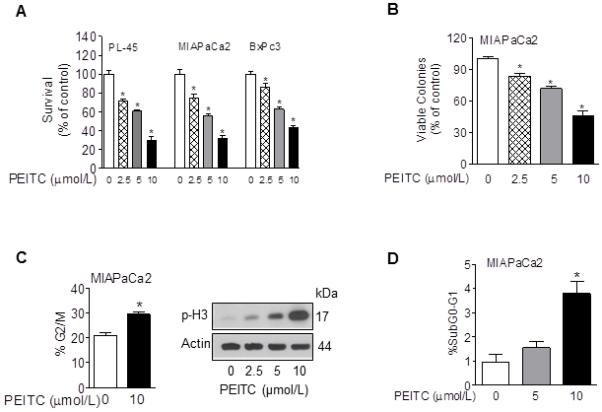
A, effect of PEITC on cell viability of pancreatic cancer cells after a 24-h exposure to DMSO or PEITC (2.5, 5, and 10 μmol/L) as determined by trypan blue dye exclusion assay; B, effect of PEITC on viable colonies of MIAPaca2 cells after a 24-h exposure to DMSO or PEITC (2.5, 5, and 10 μmol/L); C, left panel, percentage of cells in G2/M phase after a 24 h-exposure to DMSO or 10 μmol/L PEITC as determined by flow cytometry analysis; Columns, mean of two experiments performed in triplicate, Bars, SEM, *, P < 0.05. C, right panel, western immunoblotting for p-H3 using lysates from MIAPaca2 cells treated with DMSO or PEITC (2.5, 5, and 10 μmol/L) for 24 h; D, percentage of cells in SubG0/G1 phase after a 24 h-exposure to DMSO or PEITC as determined by flow cytometry analysis; Columns, mean of two experiments performed in triplicate, Bars, SEM, *, P < 0.05.
PEITC Induces G2/M Arrest in Pancreatic Cancer Cells
We further analyzed the mechanism of cell growth inhibition by PEITC in pancreatic cancer cells. We tested whether the inhibitory effect of PEITC on pancreatic cancer cells was associated with induction of cell cycle arrest. As shown in Fig. 1C, flow cytometry analysis showed that PEITC treatment (10 μmol/L) led to a statistically significant increase in the fraction of MIAPaca2 cells in G2/M phase compared with DMSO-treated (control) cells. The G2/M arrest was associated with a decrease in the percentage of cells in G0/G1 phase and an increase in the percentage of cells in SubG0/G1 and S phases (Fig. 1D and data not shown). We confirmed the mitotic arrest induced by PEITC by determining the levels of (Ser 10) phospho-histone H3, a marker for mitotic cells (24), via western blotting. As shown in Fig. 1C, PEITC treatment caused a dose-dependent increase in levels of phospho-histone H3 (Ser10) in PEITC-treated MIAPaca2 cells.
PEITC Induces Apoptosis in Pancreatic Cancer Cells
To examine the cell death induced by PEITC in pancreatic cancer cells we quantified the cytoplasmic histone-associated DNA fragmentation after exposure to DMSO (control) or PEITC (2.5, 5, and 10 μmol/L) for 24 h. As shown in Fig. 2A, PEITC at concentrations higher than 5 μmol/L induced apoptosis in pancreatic cancer cells. Exposure to 10 μmol/L PEITC increased the levels of histone-associated DNA fragmentation by 5 to 6 fold (Fig. 2A). The effect was independent of the Kras status of the cells (BxPC3 has wild type Kras while PL-45 and MIAPaca2 have mutated Kras). Moreover, flow cytometry analysis showed a significant increase in the fraction of subdiploid (apoptotic) cells after exposure to 10 μmol/L PEITC for 24 h (Fig. 1D).
FIGURE 2. PEITC induces apoptosis in pancreatic cancer cells.
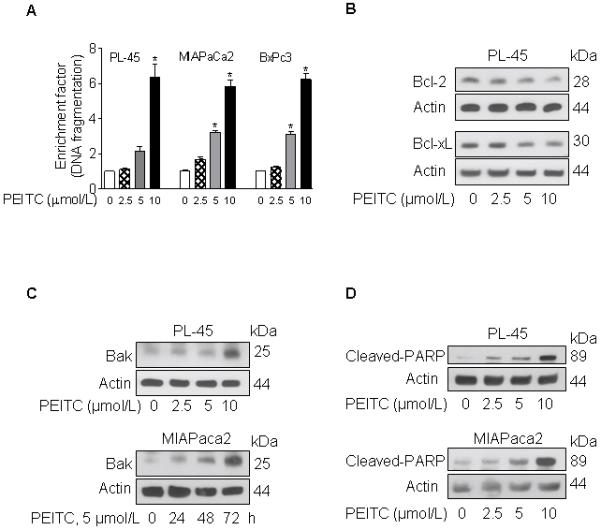
A, quantification of cytoplasmic histone-associated DNA fragmentation after a 24 h-exposure to DMSO or the indicated concentrations of PEITC; columns, mean of two experiments performed in triplicate, bars, SEM, *, P < 0.05. B, western blotting for Bcl-2 and Bcl-xL using lysates from PL-45 cells treated for 48 h with DMSO or PEITC (2.5, 5, and 10 μmol/L); C, western blotting for Bak using lysates from PL-45 cells exposed for 48 h to the indicated concentrations of PEITC or lysates from MIAPaca2 exposed to 5 μmol PEITC for the indicated time intervals; D, western blotting for cleaved-PARP using lysates from PL-45 or MIAPaca2 cells after exposure to the indicated concentrations of PEITC for 48 h.
PEITC Modulates Levels of Apoptosis Regulatory Proteins in Pancreatic Cancer Cells
To determine the effect of PEITC on levels of apoptosis regulatory proteins, PL-45 and MIAPaca2 cells were either treated with DMSO (control) or PEITC (2.5, 5, and 10 μmol/L) for 48 h, or with 5 μmol/L PEITC for various time intervals (24, 48, and 72 h). As shown in Fig. 2B-C, PEITC treatment decreased the levels of anti-apoptotic proteins Bcl-2 and Bcl-XL, and increased the levels of the pro-apoptotic protein Bak. In addition, exposure to PEITC led to cleavage of PARP in a dose-dependent manner (Fig. 2D).
PEITC Treatment Decreases Levels of Notch Proteins
Notch proteins are expressed during embryogenesis and play crucial roles in cell fate and differentiation (25). Expression of Notch proteins is absent or low in normal adult pancreas, but overexpression of Notch has been reported in pancreatic carcinogenesis (26,27). We tested whether PEITC treatment down-regulates the expression of Notch in pancreatic cancer cells. As shown in Fig. 3, PEITC treatment at concentrations higher than 5 μmol/L, decreased Notch 1 and Notch 2 levels in pancreatic cancer cells.
FIGURE 3. PEITC decreases levels of Notch proteins in pancreatic cancer cells.
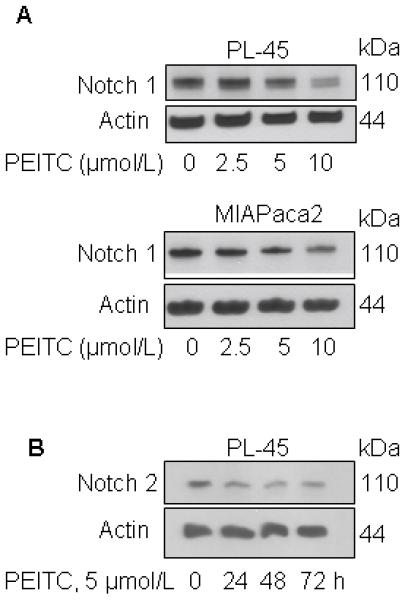
A, western blotting for Notch 1 using lysates from PL-45 or MIAPaca2 cells exposed to DMSO or PEITC (2.5, 5, and 10 μmol/L) for 48 h. B, western blotting for Notch 2 using lysates from PL-45 cells exposed to 5 μmol/L PEITC for the indicated time intervals.
PEITC Inhibits MIAPaca2 Tumor Growth in a Xenograft Animal Model Without Showing Signs of Toxicity
To test the in vivo efficacy of PEITC, MIAPaca2 tumors were established in athymic nude mice. As shown in Fig. 4A, tumors in control (vehicle-treated) mice reached 673.8 ± 118.1 mm3 (mean ± SEM) at 7 weeks after injection. In contrast, mice treated with PEITC (12 μmol/day, 5 days/week) exhibited delayed tumor growth, with 37% lower tumor volume compared with vehicle-treated mice at 7 weeks (P = 0.0255). The inhibitory effect of PEITC was also seen in average wet tumor weights from PEITC-treated mice, which were 33% lower than those of control mice (Fig. 4B, P = 0.0567). PEITC treatment was well tolerated and all mice gained weight throughout the study, with no statistically significant difference between groups (Fig. 4C). In addition, PEITC treatment did not result in toxicity or enlargement of any organs (Fig. 4D).
FIGURE 4. PEITC inhibits MIAPaca2 tumor growth in a xenograft animal model without showing signs of toxicity.
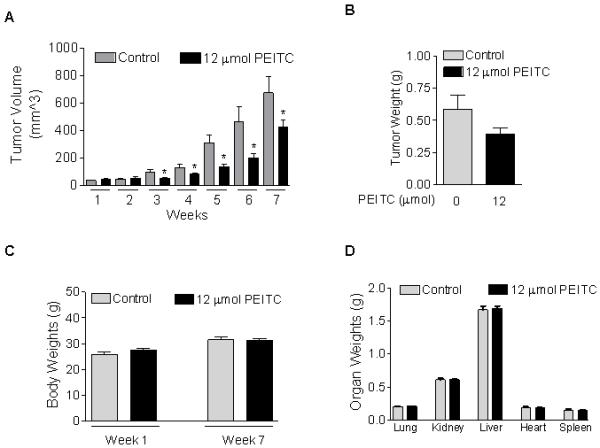
A, effect of PEITC on tumor volume in vehicle-treated (control) mice and PEITC-treated mice. B, effect of PEITC on tumor weights in control mice and PEITC-treated mice; C, effect of PEITC treatment on body weights; D, effect of PEITC treatment on organ weights; Control, n=8; PEITC, n=10; *, P < 0.05.
PEITC Reduces Proliferation and Induces Apoptosis of Pancreatic Cancer Cells in Vivo
Tumor sections were stained for the cell proliferation markers Ki-67 and PCNA. As shown in Fig. 5A-B, PEITC treatment decreased the number of Ki-67-positive cells in tumors from PEITC-treated mice compared with tumors from control mice. Quantification of Ki-67-positive cells per high power field showed that PEITC treatment reduced the number of Ki-67-positive cells by 55% compared with control mice. Moreover, PEITC treatment decreased PCNA expression in tumors by 42% compared with control (Fig. 5C-D). In addition, treatment of mice with 12 μmol PEITC provided a small (23%) but statistically significant increase in the number of TUNEL-positive cells compared with the control mice (Fig. 5E-F).
FIGURE 5. PEITC reduces proliferation and induces apoptosis of pancreatic cancer cells in vivo.
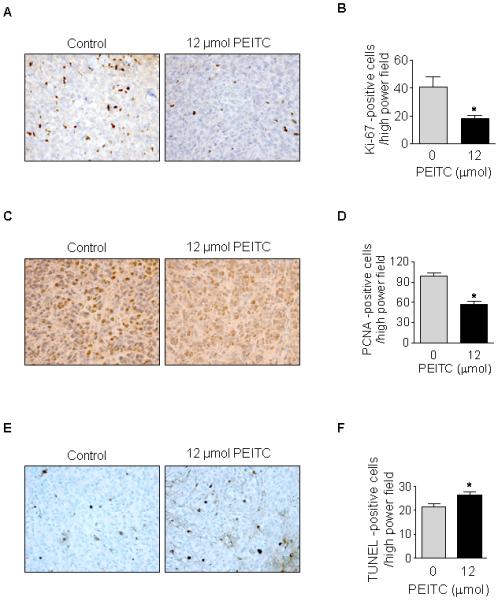
A, Ki-67 staining of tumors from vehicle-treated (control) mice and PEITC-treated mice. B, quantification of Ki-67-positive cells/high power field after scoring five 400× fields/mouse; C, PCNA staining of tumors from control mice and PEITC-treated mice; D, quantification of PCNA-positive cells/high power field after scoring five 400× fields/mouse; E, TUNEL staining of tumors from control mice and PEITC treated mice; F, quantification of TUNEL-positive cells/high power field in tumors from control mice and PEITC-treated mice after scoring five 400× fields/mouse; *, P < 0.05.
DISCUSSION
Our study demonstrates that PEITC inhibits cell growth and induces apoptosis of pancreatic cancer cells regardless of their Kras status. PEITC delays cell cycle progression of pancreatic cancer cells by arresting the cells in G2/M phase and modulates the levels of Bcl-2 family proteins. In addition, we observed that PEITC induces cleavage of PARP and DNA fragmentation in a dose-dependent manner and decreases levels of Notch 1 and 2 in human pancreatic cancer cells.
These findings are significant in several respects. Overexpression of Bcl-xL has been reported in pancreatic cancer and associated with a worse prognosis (28-30), suggesting that agents targeting Bcl-xL might prove valuable for pancreatic cancer prevention and treatment. Bak is a pro-apoptotic member of the Bcl-2 family proteins located on the outer membrane of the mitochondria; following a conformational change, Bak interacts with Bcl-xL (31) and participates in the formation of channels in the mitochondrial membrane that lead to the release of apoptogenic molecules. In this study, we show that PEITC induces apoptosis in pancreatic cancer cells by down-regulating levels of Bcl-2 and Bcl-xL and up-regulating levels of Bak. The release of cytochrome c to the cytosol activates the initiator and executioner caspases and eventually leads to cleavage of PARP and cell death (32). Cleavage of PARP is a marker of cells undergoing apoptosis and shows the completion and irreversibility of the process (33).
Similarly, activation of the Notch pathway can lead to cell proliferation (34) and inhibition of apoptosis (35). In vertebrates, the Notch family of proteins comprises four receptors (Notch 1-4). Notch pathway plays an important role in cell fate determination and differentiation (25,36), being active during early pancreatic development but normally inactive in adult pancreas cells (27). Re-activation of the Notch signaling pathway has been reported in pancreatic carcinogenesis (26) and overexpression of Notch 1 and Notch 2 receptors has been shown in PanIN lesions and pancreatic cancer (27), suggesting that inhibition of Notch pathway may be a novel target for pancreatic cancer prevention and treatment. In this study we show that PEITC treatment down-regulates the expression of Notch 1 and 2 receptors in pancreatic cancer cells. However, the contribution of Notch receptors down-regulation to the overall effect on pancreatic carcinogenesis needs further investigation.
In addition, we found that oral gavage of 12 μmol PEITC/day, 5 days/week, significantly inhibits tumor volume in a MIAPaca2 xenograft animal model without showing signs of toxicity. We did observe that the inhibitory effect of PEITC on pancreatic tumors lessened once the tumors grew larger, suggesting that PEITC might be more effective as a pancreatic cancer chemopreventive agent than as a therapeutic agent. Tumors from PEITC-treated mice showed reduced proliferation and increased apoptosis compared with tumors from control mice. Reduced proliferation was demonstrated through suppression of the proliferation markers Ki-67 and PCNA. One study has shown that lower levels of Ki-67 were associated with a better outcome in pancreatic cancer patients (37), although other studies did not find an association (38,39). The predictive value of Ki-67 was reinforced by recent studies that showed better survival or decreased tumor volume with lower levels of Ki-67 (40,41). We further confirmed the anti-proliferative effect of PEITC by PCNA staining which has been reported to correlate positively with the histological grade of pancreatic cancer (42).
Micromolar concentrations of PEITC can be achieved in vivo. For example, Ji et al. showed that the maximal plasma concentration (Cmax) of PEITC in rats reached 9.2 ± 0.6 and 42.1 ± 11.4 μmol/L after oral doses of 10 and 100 μmol/kg, respectively (43). ITCs appear to accumulate in cells by diffusion and can attain high intracellular levels reaching millimolar range (44). The bioavailability of PEITC is dependent of dose and frequency of intake (44). Following a single oral administration, the bioavailability of PEITC in rats was 77% at a dose of 0.5 mg/kg and decreased to 23% at a dose of 5.0 mg/kg (45). Intracellularly, ITCs interact with thiols such as glutathione and are metabolized through the mercapturic acid pathway to form N-acethylcysteine-conjugates which are excreted in urine (17). There is limited data on PEITC pharmacokinetics in humans. One study found that Cmax of PEITC in plasma from four volunteers who ingested 100 g of watercress ranged from 673.4 to 1155.0 nmol/L (mean, 928 ± 250.7 nmol/L) with tmax of 2.6 ± 1.1 h (45). Another study determined that the Cmax in plasma from three volunteers who ingested a single 40 mg dose of PEITC ranged from 0.64 to 1.4 μmol/L (mean 1.04 ± 0.22 μmol/L) with a tmax of 4.6 ± 0.7 h (47).
Novel chemoprevention strategies for pancreatic cancer are highly sought due to the high lethality of pancreatic cancer (6). PEITC is a promising cancer chemopreventive agent that has shown pre-clinical efficacy against different malignancies and is currently in clinical trials for lung cancer and lymphoproliferative disorders (21). PEITC has been reported to selectively target oncogenically transformed cells and cancer cells and similar concentrations did not show toxicity on normal human prostate, mammary or ovarian epithelial cells (19,48,49).
In summary, we have demonstrated that PEITC inhibits proliferation of pancreatic cancer cells in vitro and in vivo and induces G2/M phase cell cycle arrest and apoptosis in pancreatic cancer cells. Our data support further investigation of PEITC as a chemopreventive agent for pancreatic cancer, including more specific chemoprevention models for pancreatic cancer.
ACKNOWLEDGMENTS
This investigation was supported in part by the Wayne Fusaro Pancreatic Cancer Research Fund (awarded to DCW), the Shirley Hobbs Martin Memorial Fund (awarded to REB), and the NCI grant CA101753-08 (awarded to SVS). The authors thank Michelle Kienholz for editorial assistance.
REFERENCES
- 1.Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 2006;66:95–106. doi: 10.1158/0008-5472.CAN-05-2168. [DOI] [PubMed] [Google Scholar]
- 2.Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 3.Maitra A, Fukushima N, Takaori K, Hruban RH. Precursors to invasive pancreatic cancer. Adv Anat Pathol. 2005;12:81–91. doi: 10.1097/01.pap.0000155055.14238.25. [DOI] [PubMed] [Google Scholar]
- 4.Siegel R, Naishadham D, Jemal A. Cancer Statistics, 2012. CA Cancer J Clin. 2012;60:277–300. [Google Scholar]
- 5.Yachida S, Jones S, Bozic I, Antal T, Leary R, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;46:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stan SD, Singh SV, Brand RE. Chemoprevention strategies for pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2010;7:347–356. doi: 10.1038/nrgastro.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olsen GW, Mandel JS, Gibson RW, Wattenberg LW, Schuman LM. A case-control study of pancreatic cancer and cigarettes, alcohol, coffee, and diet. Am J Public Health. 1989;79:1016–1019. doi: 10.2105/ajph.79.8.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verhoeven DT, Goldbohm RA, van Poppel G, Verhagen H, van den Brandt PA. Epidemiological studies on Brassica vegetables and cancer risk. Cancer Epidemiol Biomarkers Prev. 1996;5:733–748. [PubMed] [Google Scholar]
- 9.Chan JM, Wang F, Holly EA. Vegetable and fruit intake and pancreatic cancer in a population-based case-control study in the San Francisco bay area. Cancer Epidemiol Biomarkers Prev. 2005;14:2093–2097. doi: 10.1158/1055-9965.EPI-05-0226. [DOI] [PubMed] [Google Scholar]
- 10.Higdon JV, Delage B, Williams DE, Dashwood RH. Cruciferous vegetables and human cancer risk: epidemiological evidence and mechanistic basis. Pharmacol Res. 2007;5:224–236. doi: 10.1016/j.phrs.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silverman DT, Swanson CA, Gridley G, Wacholder S, Greenberg RS, et al. Dietary and nutritional factors and pancreatic cancer: a case-control study based on direct interviews. J Natl Cancer Inst. 1998;90:1710–1719. doi: 10.1093/jnci/90.22.1710. [DOI] [PubMed] [Google Scholar]
- 12.Larsson SC, Hakansson N, Naslund I, Bergkvist L, Wolk A. Fruit and Vegetable consumption in relation to pancreatic cancer risk: a prospective study. Cancer Epidemiol Biomarkers Prev. 2006;15:301–305. doi: 10.1158/1055-9965.EPI-05-0696. [DOI] [PubMed] [Google Scholar]
- 13.Hecht SS. Inhibition of carcinogenesis by isothiocyanates. Drug Metab Rev. 2000;32:395–411. doi: 10.1081/dmr-100102342. [DOI] [PubMed] [Google Scholar]
- 14.Hayes JD, Kelleher MO, Eggleston IM. The cancer chemopreventive actions of phytochemicals derived from glucosinolates. Eur J Nutr. 2008;47(Suppl 2):73–88. doi: 10.1007/s00394-008-2009-8. [DOI] [PubMed] [Google Scholar]
- 15.Powolny AA, Bommareddy A, Hahm ER, Normolle DP, Beumer JH, et al. Chemopreventative potential of the cruciferous vegetable constituent phenethyl isothiocyanate in a mouse model of prostate cancer. J Natl Cancer Inst. 2011;103:571–584. doi: 10.1093/jnci/djr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2010;12:87–97. doi: 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y. Cancer-preventive isothiocyanates: measurement of human exposure and mechanism of action. Mutat Res. 2004;555:173–190. doi: 10.1016/j.mrfmmm.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 18.Stan SD, Kar S, Stoner GD, Singh SV. Bioactive food components and cancer risk reduction. J Cell Biochem. 2008;104:339–356. doi: 10.1002/jcb.21623. [DOI] [PubMed] [Google Scholar]
- 19.Xiao D, Powolny AA, Moura MB, Kelley EE, Bommareddy A, et al. Phenethyl isothiocyanate inhibits oxidative phosphorylation to trigger reactive oxygen species-mediated death of human prostate cancer cells. J Biol Chem. 2010;285:26558–26569. doi: 10.1074/jbc.M109.063255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bommareddy A, Hahm ER, Xiao D, Powolny AA, Fisher AL, et al. Atg5 regulates phenethyl isothiocyanate-induced autophagic and apoptotic cell death in human prostate cancer cells. Cancer Res. 2009;69:3704–3712. doi: 10.1158/0008-5472.CAN-08-4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. [accessed 2/25/2011]; www.clinicaltrials.gov. clinicaltrials.gov Identifiers: NCT00968461, NCT00005883, and NCT00691132.
- 22.Nishikawa A, Furukawa F, Uneyama C, Ikezaki S, Tanakamaru, et al. Chemopreventive effects of phenethyl isothiocyanate on lung and pancreatic tumorigenesis in N-nitrosobis(2-oxopropyl)amine-treated hamsters. Carcinogenesis. 1996;17:1381–1384. doi: 10.1093/carcin/17.6.1381. [DOI] [PubMed] [Google Scholar]
- 23.Stan SD, Zeng Y, Singh SV. Ayurvedic medicine constituent withaferin A causes G2 and M phase cell cycle arrest in human breast cancer cells. Nutr Cancer. 2008;60(S1):51–60. doi: 10.1080/01635580802381477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, et al. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 25.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 26.Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, et al. Notch mediates TGF α-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3:565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 27.Mysliwiec P, Boucher MJ. Targeting Notch signaling in pancreatic cancer patients-rationale for new therapy. Adv Med Sci. 2009;54:136–142. doi: 10.2478/v10039-009-0026-3. [DOI] [PubMed] [Google Scholar]
- 28.Miyamoto Y, Hosotani R, Wada M, Lee JU, Koshiba T, et al. Immunohistochemical analysis of Bcl-2, Bax, Bcl-X, and Mcl-1 expression in pancreatic cancers. Oncology. 1999;56:73–82. doi: 10.1159/000011933. [DOI] [PubMed] [Google Scholar]
- 29.Hinz S, Trauzold A, Boenicke L, Sandberg C, Beckmann S, et al. Bcl-XL protects pancreatic adenocarcinoma cells against CD95-and TRAIL-receptor-mediated apoptosis. Oncogene. 2000;19:5477–5486. doi: 10.1038/sj.onc.1203936. [DOI] [PubMed] [Google Scholar]
- 30.Friess H, Lu Z, Andren-Sandberg A, Berberat P, Zimmermann A, et al. Moderate activation of the apoptosis inhibitor Bcl-xL worsens the prognosis in pancreatic cancer. Ann Surg. 1998;228:780–787. doi: 10.1097/00000658-199812000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gross A, McDonnell JM, Korsmeyer SJ. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 32.Cain K, Brown DG, Langlais C, Cohen GM. Caspase activation involves the formation of the aposome, a large (approximately 700 kDa) caspase-activating complex. J Biol Chem. 1999;274:22686–22692. doi: 10.1074/jbc.274.32.22686. [DOI] [PubMed] [Google Scholar]
- 33.Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, et al. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 34.Go MJ, Eastman DS, Artavanis-Tsakonas S. Cell proliferation control by Notch signaling in Drosophila development. Development. 1998;125:2031–2040. doi: 10.1242/dev.125.11.2031. [DOI] [PubMed] [Google Scholar]
- 35.Shelly LL, Fuchs C, Miele L. Notch-1 inhibits apoptosis in murine erythroleukemia cells and is necessary for differentiation induced by hybrid polar compounds. J Cell Biochem. 1999;73:164–175. doi: 10.1002/(sici)1097-4644(19990501)73:2<164::aid-jcb3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 36.Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, et al. Rational targeting of Notch in cancer. Oncogene. 2008;27:5124–5131. doi: 10.1038/onc.2008.226. [DOI] [PubMed] [Google Scholar]
- 37.Ferrara C, Tessari G, Poletti A, Giacon C, Meggiato T, et al. Ki-67 and c-jun expression in pancreatic cancer: a prognostic marker? Oncol Rep. 1999;6:1117–1122. doi: 10.3892/or.6.5.1117. [DOI] [PubMed] [Google Scholar]
- 38.Stanton KJ, Sidner RA, Miller GA, Cummings OW, Schmidt CM, et al. Analysis of Ki-67 antigen expression, DNA proliferative fraction, and survival in resected cancer of the pancreas. Am J Surg. 2003;186:486–492. doi: 10.1016/j.amjsurg.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 39.Lebe B, Sagol O, Ulukus C, Coker A, Karademir S, et al. The importance of cyclin D1 and Ki-67 expression on the biological behavior of pancreatic adenocarcinomas. Pathol Res Pract. 2004;200:389–396. doi: 10.1016/j.prp.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 40.Karamitopoulou E, Zlobec I, Tornillo L, Carafa V, Schaffner T, et al. Differential cell cycle and proliferation marker expression in ductal pancreatic adenocarcinoma and pancreatic intraepithelial neoplasia (PanIN) Pathology. 2010;42:229–234. doi: 10.3109/00313021003631379. [DOI] [PubMed] [Google Scholar]
- 41.Aloysius MM, Hewavisenthi SJ, Bates TE, Rowlands BJ, Lobo DN, et al. Predictive value of tumor proliferation indices in periampullary cancers: Ki-67, mitotic activity index (MI) and volume corrected mitotic index (M/V) using tissue microarrays. World J Surg. 2010;34:2115–2121. doi: 10.1007/s00268-010-0681-3. [DOI] [PubMed] [Google Scholar]
- 42.Dang CX, Han Y, Qin ZY, Wang YJ. Clinical significance of expression of p21 and p53 proteins and proliferating cell nuclear antigen in pancreatic cancer. Hepatobiliary Pancreat Dis Int. 2002;1:302–305. [PubMed] [Google Scholar]
- 43.Ji Y, Kuo Y, Morris ME. Pharmacokinetics of dietary phenethyl isothiocyanate in rats. Pharm Res. 2005;22:1658–1666. doi: 10.1007/s11095-005-7097-z. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Talalay P. Mechanism of differential potencies of isothiocyanates as inducers of anticarcinogenic Phase 2 enzymes. Cancer Res. 1998;58:4632–4639. [PubMed] [Google Scholar]
- 45.Konsue N, Kirkpatrick J, Kuhnert N, King LJ, Ioannides C. Repeated oral administration modulates the pharmacokinetic behavior of the chemopreventive agent phenethyl isothiocyanate in rats. Mol Nutr Food Res. 2010;54:426–432. doi: 10.1002/mnfr.200900090. [DOI] [PubMed] [Google Scholar]
- 46.Ji Y, Morris ME. Determination of phenethyl isothiocyanate in human plasma and urine by ammonia derivatization and liquid chromatography-tandem mass spectrometry. Anal Biochem. 2003;323:39–47. doi: 10.1016/j.ab.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Liebes L, Conaway CC, Hochster H, Mendoza S, Hecht SS, et al. High-performance liquid chromatography-based determination of total isothiocyanate levels in human plasma: application to studies with 2-phenethyl isothiocyanate. Anal Biochem. 2001;291:279–289. doi: 10.1006/abio.2001.5030. [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Govind S, Sajankila SP, Mi L, Roy R, et al. Phenethyl isothiocyanate sensitizes human cervical cancer cells to apoptosis induced by cisplatin. Mol Nutr Food Res. 2011;55:1572–1581. doi: 10.1002/mnfr.201000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]