

Abstract
Type I collagen gels are routinely used in biophysical studies and bioengineering applications. The structural and mechanical properties of these fibrillar matrices depend on the conditions under which collagen fibrillogenesis proceeds, and developing a fuller understanding of this process will enhance control over gel properties. Turbidity measurements have long been the method of choice for monitoring developing gels, whereas imaging methods are regularly used to visualize fully developed gels. In this study, turbidity and confocal reflectance microscopy (CRM) were simultaneously employed to track collagen fibrillogenesis and reconcile the information reported by the two techniques, with confocal fluorescence microscopy (CFM) used to supplement information about early events in fibrillogenesis. Time-lapse images of 0.5 mg/ml, 1.0 mg/ml, and 2.0 mg/ml acid-solubilized collagen I gels forming at 27°C, 32°C, and 37°C were collected. It was found that in situ turbidity measured in a scanning transmittance configuration was interchangeable with traditional turbidity measurements using a spectrophotometer. CRM and CFM were employed to reveal the structures responsible for the turbidity that develops during collagen self-assembly. Information from CRM and transmittance images was collapsed into straightforward single variables; total intensity in CRM images tracked turbidity development closely for all collagen gels investigated, and the two techniques were similarly sensitive to fibril number and dimension. Complementary CRM, CFM, and in situ turbidity measurements revealed that fibril and network formation occurred before substantial turbidity was present, and the majority of increasing turbidity during collagen self-assembly was due to increasing fibril thickness.
Introduction
Collagen I is the most abundant protein in mammalian extracellular matrix where it is present primarily as fibrils, organized differently in different tissues (1). Given its physiological relevance, fibrillar collagen I matrices have been used both as scaffolds in tissue engineering and as three-dimensional environments for biophysical studies, especially those concerned with cell-matrix interactions (2–10). An appealing feature of collagen I matrices is their tunability. Both in vivo and in vitro, collagen I monomers self-assemble into fibrillar structures that may cross-link and/or entangle to form viscoelastic gels with varied network structures and mechanical properties. In vivo, diversity in these properties is realized through differences in monomer concentration, presence of various enzymes including those that cross-link collagen, and presence of additional extracellular matrix components that may intercalate with or otherwise affect the collagen matrix or assembly thereof (11). In vitro, network structural and mechanical properties may also be tuned through introducing cross-linkers and secondary gel components, as well as through varying temperature, pH, and ionic strength during self-assembly. Indeed, rather significant changes in both gel network structure and stiffness have been shown to result from changes in these parameters (12–16). This control opens the door to independently varying collagen I network topology and mechanical properties, allowing for studies in which cell response to these variables is probed in physiologically relevant three-dimensional environments.
Collagen network properties are sensitive to temperature, pH, and ionic strength because these conditions affect the self-assembly of collagen monomers into fibrils. Thus, a fuller understanding of fibrillogenesis will enhance matrix design. Further clarification of the self-assembly of collagen may also inform discussion of the self-assembly of other fibrillar proteins, including pathological ones (17). To date, studies investigating the mechanism and kinetics of the collagen I self-assembly process in vitro have primarily monitored turbidity, an approach first employed more than a half century ago (18). During collagen I gelation, turbidity increases as a nearly transparent solution of monomers develops into fibrils that scatter significant amounts of light. Early studies showed that the increase in turbidity during gelation tracked precipitation of collagen into fibrils (18,19). These studies and others also supported the hypothesis that collagen fibrillogenesis was a nucleation and growth process, as the turbidity curves were sigmoidal, displaying distinct lag and growth phases (18–23).
Although turbidity has been and continues to be the method of choice to monitor developing collagen networks, imaging techniques are commonly employed to reveal structure of fully developed collagen and other biopolymer gels. Collagen I networks have been visualized using confocal fluorescence microscopy (CFM) of exogenously labeled collagen (15,24,25). Unlabeled collagen fibrils can also be imaged with two-photon fluorescence and second harmonic generation microscopy (12,13,26,27). Confocal reflectance microscopy (CRM), with contrast emerging from light backscattering from collagen fibrils, has been the most commonly employed technique to image collagen gels (2–4,8–10,28,29). The primary advantage of CRM relative to other techniques is that it requires neither the presence of potentially perturbative fluorophores nor the use of high power lasers.
Despite the growing popularity of imaging to reveal details of collagen network structure, imaging techniques have only rarely been used to visualize developing networks (28–30), and turbidity studies continue to be the dominant method used to investigate the time course of collagen gelation (16,31–33). In this study, we simultaneously performed scanning transmittance microscopy (yielding in situ turbidity, IST), CRM, and CFM during self-assembly of collagen I networks of 0.5 mg/ml, 1.0 mg/ml, and 2.0 mg/ml at 27°C, 32°C, and 37°C. To our knowledge, these measurements represent the first direct visualization of the macromolecular entities responsible for the growing turbidity during collagen self-assembly. Increasing turbidity may result from increasing number and/or increasing size of collagen fibrils, and simultaneous IST measurements and confocal imaging revealed which of these processes dominate changes in turbidity at given times and under given conditions. Beyond elucidating the entities that give rise to turbidity, collapsing data from CRM into single variables revealed that IST and CRM are similarly sensitive to the number and dimension of fibrils and report similar information on the time course of collagen self-assembly.
Materials and Methods
Preparation of collagen gels
High concentration acid-solubilized rat tail collagen I (8–10 mg/ml) was obtained from BD Biosciences (San Jose, CA). Collagen gels of 0.5 mg/ml, 1.0 mg/ml, and 2.0 mg/ml were prepared by diluting the stock collagen solution with 10% Dulbecco’s modified Eagle’s medium (DMEM, Sigma, St Louis, MO) 10×, 2.5% HEPES, and distilled H2O. For fixed wavelength studies, DMEM 10× contained phenol red, and for wavelength-dependent studies, phenol red free DMEM (Life Technologies, Carlsbad, CA) 10× was employed. For gels that contain acid-solubilized fluorescein isothiocyanate (FITC)-labeled collagen I (bovine skin, Sigma, ≈1 mg/ml), a 10:1 by weight mixture of unlabeled to labeled collagen was employed. In all cases, 1N NaOH was added to each solution to achieve a pH of 7.4. Final ionic strength of all gel solutions was I ≈ 0.15. All solutions were prepared in a cold room at 4°C and neutralized immediately before measurements.
Standard turbidity assay
Standard turbidity assays were used to validate IST measurements described below. Here, turbidity of 1.0 mg/ml collagen samples forming at 27°C and 37°C was monitored at 400, 458, 488, 515, 543, and 633 nm using a dual-beam Cary 5000 spectrophotometer. In this measurement, absorbance (A) was reported, where A = −log(I/I0) = αl with I the intensity of transmitted light, I0 the intensity of transmitted light in the reference solution (the solution without collagen), α the absorption coefficient (cm−1), and l pathlength in cm. Changes to A over time during gel formation represent an increase in scattering. Turbidity was then calculated via τ = A/l ln 10 (33,34). Temperature control was maintained with a water circulator and heated sample holder. Both the sample and reference were held in 1 cm quartz cuvettes. Measurements were taken every 10 s for up to 30 min and averaged across two to three samples per wavelength.
Microscopy
A schematic diagram of the multimodal microscope is shown in Fig. S1 in the Supporting Material. Samples were illuminated in an inverted confocal laser scanning microscope (Olympus Fluoview 300) with an NA = 1.42 oil objective. The microscope was equipped with an incubator (Neue Biosciences), and the objective was modified with a homebuilt heater. In the epi-direction, descanned reflectance images were collected through a confocal pinhole on a photomultiplier tube (PMT) detector. This channel was used to collect the CRM images. For FITC-labeled collagen, CFM images were collected simultaneously on a second PMT detector. In the forward direction, nondescanned transmitted light was collected point by point on another PMT detector. These transmittance images were collapsed to provide turbidity curves as described below.
For wavelength-independent scans, an Argon ion laser at 488 nm was used to illuminate samples. The same excitation power was used for all measurements and the same detector settings were employed. For wavelength-dependent scans, measurements were performed on additional samples of 1.0 mg/ml collagen formed at 37°C using the 488 nm line of the Argon ion laser as well as two HeNe lasers at 543 nm and 633 nm. In these measurements, laser power at the sample was kept constant. Detector settings were optimized for visualization of confocal reflectance in the lowest intensity scans and were kept constant across all wavelength-dependent measurements. CRM images were corrected for the wavelength-dependent transmission of the optics in the detection path.
Upon neutralization, samples were placed in the microscope incubator preset to and equilibrated at 27°C, 32°C, or 37°C. Imaging began as soon as neutralization of the gel was complete, the sample was placed on the stage, and the desired imaging plane—≈50 μm into the sample—was found. The time between neutralization and the beginning of imaging varied between samples, from 1 to 2 min. Time zero was considered the time at which neutralization of the gel was performed. Scans were collected at 3.26 s per scan, and the time interval between imaged scans was 10 s. In all cases, scans were 1024 × 1024 pixels (235 × 235 μm), and 12 bit images were collected. Two to four samples of unlabeled 0.5 mg/ml, 1.0 mg/ml, and 2.0 mg/ml collagen gelling at 27°C, 32°C, and 37°C were imaged and analyzed for combined IST/CRM studies illuminated with the Argon ion 488nm laser. Additional samples were prepared for both wavelength-dependent scans described previously and for trimodal imaging in which CFM was also performed. A representative movie showing trimodal time-lapse images of a 1.0 mg/ml FITC-labeled collagen gel forming at 32°C is presented in Video S1.
Image analysis
Confocal reflectance images
Because CRM images had an artifact due to reflection from optical elements in the microscope, the central portion of each image was replaced by a copy of the upper left corner of the image before further analysis. From these center-replaced CRM images, total intensity and mesh size were obtained. Total intensity was obtained by summing all individual pixel values. Total intensity of the first image collected (when no features can be imaged and very little scattering is expected) was subtracted from all subsequent images to account for background.
Determination of mesh size required thresholding. Iterative isodata thresholds were set using CRM images from gels that had developed much of their ultimate structure, at a time denoted the plateau time, tPL, described below. The threshold determined from a gel image at tPL was used to analyze all images in that time series. Following threshold determination, mesh size was determined as described previously (4).
Scanning transmittance images
From scanning transmittance images, total intensity was calculated by summing the intensity at every pixel of unprocessed images. That total intensity, I, was then transformed for comparison with traditional turbidity measurements via A = −log(I/I0), with I0 the intensity of the first image collected. As in the standard turbidity measurement, turbidity was then calculated via τ = A/l ln 10. We refer to turbidity obtained in this manner as IST.
Identification of key time points from CRM and IST
IST and the CRM variables of total intensity and mesh size were plotted against time. Key time points in the gelation process were identified from both IST and CRM intensity curves. These were lag time (tLAG), inflection point (tINF), and plateau time (tPL). Two additional time points, arrest time (tARR) and the time at which the mesh size is no longer evolving (tMS), were identified from CRM images. The rates of increase of IST and CRM intensity during the growth phase (kG) were also obtained. The manner in which these values were determined is described below and depicted in Fig. S2 for a representative 2.0 mg/ml sample gelled at 37°C.
The inflection time, tINF, was defined as the time at which half the CRM intensity or IST had developed. Due to noise in the data at the end of the measurement, particularly in IST curves, this point was chosen as the maximum of the fit of the first derivative curve. tLAG was chosen as the point at which the fit to the first derivative curve had increased 5–10% over its initial value: this point also corresponded to the time at which the CRM intensity or IST curve started increasing abruptly from a near zero value. The plateau time, tPL, was determined as the time at which the fit to the first derivative curve had attained 95% of its value at the conclusion of the measurement. To identify the rate of increase of the CRM intensity or IST, kG, a linear portion of the data between tLAG and tPL was fit to a line using least squares fitting.
Arrest time, tARR, and the time at which the mesh size was set, tMS, could only be identified from CRM images rather than from both CRM and IST. tARR was defined as the time at which visualized fibrils were fixed relative to each other. For time-lapse images taken at the lower temperatures, some fibrils became fixed relative to each other before the gel structure was fixed in the field of view. In these cases, tARR was set as the time at which not only structures were set relative to others in the frame but also those same structures were seen through the remainder of the movie, as this approximated the time at which a network spanning structure was in place. Although tARR was identified through visualization of the time-lapse images, it was validated through cross-correlation analysis. Correlation coefficients between subsequent images were computed via with A(B)mn the pixel intensity value of the first (second) image in the mth row and nth column and the mean pixel value of the images. The correlation coefficient was near zero and increased steeply over several frames to a plateau. The arrest time determined visually was in close accord (within 10%) with the time at which the cross-correlation coefficient began increasing. Mesh size set time, tMS, was chosen as the point at which mesh size was 110% of its value at the conclusion of the experiment.
Results and Discussion
Qualitative description of gelation as revealed by CRM
Time-lapse images of unlabeled 0.5 mg/ml, 1.0 mg/ml, and 2.0 mg/ml acid-solubilized collagen I solutions during gelation at 27°C, 32°C, and 37°C were collected in confocal reflectance and scanning transmittance mode simultaneously. Representative time-lapse CRM imaging of the gelation process of 1.0 mg/ml collagen at each of these temperatures is presented in Video S2.
Following gelation in CRM revealed similarities across all collagen concentrations and gelation temperatures. In all cases, no structures were seen in the initial images. Later, fibrils became visible. When fibrils were first visualized, they were not locked into a network structure but instead moved relative to one another. During this phase of self-assembly, the network became denser: fibrils grew longer and more fibrils were visualized. At the same time, early visualized fibers became brighter. At arrest, fibrils were locked into position. Following arrest, previously visualized fibrils increased in brightness. Although there was no clear separation between the time scales on which fibrils became visualizable with CRM and grew in length and width, the late steps in collagen I self-assembly appeared dominated by fibrils increasing in width, as suggested by the increasing fibril brightness that continued throughout the majority of the visualized process.
Representative CRM images at the plateau time, tPL, and at the arrest time, tARR, of label-free collagen formed at each concentration and temperature investigated are shown in Fig. 1. As expected, the number of fibrils increased and mesh size decreased with increasing collagen content at each temperature. At the lower temperatures, fully formed gels were distinct from those prepared at high temperature, with fewer, longer, and thicker structures visualized. The structures may be fibrils, bundles of fibrils—termed fibers—or a combination thereof, the majority of which are expected to be <200 nm in width (13,15) and thus below the diffraction limit for optical wavelengths.
Figure 1.
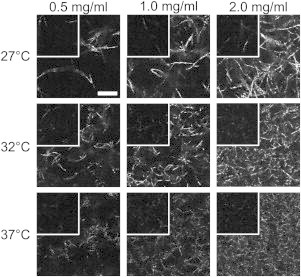
CRM images of collagen gels at plateau and arrest (overlaid insets). Scale bar is 10 μm and is the same for all images. CRM images are unprocessed but are cropped, and each shows a representative portion of the full image.
The characteristics of the gels at plateau were also present at arrest, the time at which a spanning network structure is established (Fig. 1, insets). As at plateau, at arrest lower concentration gels and those formed at lower temperatures showed sparser networks composed of fewer, longer fibrils than those of higher concentration and/or prepared at higher temperature. Although arrest and plateau gels mirrored each other, arrest images appeared more similar than did fully developed gels in terms of number of structures present and brightness of those structures, as will be discussed more fully below.
Early events of gelation as visualized by CFM
Because time-lapse CRM cannot visualize collagen monomers, oligomers, and perhaps early fibrils, its use in describing early events of collagen self-assembly is limited. To enhance understanding of these early events, time-lapse CFM images during self-assembly of FITC-labeled collagen were collected. Performed with commonly employed fluorescent tags, CFM is a more sensitive technique than CRM, under certain circumstances requiring as little as one fluorophore in the focal volume for signal to be discriminated from background. Furthermore, CFM can detect fibrils at all orientations, whereas CRM has limited sensitivity to out-of-plane oriented fibrils (35,36). CFM does have disadvantages compared to CRM for imaging collagen self-assembly: first, bleaching limits the proportion of the gelation process that can be followed in a given movie; additionally, labeling has been shown to inhibit fibril thickening and bundling in low temperature gelation (15).
At all concentrations and gelation temperatures investigated, CFM time-lapse images displayed more structure at early time points than did CRM images (Fig. 2). The earliest images revealed small isotropic features of moderate intensity against a lower intensity homogeneous background. Fig. 2 a shows the first image collected for a FITC-labeled collagen solution of 1.0 mg/ml at 32°C. At this time, the corresponding CRM image (Fig. 2 b) shows no structures. In CFM images, accretion into brighter spots on a darker background occurred within tens of seconds. At first, the bright structures were isotropic and mobile, and as their number increased they became anisotropic and fixed in position. By the arrest time (Fig. 2 c), at 5.6 min for the sample shown here, fibrils were evident. CRM images collected at this time also showed fibrils, though they appeared quite dim (Fig. 2 d).
Figure 2.

Comparison of CFM and CRM images at time points of interest. Images are from a representative 1.0 mg/ml collagen sample gelled at 32°C also shown in Video S1. Left panel: The first images collected, 88 s after neutralization. (a) The initial CFM image reveals small isotropic features of moderate intensity against a lower intensity homogeneous background, whereas (b) the earliest CRM image reveals no structures. Right panel: Images at arrest and plateau. (c) The CFM image at tARR (5.6 min) reveals many structures. (d) The CRM image at arrest displays structures of low intensity that can be better visualized when (e) contrast is enhanced. (f) The CRM image at plateau (12.5 min) reveals that many structures in the fully developed gel can already be visualized at arrest (as in (c) and (e)). Scale bar is 25 μm for all images.
Compared to CRM, CFM is expected to be relatively insensitive to fibril width. As such, complementary analysis of CFM and CRM time-lapse images of collagen self-assembly can clarify the timescales of early fibril formation and fibril thickening. At arrest, CFM images revealed more structures than CRM images. After arrest, CFM images showed very little change except for overall photobleaching. CRM images, however, appeared to have increasing numbers of fibrils and increasingly bright fibrils. Overlaying the CFM image at arrest (Fig. 2 c) with the CRM image at arrest (Fig. 2 d) shows the difference in visualized structures at that time, though enhancing contrast of the CRM image reveals a substantial number of fibrils (Fig. 2 e). Overlaying the CFM image at arrest (Fig. 2 c) with the CRM image at plateau (Fig. 2 f) shows strong colocalization. Video S1 shows time-lapse CFM and CRM imaging of the sample depicted in Fig. 2. Taken together, these images show that most fibrils are already present at arrest. Most of the change in CRM images after arrest must therefore be due to increasing width of fibrils that are already present.
CRM reveals structures responsible for turbidity
The sequence of events revealed by CRM and CFM of collagen I self-assembly—as well as the structure of the fully developed gels, with fewer and thicker fibrils at lower temperature—is broadly consistent with the nucleation and growth model of collagen self-assembly. Such a model was first proposed based on turbidity measurements showing a sigmoidal increase in turbidity during collagen self-assembly, with a lag phase of near zero turbidity followed by a growth phase with quickly increasing turbidity (19,21). In this picture, during the lag phase nucleating structures are formed, after which the growth phase—characterized by quickly increasing turbidity—begins. During the growth phase, monomers self-assemble into fibrils anchored on the nucleating structures in an entropically driven self-assembly (37). As monomers are depleted, the growths phase ends and turbidity plateaus. Implicit in this understanding of the nucleation and growth model for collagen gelation is that no large structures are present in the lag phase, though a few early studies suggested otherwise (20,38).
CRM and CFM report details of collagen gelation that can only be inferred from turbidity measurements. However, turbidity continues to be the method of choice to monitor the dynamics of biopolymer self-assembly. To compare the sequence of events revealed by CRM to those inferred from turbidity, CRM images were directly compared to turbidity measurements performed simultaneously using the multimodal microscopy set-up depicted in Fig. S1. First, traditional turbidity measurements monitoring collagen gelation in a spectrophotometer were compared to results from IST measurements. For comparison with traditional turbidity measurements, scanning transmittance images were reduced to the single variable of IST, as described in the Materials and Methods. Differences between the IST measurement and the traditional turbidity measurement as performed here include that the IST measurement used a polarized light source, a high NA objective lens, a thinner sample, and a different apparatus for temperature control. Fig. 3 shows a standard turbidity measurement and IST as measured via scanning transmittance microscopy of 1.0 mg/ml collagen samples undergoing gelation at 37°C. Although kg was very similar in these measurements, time points of interest occurred approximately a minute later in standard turbidity than in IST. The discrepancy resolved when performing IST on a thicker sample, and we suggest the time difference is due to differences in heat transfer in the two set-ups. Once corrected for this time difference, turbidity and IST curves overlapped very well, as is also evident in the wavelength dependent scans shown in Fig. S3. These measurements validated the use of IST as a measure of turbidity in a collagen solution undergoing gelation.
Figure 3.
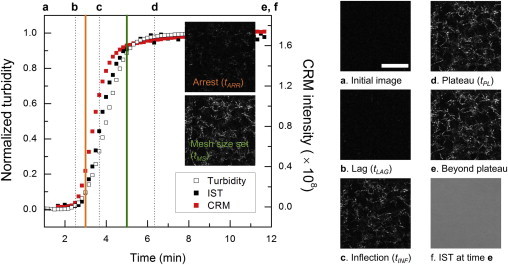
Traditional turbidity measurements track IST and CRM intensity evolution. At left, turbidity as measured with a spectrophotometer (black open squares), IST (black solid squares), and CRM intensity (red squares) from representative 1.0 mg/ml collagen samples gelled at 37°C. The standard turbidity curve was shifted by one minute as described in the text. At right, images corresponding to dotted lines at time points of interest extracted from the IST curve: (a) initial CRM image, (b) CRM image at tLAG, (c) CRM image at tINF, (d) CRM image at tPL, (e) CRM image beyond plateau, and (f) IST image beyond plateau. Images corresponding to tARR (orange line) and tMS (green line) are shown as insets at left. Scale bar is 25 μm for all images. All images are unprocessed but cropped and show the same representative portion of the full image. To see this figure in color, go online.
Having validated the IST measurement, CRM images were compared to key time points along the development of the turbidity curve during collagen self-assembly (Fig. 3). In Fig. 3, select CRM (and in one case, IST) images from the gelation of a 1.0 mg/ml collagen solution at 37°C are shown together with the corresponding time points on the IST curve, which was used to assign tLAG, tINF, and tPL. CRM images at these times revealed that at tLAG very few structures were visualized but by tINF many structures were present; in fact, most of the fibers present at tPL as well as at the end of the measurement colocalized with those present at tINF, though the structures were brighter at the later time points.
To complement information from time points obtained from turbidity, tARR and tMS, which can only be identified from CRM, were also determined. Arrest was found to occur shortly after tLAG determined from CRM for gels of all concentrations formed at all temperatures. From analysis of CRM and CFM images, it appears that most of the fibrils in the final network were already present at the arrest time even though <10% of the final turbidity was established by this time. As such, most of the increase in turbidity must be due to thickening of fibrils that were already present.
This observation is reinforced by the image at tMS, the time at which mesh size is no longer evolving. Mesh (or pore) size has emerged as a principal reporter of network structure in biopolymer gels because this quantity can be determined with ease and figures prominently in theories predicting how network structure sets material properties (15,24,39). Mesh size analysis yielded values for the fully developed gels consistent with earlier reports (12,15,40) though alternate approaches have yielded smaller mesh sizes (10,24,36). Because relative mesh size was of interest here, differences in absolute mesh size would not alter any conclusions of this study.
At early time points, when no or few fibrils were visible, mesh size could not be accurately determined, and therefore discussion of mesh size is limited to times beyond tARR. At the earliest time points assessed, between tARR and tINF, there was a steep decay in mesh size (Fig. 4a). Indeed, the great majority of mesh size evolution—from the dimensions of the sample to tens of microns—occurred before tINF, even though this point defines the time at which only half of the final turbidity had been established. This is consistent with the idea that mesh size is primarily sensitive to existence of fibrils but not their width, so long as mesh size is significantly greater than fibril width. Beyond the inflection time, CRM imaging suggested fibrils continue to thicken. This continued increase of fibril diameter resulted in slow evolution of mesh size even as turbidity continued to increase quickly, as increasing fibril width has a greater effect on turbidity than on mesh size.
Figure 4.
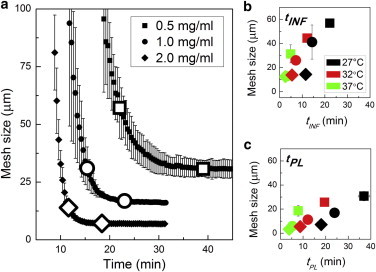
Mesh size evolution during fibrillogenesis. (a) Mesh size during gelation of 0.5 mg/ml (squares), 1.0 mg/ml (circles), and 2.0 mg/ml (diamonds) at 27°C. Every other point is shown for clarity. Large symbols indicate tINF and tPL as determined from CRM intensity curves. Mesh size at (b) tINF and (c) tPL vs. (b) tINF and (c) tPL for all gels. Error bars represent standard deviations and are shown when larger than the symbols. To see this figure in color, go online.
Plotting mesh size at tINF and tPL against time showed that time to inflection and plateau were only correlated with mesh size at a given temperature (Fig. 4, b and c). Fig. 4 c reveals that at tPL, when mesh size is no longer evolving, mesh size varied more strongly with concentration for gels formed at higher temperature than lower temperature. This is consistent with the idea that increased collagen primarily leads to more fibrils at high temperatures and supports, instead, thicker fibrils at low temperatures. The evolution of mesh size from tINF to tPL revealed a similar decrease in mesh size across gel concentrations and gelation temperatures, with mesh size of all gels decreasing by a factor of ≈2 between these time points (Fig. 4, b and c). This trend indicates that the differential in fibril thickness present at plateau was already present at tINF. CRM results at tINF—though not those earlier, at tARR—also support that conclusion, as will be discussed below.
Quantitative comparison of turbidity and CRM intensity
Comparing CRM images to points along the IST curve revealed how the number and dimension of the developing structures lead to increased turbidity. A second key goal of this study was investigating whether CRM images could be reduced to a single variable that 1) tracked turbidity development in time and 2) reflected relative turbidity of a set of gels of different collagen content and network structure. A straightforward single variable for such comparison is CRM total intensity. CRM intensity and IST curves for all gels investigated are shown in Fig. 5. It is apparent that all CRM intensity curves displayed sigmoidal shapes like the turbidity curves and that relative values of CRM and IST of the fully formed gels were similar for samples at all concentrations and gelation temperatures.
Figure 5.
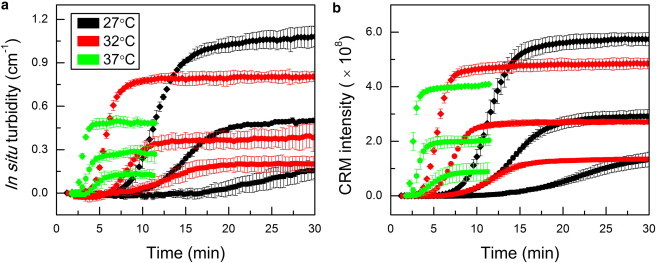
Average (a) IST and (b) CRM curves for 0.5 mg/ml (squares), 1.0 mg/ml (circles), and 2.0 mg/ml (diamonds) collagen solutions gelled at 27°C (black), 32°C (red), and 37°C (green). Every other point is shown for clarity. Error bars represent standard deviations. To see this figure in color, go online.
Turbidity and CRM intensity report similar gelation time course
Although turbidity and CRM both rely on scattering from collagen fibrils, whether turbidity and CRM intensity would reveal the same information over the full course of the self-assembly process was not immediately clear. A standard turbidity measurement and IST as performed here monitor light in the forward direction, and the decrease of that light represents the scattering in all directions excepting a small cone in the forward direction. CRM, by contrast, directly monitors only the light scattered backward that can be collected by the objective lens. For the measurements presented here, an oil objective with NA = 1.42 was employed, and light emerging in the epi-direction in a cone of ≈140° was collected. Over time, the scattering entities in a developing collagen gel change size and shape, becoming larger and more anisotropic. This is expected to alter the angular distribution of scattering over the course of gelation (41–43), potentially impacting the relative sensitivity of IST and CRM. Indeed, the proportion of scattering detected by CRM may be expected to be highest relative to that detected by turbidity measurements early in the gelation process (42–44). Similar arguments have been invoked when considering fibrin gelation as assessed by angle-dependent scattering compared to turbidity (45).
To quantitatively compare the time course of the development of IST and CRM intensity, tLAG, tINF, tPL, and kG were obtained from each curve. Fig. 6 reveals that there was clear correlation between each of the time points as defined through IST and CRM intensity at all collagen concentrations and gelation temperatures. Similarly, kG was strongly correlated between these measurements. Linear fits to the data in Fig. 6, a–c, described the data very well (R2 > 0.98) with slopes of 0.81, 0.94, and 0.96 for tLAG, tINF, and tPL, respectively. The fact that the slopes were somewhat <1 shows that the time points of interest occur earlier in CRM than IST, as is also evident in Fig. 3 (left panel). This was most obvious at the early time points, consistent with the idea that changes in angular distribution of scattering intensity during fibril development may lead to enhanced sensitivity of CRM relative to IST early in the gelation process. Indeed, we found that the full network structure was in place (tARR) shortly after tLAG as defined by CRM, when very little turbidity had developed.
Figure 6.
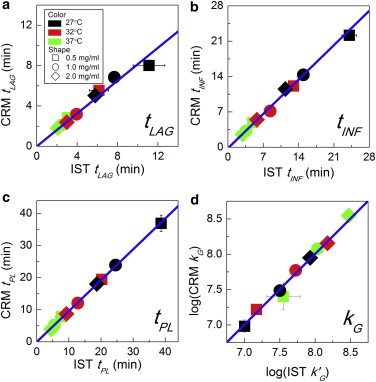
Time points of interest (tLAG, tINF, tPL) and slope of the growth phase (kG) obtained from CRM versus those obtained from IST: (a) tLAG, (b) tINF, (c) tPL, and (d) kG. Linear regression was performed with the y-intercept set to zero, yielding slopes of 0.81, 0.94, and 0.96 with R2 ≥ 0.98. In (a), if the 27°C 0.5 mg/ml point is excluded from the fit, the slope of the best-fit line is 0.88 and all R2 > 0.998. In (d), IST kG values were normalized by multiplying by final CRM intensity over final IST, yielding k′G. Error bars are standard deviations and are shown only when larger than the symbols. Colors and symbols for (a–d) are as shown in the legend in (a). To see this figure in color, go online.
Turbidity and CRM intensity are similarly sensitive to fibril number and dimension
As highlighted by Fig. 3, although CRM intensity and IST tracked each other in time, CRM contains direct information about the fibrillar structures that give rise to scattering. To investigate whether CRM and IST were similarly sensitive to fibril number and dimension, IST and CRM intensity values at particular time points during gelation were considered.
Previously, it was shown that at specific gelation conditions the ratio of turbidity to concentration of the fully developed gel, A∞ = τ∞/c, is constant (19). IST measurements at plateau were consistent with this finding, as shown in Fig. 7 a and Table S1. The values of APL = ISTPL/c, with plateau time determined from the IST curve, for a given temperature varied by no more than 15%, though IST data showed significant variation across samples of given concentration. Similar results were seen at the inflection time (AINF = ISTINF/c), when all fibrils are expected to be present but only half of the final turbidity has developed, as the fibrils have not yet reached their ultimate thickness (Fig. 7 a). Although gelation at a given temperature led to A values that were nearly constant across concentration, these values increased with decreasing gelation temperature, consistent with the idea that greater fibril width leads to increased scattering. The values of APL and AINF averaged over concentrations show AINF varied with gelation temperature to the same degree as APL (Table S1). This again indicates that differential in fibril width between structures formed at different temperatures was fully established by the inflection time, halfway through the growth phase of the turbidity curve.
Figure 7.
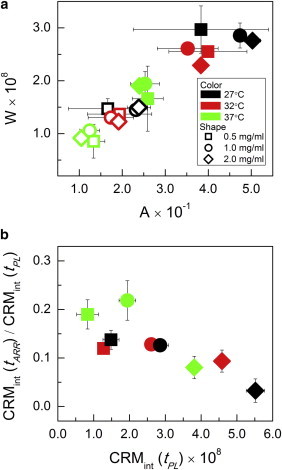
(a) The ratio of CRM intensity to concentration (W = CRMint/c) versus that of IST to concentration (A = IST/c) at tINF (open symbols) and tPL (solid symbols). Time points evaluated were determined from the CRM intensity curve for W and the IST curve for A. (b) Ratio of CRM intensity at tARR to that at tPL versus CRM intensity at tPL. Symbols and colors are as shown in (a). Error bars indicate standard deviations and are shown only when larger than the symbols. To see this figure in color, go online.
The results for CRM intensity (CRMint) mirrored those of IST, as shown in Fig. 7 a and Table S1. Indeed the values of W = CRMint/c varied slightly less for a given temperature across concentrations at both plateau and inflection than did the corresponding A values. Averaged over all samples at a given temperature, to within error W decreased with increasing gelation temperature in the same proportion as did A. Although WPL and WINF were constant at a given temperature across concentrations and decreased as a function of increasing gelation temperature, neither of these trends held at the arrest time. Indeed, CRM intensities at arrest varied little with concentration for gelation at a given temperature, consistent with arrest time being defined by the presence of a minimum spanning network. Additionally, the quantity WARR = CRMint,ARR/c showed no trend with gelation temperature, supporting the idea that fibril width differential as a function of temperature is not yet established by the arrest time, whereas it is established by the inflection time. These conclusions were strengthened by examining the proportion of plateau intensity that was present at arrest (Fig. 7 b). At a given temperature, a decreasing proportion of CRM plateau intensity was established at arrest as gel concentration increased. Additionally, for a given concentration, with decreasing gelation temperature a decreasing proportion of plateau intensity was present at arrest, consistent with the idea that most fibril thickening occurs after arrest. Fig. 7 b reveals that the fully developed gels that scattered the most, due to a large number of fibrils and/or thick fibrils, had the smallest proportion of their ultimate scattering present at arrest. This is consistent with the presence of a minimal spanning structure in which ultimate fibril diameter differential is not yet established at arrest.
To further test the hypothesis that the putative network consists of thin rods regardless of gel conditions and fully formed gel structure and to assess the relative sensitivity of the techniques employed in this study, fibril diameter during gelation was assessed using wavelength-dependent measurements and analysis based on the theory of scattering from long rods as described in the Supporting Note (Fig. S3) (34,44,46,47). Using this approach yielded an estimated fibril width of 54 nm for 1.0 mg/ml collagen gels formed at 37°C (Fig. S4), a value consistent with some results from turbidity, atomic force microscopy, and electron microscopies, though diameters up to a factor of two larger have also been reported (13,15,19,33,48,49).
Performing analysis of wavelength-dependent measurements as a function of time during gelation suggested that at arrest fibrils in 1.0 mg/ml gels forming at 37°C were ≈20 nm. The same measurement and analysis was then employed on 1.0 mg/ml gels forming at 27°C and also showed that fibrils at arrest were ≈20 nm even though this approach suggests they were nearly 100 nm in the fully formed gel (Fig. S4). Taken together, this data further supports the conclusion that the initial network spanning structure is composed of thin fibrils that are relatively insensitive to gelation conditions and that fibril thickening is the chief activity of the turbidimetric growth phase. These measurements and analysis also suggest that CRM can be employed to visualize collagen fibrils as thin as 20 nm.
Conclusion
IST measurements of collagen self-assembly performed simultaneously with confocal imaging were employed to reveal the structures that underlie the development of turbidity during collagen self-assembly. All trends seen in IST measurements across fully developed collagen gels of different concentrations and gelation temperatures were also present when evaluating CRM intensity, showing the two techniques are similarly sensitive to fibril number and dimension. The time course of IST and CRM development was also similar, with CRM revealing increased sensitivity to early scatterers. Despite the similarity of turbidity and CRM intensity measurements, taken together CRM and CFM support a somewhat different picture of the lag and growth phases of collagen self-assembly compared to that inferred from turbidity measurements alone. Our findings exclude the possibility that no large structures are present in the lag phase and that turbidity tracks precipitation through the entirety of the gelation process, demonstrate that a minimal spanning structure that is relatively insensitive to gelation conditions is present before turbidity develops, and show that the majority of the increase in turbidity is attributable to fibril thickening. In addition to clarifying details on the time course of collagen gelation, given scattering cross sections reported from different biopolymer fibers (50), we suggest that simultaneous CRM and IST measurements may also be applied to the study of gelation of a wide range of intracellular and extracellular biopolymers.
Acknowledgments
The authors thank Alana Warhit for assistance in spectrophotometric turbidity measurements and Keewook Paeng, Dat Hoang, David Reichman, Bruce Berne, and François Caton for helpful discussions.
This work was supported by the National Science Foundation via a Graduate Research Fellowship for J.Z. and by the National Science Foundation and the National Institutes of Health under grant award PESO 1227297.
Supporting Material
References
- 1.Alberts B. Garland Science; New York: 2008. Molecular Biology of the Cell. [Google Scholar]
- 2.Friedl P., Bröcker E.B. The biology of cell locomotion within three-dimensional extracellular matrix. Cell. Mol. Life Sci. 2000;57:41–64. doi: 10.1007/s000180050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petroll W.M., Ma L.S. Direct, dynamic assessment of cell-matrix interactions inside fibrillar collagen lattices. Cell Motil. Cytoskeleton. 2003;55:254–264. doi: 10.1002/cm.10126. [DOI] [PubMed] [Google Scholar]
- 4.Kaufman L.J., Brangwynne C.P., Weitz D.A. Glioma expansion in collagen I matrices: analyzing collagen concentration-dependent growth and motility patterns. Biophys. J. 2005;89:635–650. doi: 10.1529/biophysj.105.061994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaman M.H., Trapani L.M., Matsudaira P. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc. Natl. Acad. Sci. USA. 2006;103:10889–10894. doi: 10.1073/pnas.0604460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cen L., Liu W., Cao Y.L. Collagen tissue engineering: development of novel biomaterials and applications. Pediatr. Res. 2008;63:492–496. doi: 10.1203/PDR.0b013e31816c5bc3. [DOI] [PubMed] [Google Scholar]
- 7.Glowacki J., Mizuno S. Collagen scaffolds for tissue engineering. Biopolymers. 2008;89:338–344. doi: 10.1002/bip.20871. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y.L., Motte S., Kaufman L.J. Pore size variable type I collagen gels and their interaction with glioma cells. Biomaterials. 2010;31:5678–5688. doi: 10.1016/j.biomaterials.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 9.Grinnell F., Petroll W.M. Cell motility and mechanics in three-dimensional collagen matrices. Annu. Rev. Cell Dev. Biol. 2010;26:335–361. doi: 10.1146/annurev.cellbio.042308.113318. [DOI] [PubMed] [Google Scholar]
- 10.Wolf K., Te Lindert M., Friedl P. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013;201:1069–1084. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kadler K.E., Hill A., Canty-Laird E.G. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008;20:495–501. doi: 10.1016/j.ceb.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raub C.B., Unruh J., George S.C. Image correlation spectroscopy of multiphoton images correlates with collagen mechanical properties. Biophys. J. 2008;94:2361–2373. doi: 10.1529/biophysj.107.120006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raub C.B., Suresh V., George S.C. Noninvasive assessment of collagen gel microstructure and mechanics using multiphoton microscopy. Biophys. J. 2007;92:2212–2222. doi: 10.1529/biophysj.106.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roeder B.A., Kokini K., Voytik-Harbin S.L. Fibril microstructure affects strain transmission within collagen extracellular matrices. J. Biomech. Eng. 2009;131:031004. doi: 10.1115/1.3005331. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y.L., Leone L.M., Kaufman L.J. Elastic moduli of collagen gels can be predicted from two-dimensional confocal microscopy. Biophys. J. 2009;97:2051–2060. doi: 10.1016/j.bpj.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Achilli M., Mantovani D. Tailoring mechanical properties of collagen-based scaffolds for vascular tissue engineering: the effects of pH, temperature and ionic strength on gelation. Polymers. 2010;2:664–680. [Google Scholar]
- 17.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 18.Gross J., Kirk D. The heat precipitation of collagen from neutral salt solutions: some rate-regulating factors. J. Biol. Chem. 1958;233:355–360. [PubMed] [Google Scholar]
- 19.Wood G.C., Keech M.K. The formation of fibrils from collagen solutions. 1. The effect of experimental conditions: kinetic and electron-microscope studies. Biochem. J. 1960;75:588–598. doi: 10.1042/bj0750588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams B.R., Gelman R.A., Piez K.A. Collagen fibril formation. Optimal in vitro conditions and preliminary kinetic results. J. Biol. Chem. 1978;253:6578–6585. [PubMed] [Google Scholar]
- 21.Wood G.C. The formation of fibrils from collagen solutions. 2. A mechanism of collagen-fibril formation. Biochem. J. 1960;75:598–605. doi: 10.1042/bj0750598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Comper W.D., Veis A. The mechanism of nucleation for in vitro collagen fibril formation. Biopolymers. 1977;16:2113–2131. doi: 10.1002/bip.1977.360161004. [DOI] [PubMed] [Google Scholar]
- 23.Comper W.D., Veis A. Characterization of nuclei in in vitro collagen fibril formation. Biopolymers. 1977;16:2133–2142. doi: 10.1002/bip.1977.360161005. [DOI] [PubMed] [Google Scholar]
- 24.Mickel W., Münster S., Schröder-Turk G.E. Robust pore size analysis of filamentous networks from three-dimensional confocal microscopy. Biophys. J. 2008;95:6072–6080. doi: 10.1529/biophysj.108.135939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stein A.M., Vader D.A., Sander L.M. The micromechanics of three-dimensional collagen-I gels. Complexity. 2011;16:22–28. [Google Scholar]
- 26.Zoumi A., Yeh A., Tromberg B.J. Imaging cells and extracellular matrix in vivo by using second-harmonic generation and two-photon excited fluorescence. Proc. Natl. Acad. Sci. USA. 2002;99:11014–11019. doi: 10.1073/pnas.172368799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams R.M., Zipfel W.R., Webb W.W. Interpreting second-harmonic generation images of collagen I fibrils. Biophys. J. 2005;88:1377–1386. doi: 10.1529/biophysj.104.047308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brightman A.O., Rajwa B.P., Voytik-Harbin S.L. Time-lapse confocal reflection microscopy of collagen fibrillogenesis and extracellular matrix assembly in vitro. Biopolymers. 2000;54:222–234. doi: 10.1002/1097-0282(200009)54:3<222::AID-BIP80>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 29.Yang Y.L., Kaufman L.J. Rheology and confocal reflectance microscopy as probes of mechanical properties and structure during collagen and collagen/hyaluronan self-assembly. Biophys. J. 2009;96:1566–1585. doi: 10.1016/j.bpj.2008.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forgacs G., Newman S.A., Sackmann E. Assembly of collagen matrices as a phase transition revealed by structural and rheologic studies. Biophys. J. 2003;84:1272–1280. doi: 10.1016/S0006-3495(03)74942-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Margaron Y., Bostan L., Lethias C. Tenascin-X increases the stiffness of collagen gels without affecting fibrillogenesis. Biophys. Chem. 2010;147:87–91. doi: 10.1016/j.bpc.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 32.Kreger S.T., Bell B.J., Voytik-Harbin S.L. Polymerization and matrix physical properties as important design considerations for soluble collagen formulations. Biopolymers. 2010;93:690–707. doi: 10.1002/bip.21431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Wild M., Pomp W., Koenderink G.H. Thermal memory in self-assembled collagen fibril networks. Biophys. J. 2013;105:200–210. doi: 10.1016/j.bpj.2013.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carr M.E., Jr., Hermans J. Size and density of fibrin fibers from turbidity. Macromolecules. 1978;11:46–50. doi: 10.1021/ma60061a009. [DOI] [PubMed] [Google Scholar]
- 35.Jawerth L.M., Münster S., Weitz D.A. A blind spot in confocal reflection microscopy: the dependence of fiber brightness on fiber orientation in imaging biopolymer networks. Biophys. J. 2010;98:L1–L3. doi: 10.1016/j.bpj.2009.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lang N.R., Münster S., Fabry B. Estimating the 3D pore size distribution of biopolymer networks from directionally biased data. Biophys. J. 2013;105:1967–1975. doi: 10.1016/j.bpj.2013.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kadler K.E., Holmes D.F., Chapman J.A. Collagen fibril formation. Biochem. J. 1996;316:1–11. doi: 10.1042/bj3160001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silver F.H., Birk D.E. Kinetic analysis of collagen fibrillogenesis: I. Use of turbidity—time data. Coll. Relat. Res. 1983;3:393–405. doi: 10.1016/s0174-173x(83)80020-x. [DOI] [PubMed] [Google Scholar]
- 39.de Gennes P.G. Cornell University Press; Ithaca, NY: 1979. Scaling Concepts in Polymer Physics. [Google Scholar]
- 40.Banerjee P., Lenz D., Bhunia A.K. A novel and simple cell-based detection system with a collagen-encapsulated B-lymphocyte cell line as a biosensor for rapid detection of pathogens and toxins. Lab. Invest. 2008;88:196–206. doi: 10.1038/labinvest.3700703. [DOI] [PubMed] [Google Scholar]
- 41.Gelman R.A., Piez K.A. Collagen fibril formation in vitro. A quasielastic light-scattering study of early stages. J. Biol. Chem. 1980;255:8098–8102. [PubMed] [Google Scholar]
- 42.Silver F.H. Type I collagen fibrillogenesis in vitro. Additional evidence for the assembly mechanism. J. Biol. Chem. 1981;256:4973–4977. [PubMed] [Google Scholar]
- 43.Cox A.J., DeWeerd A.J., Linden J. An experiment to measure Mie and Rayleigh total scattering cross sections. Am. J. Phys. 2002;70:620–625. [Google Scholar]
- 44.Casassa E.F. Light scattering from very long rod-like particles and an application to polymerized fibrinogen. J. Chem. Phys. 1955;23:596–597. [Google Scholar]
- 45.Ferri F., Greco M., Rocco M. Growth kinetics and structure of fibrin gels. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2001;63:031401. doi: 10.1103/PhysRevE.63.031401. [DOI] [PubMed] [Google Scholar]
- 46.Berne B.J. Interpretation of the light scattering from long rods. J. Mol. Biol. 1974;89:755–758. doi: 10.1016/0022-2836(74)90049-7. [DOI] [PubMed] [Google Scholar]
- 47.Yeromonahos C., Polack B., Caton F. Nanostructure of the fibrin clot. Biophys. J. 2010;99:2018–2027. doi: 10.1016/j.bpj.2010.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piechocka I.K., van Oosten A.S.G., Koenderink G.H. Rheology of heterotypic collagen networks. Biomacromolecules. 2011;12:2797–2805. doi: 10.1021/bm200553x. [DOI] [PubMed] [Google Scholar]
- 49.Graham J.S., Vomund A.N., Grandbois M. Structural changes in human type I collagen fibrils investigated by force spectroscopy. Exp. Cell Res. 2004;299:335–342. doi: 10.1016/j.yexcr.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 50.Janmey P.A., Euteneuer U., Schliwa M. Viscoelastic properties of vimentin compared with other filamentous biopolymer networks. J. Cell Biol. 1991;113:155–160. doi: 10.1083/jcb.113.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brokaw J.L., Doillon C.J., Silver F.H. Turbidimetric and morphological-studies of type-I collagen fiber self assembly in vitro and the influence of fibronectin. Int. J. Biol. Macromol. 1985;7:135–140. [Google Scholar]
- 52.Holmes D.F., Kadler K.E. The precision of lateral size control in the assembly of corneal collagen fibrils. J. Mol. Biol. 2005;345:773–784. doi: 10.1016/j.jmb.2004.10.078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.