

Abstract
Increased understanding of inter-tumoral heterogeneity at the genomic level has led to significant advancements in the treatment of solid tumors. Functional genomic alterations conferring sensitivity to targeted therapies can take many forms, and appropriate methods and tools are needed to detect these alterations. This review provides an update on genetic variability among solid tumors of similar histologic classification, using non-small cell lung cancer (NSCLC) and melanoma as examples. We also discuss relevant technological platforms for discovery and diagnosis of clinically actionable variants and highlight the implications of specific genomic alterations for response to targeted therapy.
Keywords: Lung cancer, melanoma, tumor genotyping, personalized medicine, next-generation sequencing
Introduction
The disease ‘cancer’ is in reality a multitude of disease entities. When restricted to a single anatomical site of origin, tumors exhibit a high degree of clinical and histopathological heterogeneity. At the molecular level, such heterogeneity is even more complex. Over the past decade, classification of solid tumors has rapidly shifted from one based on histologic and anatomic characteristics to one increasingly incorporating genomic data. Compared to historical treatment with chemotherapy, targeted therapies specific for certain molecular subtypes of solid tumors have led to increased progression-free and overall survival for patients with metastatic disease, showing that genotype-directed targeted therapies hold significant promise for patients (1–4). Herein, we highlight the ways in which the depth and breadth of available genomic information has allowed for a greater understanding of the diversity of genetic profiles in human cancers. Specifically, we discuss the genetic heterogeneity that exists among large cohorts of solid tumors with common histological classifications and the implications of such variability for response to targeted therapeutics, with a focus on non-small cell lung cancer (NSCLC) and melanoma. The concept of intra-tumoral heterogeneity as well as heterogeneity among multiple lesions within one patient (a type of inter-tumoral heterogeneity) and implications for targeted therapies have been addressed elsewhere and are outside the scope of this review (5, 6).
Genetic alterations and driver mutations in solid tumors
Since the original discovery that oncogenes were mutated forms of normally expressed genes in human cells (7), somatic genomic alterations have been recognized as causative in the initiation and progression of cancer. These genetic changes can take many forms, including point mutations, insertions, deletions, combined insertions/deletions (indels), duplications, inversions, and translocations. In some cases, tumors harbor these mutations in oncogenes (in particular, tyrosine kinases and serine-threonine kinases), which render them exquisitely sensitive to targeted small-molecule inhibitors (Figure 1) (8). Despite the enormous genetic complexity present in the tumor, these specific genomic alterations, or ‘driver mutations’, cause tumors to become ‘oncogene addicted’, or ultimately reliant on specific signaling pathways such that inhibition of those pathways results in cell death (8).
Figure 1. Examples of types of ‘driver’ genomic alterations found in cancer.

Schematic representations of mutations known to occur in NSCLC, including point mutations, insertions/deletions, copy number variants, and structural variants. A, single nucleotide variant in exon 21 of EGFR (c.2573 T>G) encoding a substitution of arginine for leucine at codon 858 (L858R), and B, combined insertion/deletion (indel) in exon 19 of EGFR confer sensitivity to EGFR TKIs. Red indicates a nucleotide or amino acid that has been altered in the mutant form. C, amplification of MET and D, structural variants resulting in EML4-ALK fusions confer sensitivity to ALK TKIs.
With the recent explosion of available next-generation sequencing (NGS) technologies, we are now able to detect the whole spectrum of somatic genomic alterations in cancers using a limited number of assays and minimal amounts of tissue. However, because a solid tumor may have up to 400 mutations per megabase (Mb) (9), the task of distinguishing ‘driver’ (causative) versus ‘passenger’ (non-functional) mutations from the pool of somatic mutations observed in tumor genomes is not trivial. Thus, the most challenging task in the identification of targetable oncogenic ‘drivers’ is the integration of the diverse range of available genomic data into biologically and clinically relevant information.
In order to begin to discern potentially functional genomic alterations from the myriad of mutations and structural variants present in solid tumors, large sequencing efforts have been initiated that provide greater statistical power for discovering genomic alterations of biological importance. One such example is The Cancer Genome Atlas (TCGA), which is an initiative sponsored by the National Institutes of Health (NIH) that aims to catalog systematically genetic changes occurring in more than twenty types of human cancers, including NSCLC and melanoma (10). This analysis is made possible by the availability of fresh frozen surgically resected specimens and matched blood samples, which in most cases provide more than enough tissue for multi-platform analysis of somatic alterations at the DNA, RNA, or protein level. A relevant consideration for clinical application of widespread sequencing efforts is the limited amount and variability in quality of available tumor tissue (usually formalin-fixed and paraffin-embedded). This, along with cost of testing, issues around reimbursement policies, and the bioinformatics expertise necessary for interpretation of results are current barriers to the feasibility of translating certain genomics-based assays into the clinic.
Diagnostic platforms for molecular classification of tumors in the clinic
Despite the challenges, development of new and updated platforms for detection of single nucleotide variants (SNVs), copy number variants (CNVs), and structural variants (SVs) with minimal amounts of input genetic material is rapidly evolving. Emerging sequencing technologies have been thoroughly reviewed elsewhere (11–14); here, we discuss available technologies for molecular profiling of tumors for clinical decision making (Table 1). Notably, treating physicians need to know the strengths and limitations of the tumor profiling assays that they order for their patients.
Table 1.
Types of clinical molecular tests and variants detected.
| Molecular Methodology | Variant Types | |||
|---|---|---|---|---|
| SNVs | Small duplications, insertions, deletions, indels | Exon duplications, deletions or gene copy number changes | SVs | |
| Allele-specific PCR | ✓ | |||
| PCR and Sanger dideoxy sequencing | ✓ | ✓ | a | |
| PCR and pyrosequencing | ✓ | • | ||
| PCR and MS | ✓ | • | ||
| PCR and single base extension | ✓ | |||
| MLPA | ✓ | ✓ | ||
| FISH | b | ✓ | ||
| NGS – custom panels (amplicon capture) | ✓ | ✓ | ||
| NGS – custom panels (hybridization capture) | ✓ | ✓ | ✓ | • |
| NGS – whole exome sequencing | ✓ | ✓ | ✓ | • |
| NGS – whole genome sequencing | ✓ | ✓ | ✓ | ✓ |
✓ Variant detected. • Variant detected with difficulty.
Variant detected if fusion RNA is extracted first.
Variant in gene copy number only.
FISH = fluorescence in situ hybridization; indels = mutations including both insertions and deletions; MLPA= multiplex ligation-dependent probe amplification; MS = mass spectrometry; NGS = next-generation sequencing; PCR = polymerase chain reaction; SNVs = single nucleotide variants; SVs = structural variants. Adapted from Vnencak-Jones et al. [97]. “Types of Molecular Testing.” My Cancer Genome, http://www.mycancergenome.org/content/other/molecular-medicine/types-of-molecular-tumor-testing. © Copyright 2013 Vanderbilt University.
For SNVs and small insertions, deletions, or indels, PCR followed by dideoxynucleotide sequencing remains a cost-effective, reliable method for detection of known variants. However, direct sequencing is low-throughput as well as limited in its sensitivity, detecting only variant alleles present at a frequency of at least 20–25%. By contrast, multiplexed assays such as SNaPshot and Sequenom mass ARRAY can query already known mutations in several genes at once, detecting variant alleles present at frequencies as low as 1.56% (15–18). NGS, in the form of targeted/custom panels, whole exome sequencing (WES), or whole genome sequencing (WGS) offers deep coverage (i.e. high sensitivity) and the highest possible throughput in terms of detecting many somatic SNVs, small insertions and/or deletions at once. However, the use of NGS does not necessarily imply comprehensiveness; for example, the Illumina Truseq Amplicon Cancer Panel (TSACP), a multiplexed amplicon-based targeted re-sequencing assay that encompasses a panel of cancer-associated genes, interrogates only specific exons and may therefore miss detection of certain novel mutations in other locations. Capture-based targeted re-sequencing methods have similar drawbacks; thus, data outputs from these assays must be carefully interpreted and not assumed to be exhaustive in their detection of potentially functional genomic alterations.
While fluorescence in situ hybridization (FISH) and multiplex ligation-dependent probe amplification (MLPA) remain the clinical standard for targeted detection of CNVs, NGS technologies afford higher throughput and unbiased detection of CNVs. Finally, NGS, in particular WGS, provides a mechanism for genome-wide detection of structural variants, eliminating the need for previous knowledge of potential fusions or input of RNA (more easily degraded and thus logistically more difficult to obtain and preserve on a large scale).
Management, analysis, and reporting of NGS data back to the treating physician all remain significant hurdles to widespread adoption of NGS technologies, but algorithms for more automated and accurate variant calling are improving. A relevant consideration regarding the practicality and ethics of NGS is the question of ‘how much is too much?’ These platforms are invaluable for discovery, but adoption into routine patient care will require careful stewardship and meticulous, integrated analysis of these large datasets.
Routine molecular subtyping of solid tumors by known driver mutations
Although many tumors have a broad spectrum of genetic alterations, some of these changes already have well-understood implications for existing and emerging targeted therapies. In these cases, translation to the clinic can be achieved by the design of targeted molecular genotyping assays that are accurate, sensitive, timely, and cost-effective for cancer patients. Indeed, many such academic, commercial, and government-sponsored targeted genotyping efforts are in progress at centers all over the world. For example, in 2009 the French National Cancer Institute (NCI) implemented a program to provide free tumor genotyping for certain cancer subtypes in 28 public hospitals across the country. The Lung Cancer Mutation Consortium (LCMC) is a similar effort at 14 academic institutions across the United States that was founded with the goal of increasing genotype-driven therapy for lung cancer while laying the foundation for widespread implementation of tumor genotyping across the country (19). Encouragingly, the multitude of clinical tumor genotyping efforts currently underway are too numerous to name, and they are generating an enormous amount of data about the genetic landscape of solid tumors while also facilitating access of patients to personalized cancer therapy.
Here, as a representative use case, we present updated results of molecular profiling from the Vanderbilt Personalized Cancer Medicine Initiative (PCMI). We implemented SNaPshot analysis at the Vanderbilt-Ingram Cancer Center in July, 2010, for routine stratification of NSCLCs and melanomas into clinically relevant molecular subtypes (15, 20). Genotyping is performed on DNA extracted from formalin fixed, paraffin-embedded (FFPE) specimens to assay for common (1% or more frequency in the Catalog of Somatic Mutations in Cancer [COSMIC]) somatic mutations across multiple cancer-associated genes. Together, these SNaPshot panels, which can detect ~40 mutations in 6–9 genes, have been used to inform treatment decisions for nearly 2,000 cancer patients at Vanderbilt (as of August 2013). The data generated from these panels is stored in structured format in the permanent electronic health record and also in the Vanderbilt Research Derivative (RD) for consented patients. The RD is a comprehensive data warehouse of over 3,000,000 individual patient records that is updated in near-real-time, enabling automated collection and reporting of SNaPshot information.
Using SNaPshot data to inform clinical care: NSCLC and melanoma as model systems
NSCLCs are histologically subdivided into adenocarcinomas, squamous cell carcinomas, and large cell carcinomas. Previously lumped together clinically (21), NSCLCs have been shown to harbor recurrent alterations in multiple oncogenes. Among 1,003 NSCLC specimens (predominantly adenocarcinoma) genotyped at Vanderbilt University Medical Center (VUMC) between 2010–2013, 424 harbored known driver mutations, including KRAS (22.9%), EGFR (14.8%), PIK3CA (2.1%), BRAF (1.9%), ERBB2 (0.9%), MEK1 (0.8%), NRAS (0.5%), AKT1 (0.3%), and PTEN (0.2%) (Figure 2A). This breakdown is similar to what has been published in the literature for each of these mutations. Testing for fusions, such as those involving ALK and in some cases, ROS1 and RET, was performed separately. Such ‘driver’ mutations are typically mutually exclusive and serve as a mechanism by which tumors can be sub-classified regarding their likelihood of response to pharmacological inhibition of their activated pathways.
Figure 2. Routine molecular subtyping of non-small cell lung cancers and melanomas.

Frequency of mutations identified by SNaPshot genotyping through Vanderbilt’s PCMI from July 2010 to August 2013. A, frequency of lung-cancer associated mutations. 34 lung tumor specimens contained multiple mutations in the genes listed. *Note: ALK FISH performed separately. B, spectrum of mutations identified in EGFR. 27 lung tumor specimens contained multiple mutations in EGFR. C, frequency of melanoma-associated mutations. 23 melanoma specimens contained multiple mutations in the genes listed. D, spectrum of mutations identified in BRAF. 1 melanoma specimen contained multiple mutations in BRAF. N = number of tested specimens containing at least one mutation in the genes listed. Frequencies are listed as percentages of total mutations identified.
However, even among molecular subsets defined for NSCLCs, heterogeneity exists (Figure 2B). For example, mutations in EGFR are known to be present in 10–35% of NSCLCs (most frequently in adenocarcinomas from former light smokers or never smokers—i.e., fewer than 100 cigarettes over a lifetime) and confer sensitivity to the EGFR TKIs erlotinib, gefitinib, and afatinib (22–25) (Table 2). The majority of these activating mutations in EGFR occur either as multi-nucleotide in-frame deletions in exon 19 or as single-nucleotide substitutions (L858R) in exon 21; less frequent lung cancer-associated EGFR mutations that also confer TKI sensitivity include G719A/G719C/G719S (exon 18) and L861Q (exon 21) (26) (Figure 2B). The EGFR T790M mutation (exon 20) is present in 50% of EGFR-mutant tumors with acquired resistance to first-generation TKIs (erlotinib and gefitinib) and may be a biomarker of sensitivity to third-generation TKIs such as C0-1686 and AZD9291(27–29).
Table 2.
Alterations in signaling enzymes known to be associated with sensitivity to available targeted therapies.
| Kinase-targeted therapies in NSCLC | ||||
|---|---|---|---|---|
| Oncogene | Targeted therapy | Level of evidence | Associated publications | |
| ALK fusions | Crizotinib | Phase III trial | Shaw et al. [36] | |
| IPI-504 | Phase II trial | Sequist et al. [52] | ||
| Ganetespib | Phase II trial | Socinski et al. [79] | ||
| LDK378 | Phase I trial | Shaw et al. [77] | ||
| AP26113 | Phase I trial | Camidge et al. [78] | ||
| CH5424802 | Phase I/II trial | Seto et al. [84] | ||
| ALK | Fusion + G1269A | Ganetespib | Case report | Sang et al. [49] |
| BRAF | Y472C | Dasatinib | Case report | Sen et al. [37] |
| V600E | Vemurafenib | Case report | Gautschi et al. [53] | |
| Dabrafenib | Case report | Rudin et al. [38] | ||
| DDR2 | S768R | Dasatinib | Case report | Pitini et al. [39] |
| Dasatinib + erlotinib | Case report | Hammerman et al. [54] | ||
| EGFR | Exon19del/L858R | Gefitinib | Phase III trial | Mok et al. [3] Mitsudomi et al. [40] Maemondo et al. [80] |
| Erlotinib | Phase III trial | Zhou et al. [4] Rosell et al. [41] |
||
| Afatinib | Phase III trial | Sequist et al. [25] | ||
| Dacomitinib | Phase II trial | Ramalingam et al. [42] | ||
| Exon 19ins | Erlotinib Gefitinib Afatinib |
Retrospective analysis | He et al. [51] | |
| G719A/S | Neratinib | Phase II trial | Sequist et al. [43] | |
| L861Q | Gefitinib Erlotinib |
Case reports | Yeh et al. [50] | |
| T790M | CO-1686 | Phase I trial | Sequist et al. [86] | |
| AZD9291 | Phase I trial | Ranson et al. [29] | ||
| ERBB2 | Exon20ins/G776L | Afatinib | Phase II trial | De Greve et al. [55] |
| Trastuzumab + paclitaxel | Case report | Cappuzzo et al. [56] | ||
| Trastuzumab + chemo | Retrospective analysis | Mazieres et al. [57] | ||
| FGFR1 | Amplification | BGJ398 | Case report | Malchers et al. [81] |
| KRAS |
a G12A/R/D/C/S/V a G13D |
Selumetinib + docetaxel | Phase II trial | Janne et al. [31] |
| MEK1 | K57N | Selumetinib | Preclinical data | Marks et al. [58] |
| MET | Amplification | Crizotinib | Case report | Ou et al. [59] |
| NRAS | b Q61L/R/K | Selumetinib Trametinib |
Preclinical data | Ohashi et al. [60] |
| PIK3CA | H1047R | BKM120 | Phase I trial | Bendell et al. [61] |
| RET fusions | Cabozantinib | Phase II trial | Drilon et al. [62] | |
| Vandetinib | Case report | Gautschi et al. [63] | ||
| Ganestespib | Preclinical data | Sang et al. [49] | ||
| ROS1 fusions | Crizotinib | Case report | Bergethon et al. [64] | |
| Case report | Davies et al. [65] | |||
| Ganestespib | Preclinical data | Sang et al. [49] | ||
| Kinase-targeted therapies in melanoma | ||||
|---|---|---|---|---|
| Oncogene | Targeted therapy | Level of evidence | Associated publications | |
| BRAF | L597S | TAK-733 | Case report | Dahlman et al. [66] |
| L597R | Vemurafenib | Case report | Bahadoran et al. [67] | |
| L597V | Trametinib | Phase I/II trial | Falchook et al. [45] | |
| c V600E/K/M/R/D | Vemurafenib | Phase III trial | Chapman et al. [1] | |
| Dabrafenib | Phase III trial | Hauschild et al. [35] | ||
| Trametinib | Phase III trial | Flaherty et al. [46] | ||
| Dabrafenib + trametinib | Phase I/II trial | Flaherty et al. [68] | ||
| K601E | Trametinib | Case report | Kim et al. [82] | |
| KIT | L576P K642E |
Imatinib | Phase II trial Phase II trial |
Carvajal et al. [70] Hodi et al. [85] |
| V559A | Imatinib | Case report | Terheyden et al. [71] | |
| W557R V559D |
Imatinib | Preclinical data | Beadling et al. [72] | |
| L576P | Dasatinib | Case report | Woodman et al. [73] | |
| L576P W557G |
Sunitinib | Retrospective analysis | Minor et al. [74] | |
| V560D | Sorafenib | Case report | Quintas-Cardama et al. [75] | |
| L576P V559A |
Nilotinib | Phase II Trial | Cho et al. [83] | |
| NRAS | d Q61L/K/R | MEK162 | Phase II trial | Ascierto et al. [76] |
| Selumetinib | Phase I trial | Adjei et al. [47] | ||
| Trametinib + PD-0332991 | Preclinical data | Kwong et al. [48] | ||
Specific mutations listed are those found in references cited. Other substitutions in KRAS codons 12, 13, and 61 have been found in NSCLC; differences in their sensitivity to selumetinib + docetaxel are unknown at this time.
Specific mutations listed are those found in references cited. Other substitutions in NRAS codons 12, 13, and 61 have been found in NSCLC; differences in their sensitivity to selumetinib and trametinib are unknown at this time.
V600M/R/D substitutions occur less frequently than V600E/K; specific outcomes for V600M/R/D are not included in these clinical trial data; sensitivity is predicted based on preclinical data and case reports (105–107).
Specific mutations listed are those found in references cited. Other substitutions in NRAS codons 12, 13, and 61 have been found in melanoma; differences in their sensitivity to MEK inhibitors are unknown at this time
In addition to EGFR alterations, heterogeneity is seen in other common lung cancer-associated ‘driver’ oncogenes. For example, KRAS-activating mutations, which are found in ~20% of lung adenocarcinomas, can occur at positions G12, G13, and Q61 (30). Though currently less ‘targetable’ than EGFR mutations, KRAS mutations are almost exclusively present in the setting of wild-type EGFR and are thus predictive of insensitivity to EGFR TKIs (30). Recent data from a phase II trial indicate that addition of the MEK inhibitor, selumetinib, to docetaxel increased progression-free survival by 3.2 months compared to docetaxel alone in patients with KRAS-mutant lung cancer (31). A phase III trial evaluating this combination is planned. As more promising targeted therapies become available for KRAS-mutant lung cancer, it may be necessary to elucidate clinically relevant differences among the molecular subsets stratified by location of mutations in the protein.
Recent advances have also been made in our understanding of the biology of melanoma, resulting in similar sub-classifications according to ‘driver’ mutations (Figure 2C) (32). Substitutions of various amino acids for valine 600 (V600) in the kinase domain of BRAF, the most common of such ‘driver’ mutations, are found in approximately 50% of melanomas (most often in primary tumors located on body surfaces with intermittent intense sun exposure). These BRAF V600 mutations are known to lead to activation of the mitogen activated protein kinase (MAPK) pathway (33); they have also been shown to confer sensitivity to the small molecule BRAF serine-threonine kinase inhibitors, vemurafenib and dabrafenib (1, 33–35). Of the BRAF V600 mutations identified to date in melanoma, the V600E substitution comprises approximately 80%; the remaining fraction is made up of V600K (15%), V600M, V600D, and V600R substitutions (all less than 5%) (32) (Figure 2D). Non-V600 mutations in BRAF have also been identified as a distinct molecular subtype (Table 2); specific implications for targeted therapies across the range of BRAF mutations are discussed in greater detail below. In addition to BRAF mutations, other recurrent oncogenic ‘driver’ mutations that have been identified in melanoma include mutations in GNA11, GNAQ (both predominantly found in uveal melanomas), KIT (predominantly mucosal and acral melanomas), NRAS (all sites except uveal), and CTNNB1 (32). Among 955 melanoma specimens genotyped at Vanderbilt between 2010–2013, 610 harbored known recurrent mutations in genes including BRAF (39.1%), NRAS (18.1%), GNAQ (2.8%), GNA11 (1.3%), KIT (2.3%), and CTNNB1 (1.1%) (32) (Figure 2C). A more comprehensive list of targetable mutations in melanoma and NSCLC can be found in Table 2 (1, 3, 4, 25, 29, 31, 35–86).
Because of substantial international efforts to increase the collective knowledge base regarding the frequencies of actionable genomic alterations in solid tumors, equal efforts are necessary to curate this information such that it can be delivered in a reliable, easily-accessible format. In an attempt to address this need for more efficient translation of scientific progress to clinical application, Vanderbilt created My Cancer Genome (MCG) as an online resource for clinicians and patients worldwide. Routinely edited by field experts around the globe, MCG is a unique database that links scientific literature about known oncogenic ‘drivers’ to information about available clinical trials. As the technology advances for routine tumor genotyping in the clinic, resources like MCG will be critical to facilitate mainstream assimilation of personalized cancer medicine worldwide.
BRAF-omas and ALK-omas: characterizing cancer based on driver mutations rather than histology or site of origin
Despite significant progress in the molecular sub-classification of tumors based on integrated genomic information, the initial categorization of cancer types is still determined by tissue of origin. In other words, the current paradigm dictates that we refer to ‘EGFR-mutant lung tumors’ or ‘BRAF-mutant melanomas’, rather than classifying cancers solely based on their genetic makeup or other molecular features. This phenomenon is the result of decades of organ-centric clinical tradition. However, as large-scale genomic information is now available for many different kinds of cancers, we have seen that some ‘driver’ mutations are shared across tumor types (87). The potential implications of defining tumors by their driver mutations, e.g. “BRAF-omas”, are paradigm-shifting.
Even so, the presence of a ‘driver’ mutation such as mutant BRAF V600E does not automatically mean that tumors are sensitive to BRAF inhibition alone. According to data from COSMIC, lung adenocarcinomas, colorectal adenocarcinomas (CRCs), melanomas, and papillary thyroid cancers (PTCs) are all known to harbor BRAF mutations in approximately 3%, 14%, 45%, and 50% of cases, respectively (Figure 3A). However, the response of these ‘BRAF-omas’ to vemurafenib varies substantially based on tissue of origin (Table 2). While BRAF inhibitors vemurafenib and dabrafenib have achieved overall response rates greater than 50% in clinical trials of patients with V600-mutant BRAF (1, 35, 68, 88, 89) and show early promise in PTCs with BRAF V600E mutations (partial regression or stable disease in 3 of 3 BRAF-mutant tumors in a phase I trial) (90), the efficacy of these inhibitors appears significantly lower in BRAF-mutant CRCs (91, 92). In addition, while V600 substitutions are the most common BRAF alterations found in melanoma, CRC, and PTC, BRAF mutations in lung cancer commonly occur at non-V600 locations, such as G466, G469, D594 and L597 (93, 94) (Figure 3B). The response rate of lung tumors harboring non-V600 BRAF mutations to BRAF and MEK targeted therapies is currently unknown; however, recent retrospective analyses suggest that NSCLCs harboring non-V600 BRAF mutations may portend a more favorable prognosis than those harboring BRAF V600 mutations (95). Metastatic melanomas harboring L597 mutations have been reported to respond to trametinib and TAK-733 (MEK inhibitors) (66) and potentially vemurafenib (67).
Figure 3. “BRAF-omas”: location and relative frequency of BRAF mutations across multiple different solid tumors.
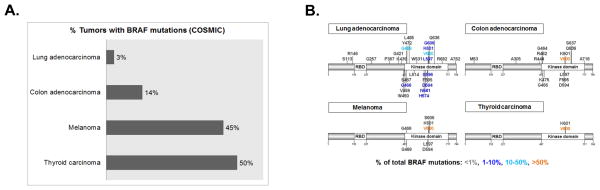
Indications for targeted therapies differ based on anatomical site (see text for details). A, percentage of lung adenocarcinoma, colon adenocarcinoma, melanoma, and thyroid carcinoma samples with documented BRAF mutations in the Catalog of Somatic Mutations in Cancer (COSMIC) database. B, codon location of non-synonymous BRAF mutations with frequencies of greater than 0.1% in COSMIC database for each tumor type listed in A.
Another example of a targetable ‘driver’ genomic alteration that has been found in multiple different solid tumor types is the rearrangement of the anaplastic lymphoma kinase (ALK). ALK fusions are found in 2–7% of NSCLCs (96) and have also been found in other solid tumors including inflammatory myofibroblastic tumor (IMT), rhabdomyosarcoma, serous ovarian carcinoma, colorectal carcinoma, breast carcinoma, renal medullary carcinoma, renal cell carcinoma, and esophageal squamous cell carcinoma (97). ALK TKIs have proven effective at treating ALK-positive NSCLCs (96), and early clinical studies suggest potential benefit in IMT (98); however their efficacy in other solid tumors with ALK translocations is currently unknown. Furthermore, ALK fusions themselves are heterogeneous. All ALK fusions defined to date contain the entire ALK kinase domain; however, the 5′ gene fusion partner varies. In NSCLC, more than 10 different ALK fusion variants have been described to date. Whether the specific ALK fusion present within the tumor alters sensitivity to ALK TKIs remains to be determined. This heterogeneity even among tumors sharing common ‘drivers’ such as BRAF mutations and ALK rearrangements presents a challenge for the development of targeted therapeutics based on genetic characterization. Further elucidation of the range of drug sensitivities across genetic subtypes within tumors of common and distinct anatomic locations will be necessary in order to make sense of this biologic diversity and more accurately predict proper therapies for patients.
An example of a new initiative along these lines is the so-called ‘basket trial’, in which inclusion criteria do not require a specific disease subtype but rather allow treatment of multiple cohorts of patients with cancers of different origins that share BRAF V600E mutations in common (99). In stark contrast to the traditional clinical trial methodology requiring large-scale, serially executed trials, the basket trial is designed to facilitate investigation of treatment efficacy in multiple independent cohorts at once. In addition, the trial design promotes real-time evaluation of outcomes such that treatment efficacy data combining tumor type with genetic profiling can be compiled and used to expand promising cohorts. The hope is that, for the responding cohorts, the initial basket trial will then evolve into larger trials, leading to FDA approval and clinical application with minimal turnaround time.
This kind of effort is also part of a broader initiative announced by the NCI National Clinical Trials Network (NCTN) to treat 1,000 tumors with specific targeted therapies on the basis of identified molecular alterations. Termed the NCI-MATCH (Molecular Analysis for Therapy Choice Trial), the goal of this trial is to learn more efficiently about the genetics of cancer from clinical data and to challenge the current standards for treatment indications in cancer therapy (100). Specifically, tumors from patients who have failed standard therapies will be evaluated for known ‘actionable’ genomic alterations and subsequently placed on targeted therapies for which preclinical or early clinical data suggest efficacy for the specific genotype identified. While these trials represent a promising step forward in the field of personalized cancer medicine, there are significant challenges inherent in their design, which will need to be carefully considered in both study implementation and data interpretation. Coordination among multiple centers will be necessary to gain requisite patient accrual, especially for tumors of rare genotypes. Along those lines, obtaining sufficient statistical power for analysis and establishing robust controls for efficacy comparisons are non-trivial challenges presented by such studies. With such considerations in mind, the proposed study design for NCI-MATCH follows a Simon two-stage design within each drug-by-mutation arm with 30 patients per arm. The dual primary endpoints are proposed to be overall response rate (RR) of 5% versus 25% or PFS of 15% versus 35% at six months (100).
Moving forward, we can also learn from the past: the NCI has recently announced an initiative to analyze tissue of ‘exceptional responders’ of failed past clinical trials from the pre-genomic era, with the hope of expanding our repertoire of paired pharmacologic inhibitors and specific genomic alterations that confer sensitivity. This model further emphasizes the importance of fundamental changes in standard clinical trial design; before biomarker-driven enrollment in trials, potentially useful anti-cancer pharmacologic agents in development for solid tumors may have ‘failed’ approval because they were tested in a cohort of patients whose tumors harbored too much genetic heterogeneity to achieve any statistically significant clinical response or discern in which specific subset(s) the agent shows most efficacy. This initiative is a mechanism by which previously unrecognized tumor heterogeneity is now potentially informative in combination with current sequencing technology.
Heterogeneity of mechanisms of acquired resistance to targeted therapies
Despite progress in targeting molecular subsets of NSCLC and melanoma, those patients who initially experience clinical responses to targeted therapy ultimately develop progressive disease, usually within 6–12 months (101, 102). Known mechanisms of resistance to EGFR, ALK, and BRAF kinase inhibitors have been discussed elsewhere (101–104) and are outside the scope of this review. However, the data so far suggest an equal, if not greater, amount of heterogeneity and complexity and emphasize the importance of rebiopsy for all patients at the time of disease progression to advance our understanding of resistance mechanisms.
Conclusion
Continued integration of large (e.g., TCGA) and small (e.g., case reports) datasets combined with advancements in sequencing and informatics technology will deepen our understanding of the heterogeneity of human cancers. Of note, this review focused primarily on heterogeneity at the DNA level, but epigenetic, transcriptional, and post-transcriptional changes may also contribute to heterogeneity among solid tumors. Ultimately, enhancement of our understanding of tumor heterogeneity will provide us with more distinct, clinically relevant molecular subsets of cancer that can be treated with increasing efficacy and lead to further improvement in patient outcomes.
Acknowledgments
We would like to thank Lucy Wang, Joseph Burden, Scott Sobecki, and Pam Carney for their help in creating the reporting tool to extract and visualize the Vanderbilt PCMI data reported in this manuscript. We would also like to thank Donald Hucks, Winston Tan, and the VUMC Molecular Diagnostics lab staff for technical assistance with acquisition of data.
Financial support:
WP received support from the V Foundation and the NCI (R01-CA121210, P01-CA129243, U54-CA143798, Vanderbilt’s Specialized Program of Research Excellence in Lung Cancer grant (CA90949), and the VICC Cancer Center Core grant (P30-CA68485). CML was supported by a Damon Runyon Clinical Investigator Award. CML and DBJ were supported by a K12 grant (K12 CA 0906525). JAS received support from 5K24 CA097588-09, the Mary Hendrickson-Johnson ACS Professorship: Melanoma Professorship, and the Melanoma Research Alliance Team Science Award. ZZ received funding from RO1LM011177 and P30CA68485. CBM was supported by Public Health Service Award T32 GM07347 from the National Institute of General Medical Studies for the Vanderbilt Medical-Scientist Training Program. Additional support was provided by the TJ Martell Foundation, the Kleberg Foundation, the James C. Bradford Family Foundation, the Joyce Family Foundation, and an anonymous donor.
Footnotes
Conflicts of interest:
CML: Consulting/Advisory Board: Pfizer; Honoraria from Speakers Bureau: Qiagen, Abbott Molecular.
LH: Consultant/advisory board: Bristol-Meyers Squibb and Clovis (compensated) and PUMA (uncompensated); Honoraria: Boehringer Ingelheim; Commercial research funding: Astellas.
JAS: Commercial research grant: Novartis and Bristol-Myers Squibb; Consultant/Advisory board: Amgen, GlaxoSmithKline, Roche.
WP: Consultant: MolecularMD, AstraZeneca, Bristol-Myers Squibb, Symphony Evolution, Clovis Oncology, Exelixis, and Clarient; Commercial research grants: Enzon, Xcovery, AstraZeneca, Symphogen, Clovis Oncology, and Bristol-Myers Squibb; Other: Rights to EGFR T790M testing were licensed on behalf of WP and others by MSKCC to MolecularMD.
The other authors have no conflicts of interest to report.
References
- 1.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 4.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. The lancet oncology. 2011;12:735–42. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 5.Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C. Intratumor heterogeneity: seeing the wood for the trees. Sci Transl Med. 2012;4:127ps10. doi: 10.1126/scitranslmed.3003854. [DOI] [PubMed] [Google Scholar]
- 6.Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–22. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 7.Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170–3. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- 8.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077–80. doi: 10.1158/0008-5472.CAN-07-3293. discussion 80. [DOI] [PubMed] [Google Scholar]
- 9.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins FS, Barker AD. Mapping the cancer genome. Pinpointing the genes involved in cancer will help chart a new course across the complex landscape of human malignancies. Sci Am. 2007;296:50–7. [PubMed] [Google Scholar]
- 11.MacConaill LE. Existing and emerging technologies for tumor genomic profiling. J Clin Oncol. 2013;31:1815–24. doi: 10.1200/JCO.2012.46.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 13.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–96. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 14.Chmielecki J, Meyerson M. DNA Sequencing of Cancer: What Have We Learned? Annu Rev Med. 2013 doi: 10.1146/annurev-med-060712-200152. [DOI] [PubMed] [Google Scholar]
- 15.Su Z, Dias-Santagata D, Duke M, Hutchinson K, Lin YL, Borger DR, et al. A platform for rapid detection of multiple oncogenic mutations with relevance to targeted therapy in non-small-cell lung cancer. J Mol Diagn. 2011;13:74–84. doi: 10.1016/j.jmoldx.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2:146–58. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rekhtman N, Paik PK, Arcila ME, Tafe LJ, Oxnard GR, Moreira AL, et al. Clarifying the spectrum of driver oncogene mutations in biomarker-verified squamous carcinoma of lung: lack of EGFR/KRAS and presence of PIK3CA/AKT1 mutations. Clin Cancer Res. 2012;18:1167–76. doi: 10.1158/1078-0432.CCR-11-2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arcila M, Lau C, Nafa K, Ladanyi M. Detection of KRAS and BRAF mutations in colorectal carcinoma roles for high-sensitivity locked nucleic acid-PCR sequencing and broad-spectrum mass spectrometry genotyping. J Mol Diagn. 2011;13:64–73. doi: 10.1016/j.jmoldx.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson BE, Kris MG, Berry LD, Kwiatkowski DJ, Iafrate AJ, Varella-Garcia M, Wistuba II, Franklin WA, Ladanyi M, Su P-F, Sequist LV, Kuri FR, Garon EB, Pao W, Rudin CM, Schiller JH, Haura EB, Giaccone G, Minna JD, Bunn PA. A multicenter effort to identify driver mutations and employ targeted therapy in patients with lung adenocarcinomas: The Lung Cancer Mutation Consortium (LCMC); ASCO Annual Meeting Proceedings; 2013. [Google Scholar]
- 20.Levy MA, Lovly CM, Pao W. Translating genomic information into clinical medicine: lung cancer as a paradigm. Genome Res. 2012;22:2101–8. doi: 10.1101/gr.131128.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92–8. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 22.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 23.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 24.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients With Metastatic Lung Adenocarcinoma With EGFR Mutations. J Clin Oncol. 2013;31:3327–34. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 26.Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR–targeted therapy and beyond. Mod Pathol. 2008;21 (Suppl 2):S16–22. doi: 10.1038/modpathol.3801018. [DOI] [PubMed] [Google Scholar]
- 27.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walter AO, Tjin Tham Sjin R, Haringsma HJ, Ohashi K, Sun J, Lee K, et al. Discovery of a mutant–selective covalent inhibitor of EGFR that overcomes T790M–mediated resistance in NSCLC. Cancer discovery. 2013 doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ranson M, Pao W, Kim DW, Kim SW, Ohe Y, Felip E, Planchard D, Ghiorghiu S, Cantarini M, Janne PA. Preliminary results from a Phase I study with AZD9291: An irreversible inhibitor of epidermal growth factor receptor (EGFR) activating and resistance mutations in non-small-cell lung cancer (NSCLC). European Cancer Congress; 2013. Abstract #33. [Google Scholar]
- 30.Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc. 2009;6:201–5. doi: 10.1513/pats.200809-107LC. [DOI] [PubMed] [Google Scholar]
- 31.Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. The lancet oncology. 2013;14:38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 32.Lovly CM, Dahlman KB, Fohn LE, Su Z, Dias-Santagata D, Hicks DJ, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PloS one. 2012;7:e35309. doi: 10.1371/journal.pone.0035309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 34.Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, et al. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003;95:1878–90. doi: 10.1093/jnci/djg123. [DOI] [PubMed] [Google Scholar]
- 35.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 36.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 37.Sen B, Peng S, Tang X, Erickson HS, Galindo H, Mazumdar T, et al. Kinase-impaired BRAF mutations in lung cancer confer sensitivity to dasatinib. Sci Transl Med. 2012;4:136ra70. doi: 10.1126/scitranslmed.3003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rudin CM, Hong K, Streit M. Molecular characterization of acquired resistance to the BRAF inhibitor dabrafenib in a patient with BRAF-mutant non-small-cell lung cancer. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2013;8:e41–2. doi: 10.1097/JTO.0b013e31828bb1b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pitini V, Arrigo C, Di Mirto C, Mondello P, Altavilla G. Response to dasatinib in a patient with SQCC of the lung harboring a discoid-receptor-2 and synchronous chronic myelogenous leukemia. Lung Cancer. 2013;82:171–2. doi: 10.1016/j.lungcan.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 40.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. The lancet oncology. 2010;11:121–8. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 41.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. The lancet oncology. 2012;13:239–46. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 42.Ramalingam SS, Blackhall F, Krzakowski M, Barrios CH, Park K, Bover I, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2012;30:3337–44. doi: 10.1200/JCO.2011.40.9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sequist LV, Besse B, Lynch TJ, Miller VA, Wong KK, Gitlitz B, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:3076–83. doi: 10.1200/JCO.2009.27.9414. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J, Zhang L, Su X, Li M, Xie L, Malchers F, et al. Translating the therapeutic potential of AZD4547 in FGFR1-amplified non-small cell lung cancer through the use of patient-derived tumor xenograft models. Clin Cancer Res. 2012;18:6658–67. doi: 10.1158/1078-0432.CCR-12-2694. [DOI] [PubMed] [Google Scholar]
- 45.Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. The lancet oncology. 2012;13:782–9. doi: 10.1016/S1470-2045(12)70269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 47.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503–10. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer discovery. 2013;3:430–43. doi: 10.1158/2159-8290.CD-12-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yeh P, Chen H, Andrews J, Naser R, Pao W, Horn L. DNA-Mutation Inventory to Refine and Enhance Cancer Treatment (DIRECT): a catalog of clinically relevant cancer mutations to enable genome-directed anticancer therapy. Clin Cancer Res. 2013;19:1894–901. doi: 10.1158/1078-0432.CCR-12-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He M, Capelletti M, Nafa K, Yun CH, Arcila ME, Miller VA, et al. EGFR exon 19 insertions: a new family of sensitizing EGFR mutations in lung adenocarcinoma. Clin Cancer Res. 2012;18:1790–7. doi: 10.1158/1078-0432.CCR-11-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sequist LV, Gettinger S, Senzer NN, Martins RG, Janne PA, Lilenbaum R, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol. 2010;28:4953–60. doi: 10.1200/JCO.2010.30.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gautschi O, Pauli C, Strobel K, Hirschmann A, Printzen G, Aebi S, et al. A patient with BRAF V600E lung adenocarcinoma responding to vemurafenib. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2012;7:e23–4. doi: 10.1097/JTO.0b013e3182629903. [DOI] [PubMed] [Google Scholar]
- 54.Hammerman PS, Sos ML, Ramos AH, Xu C, Dutt A, Zhou W, et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer discovery. 2011;1:78–89. doi: 10.1158/2159-8274.CD-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Greve J, Teugels E, Geers C, Decoster L, Galdermans D, De Mey J, et al. Clinical activity of afatinib (BIBW 2992) in patients with lung adenocarcinoma with mutations in the kinase domain of HER2/neu. Lung Cancer. 2012;76:123–7. doi: 10.1016/j.lungcan.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 56.Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med. 2006;354:2619–21. doi: 10.1056/NEJMc060020. [DOI] [PubMed] [Google Scholar]
- 57.Mazieres J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013;31:1997–2003. doi: 10.1200/JCO.2012.45.6095. [DOI] [PubMed] [Google Scholar]
- 58.Marks JL, Gong Y, Chitale D, Golas B, McLellan MD, Kasai Y, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008;68:5524–8. doi: 10.1158/0008-5472.CAN-08-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ou SH, Kwak EL, Siwak-Tapp C, Dy J, Bergethon K, Clark JW, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2011;6:942–6. doi: 10.1097/JTO.0b013e31821528d3. [DOI] [PubMed] [Google Scholar]
- 60.Ohashi K, Sequist LV, Arcila ME, Lovly CM, Chen X, Rudin CM, et al. Characteristics of lung cancers harboring NRAS mutations. Clin Cancer Res. 2013;19:2584–91. doi: 10.1158/1078-0432.CCR-12-3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30:282–90. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 62.Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, Stephens P, et al. Response to Cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer discovery. 2013;3:630–5. doi: 10.1158/2159-8290.CD-13-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gautschi O, Zander T, Keller FA, Strobel K, Hirschmann A, Aebi S, et al. A patient with lung adenocarcinoma and RET fusion treated with vandetanib. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2013;8:e43–4. doi: 10.1097/JTO.0b013e31828a4d07. [DOI] [PubMed] [Google Scholar]
- 64.Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–70. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davies KD, Le AT, Theodoro MF, Skokan MC, Aisner DL, Berge EM, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. 2012;18:4570–9. doi: 10.1158/1078-0432.CCR-12-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, Jia P, et al. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer discovery. 2012;2:791–7. doi: 10.1158/2159-8290.CD-12-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bahadoran P, Allegra M, Le Duff F, Long-Mira E, Hofman P, Giacchero D, et al. Major clinical response to a BRAF inhibitor in a patient with a BRAF L597R-mutated melanoma. J Clin Oncol. 2013;31:e324–6. doi: 10.1200/JCO.2012.46.1061. [DOI] [PubMed] [Google Scholar]
- 68.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305:2327–34. doi: 10.1001/jama.2011.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Terheyden P, Houben R, Pajouh P, Thorns C, Zillikens D, Becker JC. Response to imatinib mesylate depends on the presence of the V559A-mutated KIT oncogene. J Invest Dermatol. 2010;130:314–6. doi: 10.1038/jid.2009.197. [DOI] [PubMed] [Google Scholar]
- 72.Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14:6821–8. doi: 10.1158/1078-0432.CCR-08-0575. [DOI] [PubMed] [Google Scholar]
- 73.Woodman SE, Trent JC, Stemke-Hale K, Lazar AJ, Pricl S, Pavan GM, et al. Activity of dasatinib against L576P KIT mutant melanoma: molecular, cellular, and clinical correlates. Mol Cancer Ther. 2009;8:2079–85. doi: 10.1158/1535-7163.MCT-09-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Minor DR, Kashani-Sabet M, Garrido M, O’Day SJ, Hamid O, Bastian BC. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res. 2012;18:1457–63. doi: 10.1158/1078-0432.CCR-11-1987. [DOI] [PubMed] [Google Scholar]
- 75.Quintas-Cardama A, Lazar AJ, Woodman SE, Kim K, Ross M, Hwu P. Complete response of stage IV anal mucosal melanoma expressing KIT Val560Asp to the multikinase inhibitor sorafenib. Nat Clin Pract Oncol. 2008;5:737–40. doi: 10.1038/ncponc1251. [DOI] [PubMed] [Google Scholar]
- 76.Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. The lancet oncology. 2013;14:249–56. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- 77.Shaw AT, Mok T, Spigel DR, Nishio M, Felip E, Tan DS-W, Garcia-Campelo MR, Groen HJM, Shaker RD, Schaefer ES, Farrell NJ, Blakesley RE, Weir A, Ristic M, Selvaggi G, Scagliotti G. A phase II single-arm study of LDK378 in patients with ALK-activated (ALK+) non-small cell lung cancer (NSCLC) previously treated with chemotherapy and crizotinib (CRZ). ASCO Annual Meeting Proceedings; 2013. Abstract # TPS8119. [Google Scholar]
- 78.Camidge DR, Bazhenova L, Salgia R, Weiss GJ, Langer CJ, Shaw AT, Narasimhan NI, Dorer DJ, Rivera VM, Zhang J, Clackson T, Haluska FG, Gettinger SN. First-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies: Updated results. ASCO Annual Meeting Proceedings; 2013. Abstract #8031. [Google Scholar]
- 79.Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Cancer Res. 2013;19:3068–77. doi: 10.1158/1078-0432.CCR-12-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–8. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 81.Malchers F, Dietlein F, Schottle J, Lu X, Nogova L, Albus K, et al. Cell-Autonomous and Non-Cell-Autonomous Mechanisms of Transformation by Amplified FGFR1 in Lung Cancer. Cancer discovery. 2014;4:246–57. doi: 10.1158/2159-8290.CD-13-0323. [DOI] [PubMed] [Google Scholar]
- 82.Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31:482–9. doi: 10.1200/JCO.2012.43.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cho JH, Kim KM, Kwon M, Kim JH, Lee J. Nilotinib in patients with metastatic melanoma harboring KIT gene aberration. Invest New Drugs. 2012;30:2008–14. doi: 10.1007/s10637-011-9763-9. [DOI] [PubMed] [Google Scholar]
- 84.Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1–2 study. The lancet oncology. 2013;14:590–8. doi: 10.1016/S1470-2045(13)70142-6. [DOI] [PubMed] [Google Scholar]
- 85.Hodi FS, Corless CL, Giobbie-Hurder A, Fletcher JA, Zhu M, Marino-Enriquez A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31:3182–90. doi: 10.1200/JCO.2012.47.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sequist LV, Soria JC, Gadgeel SM, Wakelee HA, Camidge DR, Varga A, Fidias P, Wozniak AJ, Neal JW, Doebele RC, Garon EB, Jaw-Tsai SS, Stern JC, Allen AR, Goldman JW. First-in-human evaluation of CO-1686, an irreversible, selective, and potent tyrosine kinase inhibitor of EGFR T790M. ASCO Annual Meeting Proceedings; 2013. Abstract #2524. [Google Scholar]
- 87.Chang K, Creighton CJ, Davis C, Donehower L, Drummond J, Wheeler D, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379:1893–901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim KB, Cabanillas ME, Lazar AJ, Williams MD, Sanders DL, Ilagan JL, et al. Clinical Responses to Vemurafenib in Patients with Metastatic Papillary Thyroid Cancer Harboring BRAF(V600E) Mutation. Thyroid. 2013;23:1277–83. doi: 10.1089/thy.2013.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer discovery. 2012;2:227–35. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–3. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 93.Paik PK, Arcila ME, Fara M, Sima CS, Miller VA, Kris MG, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29:2046–51. doi: 10.1200/JCO.2010.33.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pratilas CA, Hanrahan AJ, Halilovic E, Persaud Y, Soh J, Chitale D, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68:9375–83. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cardarella S, Ogino A, Nishino M, Butaney M, Shen J, Lydon C, et al. Clinical, Pathologic, and Biologic Features Associated with BRAF Mutations in Non-Small Cell Lung Cancer. Clin Cancer Res. 2013;19:4532–40. doi: 10.1158/1078-0432.CCR-13-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013;13:685–700. doi: 10.1038/nrc3580. [DOI] [PubMed] [Google Scholar]
- 98.Butrynski JE, D’Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363:1727–33. doi: 10.1056/NEJMoa1007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Willyard C. ‘Basket studies’ will hold intricate data for cancer drug approvals. Nat Med. 2013;19:655. doi: 10.1038/nm0613-655. [DOI] [PubMed] [Google Scholar]
- 100.Kaiser J. Biomedicine. Rare cancer successes spawn ‘exceptional’ research efforts. Science. 2013;340:263. doi: 10.1126/science.340.6130.263. [DOI] [PubMed] [Google Scholar]
- 101.Salama AK, Flaherty KT. BRAF in Melanoma: Current Strategies and Future Directions. Clin Cancer Res. 2013;19:4326–34. doi: 10.1158/1078-0432.CCR-13-0779. [DOI] [PubMed] [Google Scholar]
- 102.Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31:1070–80. doi: 10.1200/JCO.2012.43.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013;49:1297–304. doi: 10.1016/j.ejca.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 104.Lovly CM, Pao W. Escaping ALK inhibition: mechanisms of and strategies to overcome resistance. Sci Transl Med. 2012;4:120ps2. doi: 10.1126/scitranslmed.3003728. [DOI] [PubMed] [Google Scholar]
- 105.Klein O, Clements A, Menzies AM, O’Toole S, Kefford RF, Long GV. BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer. 2013;49:1073–9. doi: 10.1016/j.ejca.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 106.Ponti G, Pellacani G, Tomasi A, Gelsomino F, Spallanzani A, Depenni R, et al. The somatic affairs of BRAF: tailored therapies for advanced malignant melanoma and orphan non-V600E (V600R-M) mutations. J Clin Pathol. 2013;66:441–5. doi: 10.1136/jclinpath-2012-201345. [DOI] [PubMed] [Google Scholar]
- 107.Ponti G, Tomasi A, Pellacani G. Overwhelming response to Dabrafenib in a patient with double BRAF mutation (V600E; V600M) metastatic malignant melanoma. J Hematol Oncol. 2012;5:60. doi: 10.1186/1756-8722-5-60. [DOI] [PMC free article] [PubMed] [Google Scholar]