

Abstract
There is a growing interest in understanding the properties of intrinsically disordered proteins (IDPs); however, the characterization of these states remains an open challenge. IDPs appear to have functional roles that diverge from those of folded proteins and revolve around their ability to act as hubs for protein-protein interactions. To gain a better understanding of the modes of binding of IDPs, we combined statistical mechanics, calorimetry, and NMR spectroscopy to investigate the recognition and binding of a fragment from the disordered protein Gab2 by the growth factor receptor-bound protein 2 (Grb2), a key interaction for normal cell signaling and cancer development. Structural ensemble refinement by NMR chemical shifts, thermodynamics measurements, and analysis of point mutations indicated that the population of preexisting bound conformations in the free-state ensemble of Gab2 is an essential determinant for recognition and binding by Grb2. A key role was found for transient polyproline II (PPII) structures and extended conformations. Our findings are likely to have very general implications for the biological behavior of IDPs in light of the evidence that a large fraction of these proteins possess a specific propensity to form PPII and to adopt conformations that are more extended than the typical random-coil states.
Introduction
Protein-protein interactions (PPint) regulate a large number of biomolecular processes. It is becoming increasingly evident that to achieve a comprehensive understanding of partner recognition and binding affinity in PPint, in addition to the structures of the macromolecular complexes, it is necessary to characterize in detail the conformations that proteins and peptides adopt before binding (1). Dynamical behavior is indeed a key modulator of the energetics of macromolecular interactions. Biomolecular structural fluctuations of backbone and side chain atoms are finely tuned to allow processes such as partner selection (2), allosteric modulation (3), and cellular signaling (4–8). Structural dynamics are particularly relevant in weak bindings and in molecular interactions that involve intrinsically disordered proteins (IDPs) (9–11). It is now clear that disordered protein states are highly abundant in all living organisms and have biological activities that can be distinct from those of folded proteins (12–15). In this context, understanding the mechanisms by which IDPs recognize and interact with their partners remains a major challenge. This task requires a structural characterization based on probability distributions to account for the conformational heterogeneity of IDPs rather than single structures.
Efforts to define a relationship among the local dynamics, residual structure, and binding properties of IDPs are hampered by the intrinsic limits of standard experimental techniques. High-resolution structural techniques such as x-ray crystallography are able to indirectly imply structural disorder, but they cannot quantitatively characterize transient structural states and populations adopted in IDP ensembles. Such information can be partially obtained from single-molecule experiments (16), but at a significantly lower resolution than atomic details. Recent advances in biomolecular NMR have enabled new approaches to probe the conformational preferences of disordered protein states (17–21). These methods provide new opportunities to characterize transient structures and their populations in the conformational ensembles of IDPs, with significant accuracy (22–24). This study illustrates the latter point very clearly by addressing the solution properties of a disordered protein fragment and by defining their relationship with the thermodynamic affinity for a binding partner. We obtained this result by measuring backbone chemical shifts (CSs) and employing the resulting data to study transient secondary-structure populations. To that end, we used the δ2D method (22,25) and backbone dynamics (using the random coil index (RCI)) (26), as well as ensemble-averaged restrained molecular-dynamics (MD) simulations with full representation of all protein and solvent atoms. Using this approach, we characterized the conformational determinants of complex formation between the growth factor receptor-bound protein 2 (Grb2) and the disordered protein Grb2-associated binder 2 (Gab2) (27–31).
Grb2 is a 25 kDa adaptor protein that is involved in signal transduction and cell communication, and is composed of an SH2 domain flanked by N- and C-terminal SH3 domains. Gab proteins belong to a family of large multisite docking (LMD) proteins that play scaffolding roles in the assembly of large multimolecular complexes (32–34). These proteins are often implicated in signal integration and serve as processing platforms for several signaling pathways. We focused on Gab2, a largely intrinsically disordered protein of ∼74 kDa with a folded N-terminal PH domain whose interaction with Grb2 has been implicated in normal cell signaling (35) and cancer development (27). Peptide arrays showed two binding sites in Gab2 (Gab2a and Gab2b) interacting with the C-terminal domain of Grb2 (Grb2 SH3C). Both epitopes host an RxxK motif, which is essential for Grb2 SH3C binding, with the highest affinity found in the Gab2b motif (27).
We employed the fragment 503–524 of Gab2 (Gab2503-524), i.e., spanning the Gab2b motif, to characterize the binding affinity for the SH3C domain of Grb2, which occurs with a low micromolar affinity (Table 1). Our NMR CS analyses showed that Gab2503-524 is mainly disordered in solution, which is in agreement with the conformational properties adopted in the full-length Gab2 sequence (36), but adopts some residual structure of extended β and polyproline type II (PPII) character. Structural ensemble refinement, isothermal titration calorimetry (ITC), and analysis of the effects of point mutations revealed that the population of preexisting bound conformations in the free-state ensemble is an essential factor in the recognition and binding of Gab2b by Grb2 SH3C. Transient PPII segments were shown to play a key role in the affinity of this PPint. This structural motif is a well-known recognition element for SH3 domains and has been implicated in the interaction properties of IDPs. The data presented here therefore address the molecular determinants of binding modes in IDPs and show how a description of their structural propensities as probability distributions derived from the information contained in NMR data can lead to a thorough understanding of their biological properties.
Table 1.
ITC measurements of Grb2 SH3C interactions with WT and mutant peptides derived from Gab2
| Sequence | na | Kd (μM) | ΔH (kcal·mol−1) | −TΔS (kcal·mol−1) | |
|---|---|---|---|---|---|
| WT | SRGSEIQPPPVNRNLKPDRKAK | 0.91 ± 0.06 | 3.95 ± 0.51 | −11.8 ± 1.33 | 4.46 ± 1.42 |
| P510A | SRGSEIQAPPVNRNLKPDRKAK | 1.11 ± 0.07 | 5.09 ± 0.34 | −10.4 ± 0.94 | 3.15 ± 0.99 |
| P511A | SRGSEIQPAPVNRNLKPDRKAK | – | TLQb | – | – |
| P512A | SRGSEIQPPAVNRNLKPDRKAK | 0.99 ± 0.06 | 6.00 ± 0.25 | −11.3 ± 0.34 | 4.22 ± 0.31 |
| R515A | SRGSEIQPPPVNANLKPDRKAK | – | NBDc | – | – |
| K518A | SRGSEIQPPPVNRNLAPDRKAK | – | NBDc | – | – |
| P519A | SRGSEIQPPPVNRNLKADRKAK | 0.84 ± 0.01 | 57.85 ± 1.7 | −17.2 ± 0.36 | 11.4 ± 0.39 |
Stoichiometry.
Affinity was too low for exact quantification.
No binding was detected.
Materials and Methods
Peptide synthesis and purification
Peptides spanning the wild-type (WT) sequence (SRGSEIQPPPVNRNLKPDRKAK) of Gab2503-524 or point mutations (see Table 1) were produced by 9-fluorenylmethoxycarbonyl (Fmoc) solid-phase peptide synthesis (37) using a Respep SL tabletop peptide synthesizer (Intavis Bioanalytical Instruments, Koeln, Germany). Briefly, amino acids with amines protected by Fmoc groups were coupled via their carboxyl groups to the amino groups of a growing chain starting with an Fmoc-lysine Wang resin (two wells, each containing 20 μmol). Before each round of coupling, the unreacted amino acids were washed out and the N-terminus was deprotected with piperidine/dimethylformamide. After the final deprotection step, the resins were removed from the synthesizer and the peptides were cleaved off with trifluoroacetic acid (TFA) in a sealed syringe.
TFA cleavage products were precipitated with ice-cold tert-butyl methyl ether (TBME) and centrifuged three times, with replacement of the supernatant with fresh TBME at 6000 × g for 10 min to wash away the TFA. These pellets were then desiccated overnight and redissolved in 4 ml of a 12.5% methanol/water solution. Then 20 μl of these solutions was analyzed by electrospray ionization (ESI) coupled with liquid chromatography/mass spectrometry (LC-MS) on a system comprised of a Waters (Waters, Milford, MA) 515 HPLC pump, a Waters Micromass ZQ MS detector, a Waters 600 controller, a Waters 2998 photodiode array (PDA) detector, and a Waters sample manager. Then 4 ml was taken for preparative LC-MS and used with mass-triggered collection of LC fractions for peptide purification. Methanol was removed from the purified peptide solutions on an EZ-2 plus centrifugal evaporator (Genevac, Ipswich, UK) and water was removed by lyophilization in preweighed microcentrifuge tubes (Eppendorf, Hamburg, Germany).
Purification of Grb2 SH3C
The pGEX vector encoding Grb2 SH3C has been described elsewhere (38). For protein expression, bacteria were grown in terrific broth with 100 μg/ml ampicillin. When OD600 reached 0.8, expression was induced with 0.05 mM isopropyl-β-D-thiogalactopyranoside overnight at 18°C, and bacteria were then sedimented by centrifugation. To generate protein for ITC, chilled bacterial pellets were lysed in cold TPE (1% (v/v) Triton X-100, PBS (pH 7.4), 100 mM EDTA, 10 μg/ml aprotinin, 0.7 μg/ml pepstatin A, 0.5 μg/ml leupeptin, and 5 μg/ml antipain) and sonicated, and the lysate was clarified by centrifugation at 20,000 × g for 1 h at 4°C. GST-fusion protein was purified by coincubation with glutathione (GSH)-sepharose beads overnight at 4°C on a nutator. Beads were then washed extensively with 50 mM TrisHCl (pH 7.5), 100 mM EDTA, and 0.1% (v/v) Tween 20. Bound GST-fusion protein was eluted with 100 mM GSH, pH adjusted to approximately ∼7.5 with TrisHCl (pH 8.8), and the eluate was dialyzed against 5 mM TrisHCl (pH 7.5). The integrity of the dialyzed protein was analyzed by SDS-PAGE and Coomassie Blue staining, and the protein concentration was assayed by the Bradford method. Purified GST-Grb2 SH3C fusion protein was snap-frozen in aliquots and stored at −80°C until further use in ITC.
ITC
ITC was performed with a VP-ITC MicroCalorimeter (MicroCal, Northampton, MA). Peptides were dissolved at 250 μM (750 μM for the P519A peptide) in ITC buffer (25 mM HEPES (pH 7.5, adjusted with potassium hydroxide), 100 mM potassium acetate, and 5 mM magnesium acetate), clarified by centrifugation for 10 min at 20,000 × g before use, degassed (ThermoVac; MicroCal), and titrated from a syringe (300 μl total volume) into a sample chamber holding 1.43 ml of clarified and degassed 25 μM GST-Grb2 SH3C (75 μM when analyzing the P519A peptide). Upon reaching the equilibrium temperature of 25°C, peptide solutions were titrated into the sample chamber by 18 injections of 15 μl each. The resulting peaks of measured deviations from the equilibrium temperature were integrated to yield the quantity of heat generated. The best fit to the data to calculate the binding affinity Kd was obtained using χ2 minimization on a model assuming a single set of binding sites. All steps of the data analysis were performed using ORIGIN (V5.0) software provided by the manufacturer.
NMR spectroscopy
Initial assignment of peptide resonances (Cα, Cβ, Hα, HN, and N) was performed under acidic conditions by dissolving the peptides in H2O using 10% v/v D2O for resonance locking. The acidic pH improved the assignment convergence owing to the limited broadening of the resonances of exchangeable protons. Subsequently, the assignments were transferred to spectra recorded under buffer conditions relevant to this investigation (25 mM HEPES pH 7.5 (adjusted with potassium hydroxide), 100 mM potassium acetate) at a temperature of 303 K. All NMR spectra were referenced using the resonance of the 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) molecule.
Experiments were carried out using 600 MHz and 800 MHz spectrometers (Bruker Avance III, Bruker Biospin, Billerica, MA). Proton assignment was carried out using a combination of total correlation spectroscopy (TOCSY) and rotating-frame nuclear Overhauser enhancement spectroscopy (ROESY) spectra. These were used for resonance assignment according to highly established protocols (39). Having completed the proton assignment, we were able to assign carbon and nitrogen resonances by using natural abundance 13C and 15N and recording heteronuclear single quantum coherence (HSQC) spectra. Random-coil CSs from the CamCoil method (25) were used to help assign the 13C CSs.
Determination of the secondary-structure populations and dynamics of IDPs
To address the secondary-structure populations of IDPs, we employed the δ2D method (22,25), which is based on the structural information provided by NMR CSs, by using the measured backbone CSs (Cα, Cβ, Hα, HN, and N). CSs were also employed to infer the local dynamics using the RCI (26).
Structural ensemble refinement by CS-restrained MD
To refine accurate structural ensembles of Gab2503-524, we employed CSs in restrained MD simulations using the CamShift method (40). Briefly, CS restraints were imposed by adding a pseudo-energy term (ECS) to a standard molecular-mechanics force field (EFF):
| (1) |
The resulting force field (ETot) was employed in MD simulations, where the pseudo-energy term is given by
| (2) |
where the i sum runs over all the CSs employed in the refinement, α is the weight of the restraint term, and δexp and δcalc are the experimental and calculated CSs, respectively. We employed the replica-averaged scheme, which implies that a given CS is calculated by
| (3) |
where m runs over four replicas and δmCalc is the CS of replica m.
We performed replica-averaged restrained MD simulations by using an implementation of GROMACS (41) as described previously (42) and employing a recent protocol based on four replicas (43). Each of the four replicas was equilibrated separately. Starting conformations were modeled as linear chains and accommodated in a dodecahedron box volume of 276 nm3 of volume. The box was filled with explicit waters and energy minimized. For each replica, the system was thermalized during an NVT simulation of 250 ps in which the temperature was increased up to 300 K. Subsequently, the pressure was equilibrated using a MD simulation of 200 ps under the Berendsen barostat. Finally, the individual replicas were equilibrated for 1-ns-long simulations.
The scheme of the replica-averaged restrained MD simulations is as follows: The four replicas evolve through a series of annealing cycles between 300 K and 400 K, with each cycle being composed of 100 ps at 300 K, 100 ps of linear increase in the temperature up to 400 K, 100 ps of constant-temperature MD simulations at 400 K, and 300 ps of a linear decrease in the temperature to 300 K. During these cycles, the experimental restraints are imposed as averages over the four replicas according to Eqs. 1–3. The total amount of sampling in each Gab2b peptide analyzed was 1 μs (250.2 ns per replica equivalent to 417 cycles).
The simulations were carried out using the Amber03W force field (44) and the TIP4P2005 (45) water model. This force-field setting was optimized to account for the conformational properties of IDPs. The protonation states of pH-sensitive residues were as follows: Arg and Lys were positively charged, Asp and Glu were negatively charged, and His was neutral. The net charge of the system was neutralized by the addition of four Cl− ions (three in the case of R515A and K518A). A time step of 2 fs was used together with LINCS constraints (46). van der Waals and electrostatic interactions were cut off at 0.9 nm, and long-range electrostatic effects were treated with the particle mesh Ewald method (47). All of the simulations were done in the canonical ensemble by keeping the volume fixed and setting the system temperature with a V-rescale thermostat (48).
We collected the final samplings from the 300 K portions of the replica simulations after discarding the initial 50 ns in each replica (in this time lapse, the simulations are allowed to converge). The total number of conformations collected in each sampling was 13,320. Convergence of the simulations is reported in Fig. S4 in the Supporting Material.
Results
Conformational properties of Gab2503-524
We used NMR spectroscopy, calorimetry, and statistical thermodynamics to characterize the molecular determinants of the Gab2503-524/Grb2 SH3C interaction. Multiple lines of evidence have shown that the binding between these two domains is a key event in the interaction of the respective full-length proteins (27,36). ITC measurements show that the Gab2503-524 fragment, which includes the RxxK binding motif of Gab2b, has micromolar affinity for Grb2 SH3C (see Table 1, Fig. S1, and the Materials and Methods section for details regarding the peptide synthesis and ITC measurements). Previous x-ray studies revealed that the conformation adopted by the Gab2b motif when complexed to Grb2 SH3C (27) includes a short 310 helix (residues 515–517) positioning the arginine and lysine of the RxxK motif in parallel orientation and a PPII fragment (residues 510–512; Fig. 1). Although the conformation of Gab2b bound to Grb2 SH3C is known, we focused on the conformational properties of Gab2503-524 when isolated in solution to gain insight into the determinants for the binding affinity. To this end, we measured NMR CSs of backbone atoms (in particular Cα, Cβ, Hα, HN, and N) for Gab2503-524 at pH 7.5 and 303 K (Fig. S2). NMR CSs of Cα, Cβ, and Hα atoms, which are fine probes of secondary structures in proteins, show values that are in the range of theoretical random-coil standards, as calculated using the CamCoil method (25) (Fig. S3). This finding indicates that Gab2503-524 maintains the unstructured nature that it adopts within the full-length sequence (36). Although unstructured proteins and peptides are difficult to characterize with standard techniques of structural biology (i.e., x-ray crystallography and NOE/3J-coupling-based NMR (49–52)), recent approaches exploiting the information extracted from CSs allow us to address, with residue-specific resolution, the population of transient secondary structures and coil regions.
Figure 1.
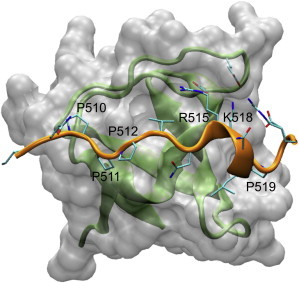
Binding interface between Grb2 SH3C (represented as a white surface/green cartoon) and Gab2b peptide (orange cartoon). The figure was produced using the x-ray coordinates (PDB code: 2vwf). To see this figure in color, go online.
We exploited the information contained in the CSs using the δ2D method (22,25), which has proved to be a powerful tool for studying the conformational properties of IDPs (53–58), and calculated in this manner the secondary-structure populations in the disordered Gab2503-524 fragment. This analysis revealed nonnegligible amounts of residual structure in the free state (Fig. 2). In particular, a region spanning residues 508–511 (designated throughout this article as residual structure 1 (RS1)), adopted partial PPII and extended β character with population peaks of up to 20% of the ensemble (Fig. 2 A). This finding suggests that, although RS1 is flexible and unstructured, it has a significant propensity to be highly extended when unbound in solution. Because of the extended β and PPII characters, RS1, which also shows a minor helical population (which is considered negligible because it is at the limit of the methodological error), is associated with a reduced coil character (Fig. 2 B). This finding was independently confirmed by the RCI (26) analysis (Fig. 2 B). Another region of the fragment that showed a significant content of residual PPII structure and partial character of an extended β conformation, namely RS2, spanned residues 517–521.
Figure 2.
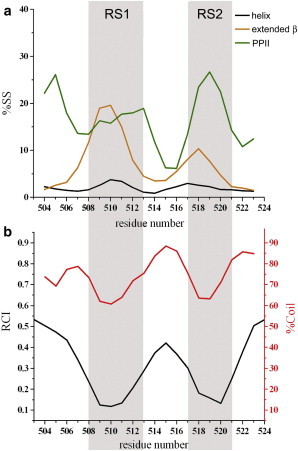
Conformational properties of the free state of Gab2503-524 by analysis of CSs. (A) Populations of secondary-structure elements using δ2D (22,25). Black, orange, and green lines report the populations of helix, extended β, and PPII conformations, respectively. (B) Contents of random-coil regions estimated by δ2D (red) and RCI (26) (black line). The scale for the δ2D profile is reported on the right side (red) and the scale for RCI appears on the left side (black). To see this figure in color, go online.
Residual structure in both RS1 and RS2 might have relevance in the context of binding the Grb2 SH3C. In RS1, the bound state of Gab2b consists of a PPII fragment (residues 510–512) that follows residue Q509, which is in an extended β conformation (backbone ϕ and ψ angles of −158.1 and 138.5, respectively). On the other hand, a population of residual PPII in RS2 might play a role in the bound state by favoring an important kink starting at residue 518, and allowing R521 to fold back in the binding cavity to establish a key salt bridge with Grb2 SH3C (Fig. 1). Notably K518 adopts backbone ϕ and ψ angles of −68.65 and 140.06, respectively, which are typical values for the PPII region. Accordingly, the residual PPII conformation in the unbound state might preorganize the formation of the kink that stabilizes the complex.
Effect of point mutations on conformational selection
To further investigate the role that residual structure in the unbound state plays in the recognition of Gab2503-524, we performed extensive refinement of its structural ensemble by using CS-restrained MD (42). The samplings (totaling 1 μs for each construct) were performed by imposing CSs as ensemble averages over four replicas and were carried out until convergence was proved (Fig. S4). The resulting ensemble showed high agreement between experimental CSs and those calculated using an independent and highly accurate predictor, SPARTA+ (59), which is based on a totally different approach compared with the CamShift method (42) used in our structural refinement (Fig. S5). This back validation suggests that the refined ensembles are highly accurate and match the experimental data with standard deviations (SDs) that are lower than the statistical error of SPARTA+ (Fig. S5).
Our analyses focused on a comparison between the conformations of the free state (NMR ensemble) and those of the bound state (x-ray structure). We computed a free-energy projection of 13,320 conformations collected from the NMR sampling by using two independent coordinates. The first was built by calculating the root mean-square deviation (RMSD) of the ϕ and ψ backbone angles between the free-state structures and the bound conformation. The second coordinate was based on the gyration radius calculated using the Cα atoms. To be consistent with the x-ray structure, only residues 508–521 were employed in this calculation. The analysis resulted in a spread free-energy landscape (Fig. 3 A), with the first coordinate ranging from approximately 20° to 100° in dihedral RMSD, and the second coordinate (gyration radius) ranging from 5.5 to 11.5 Å. In addition to a main basin of random-coil conformations (basin b1), the energy landscape showed a second minimum (basin b2) including conformations with low dihedral RMSD values (∼40°) and gyration radii similar to that adopted in the bound structure (8.5 Å). The conformations corresponding to basin b2 are highly similar to that of the bound state (Fig. 3 A). Accordingly, in line with the CS analyses of δ2D (22,25) and RCI (26), the energy landscape showed a set of preorganized conformations populating the free-state ensemble.
Figure 3.
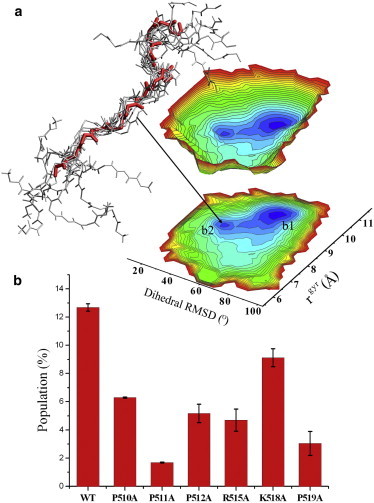
Structural ensembles of the free state of Gab2503-524 sampled by CS-restrained simulations. (A) Energy landscape of the WT sequence projected on two coordinates: the dihedral RMSD from the x-ray structure of the bound state and the gyration radius. The landscape is evaluated in discrete points using the Boltzmann equation and then interpolated using spline functions (see De Simone et al. (2,60) for additional details regarding the free-energy projections). The plot shows a basin composed of structures that are very similar to the bound state, corresponding to a low RMSD and a radius of gyration that is close to 8.5 Å (the value adopted in the bound structure). A bundle of conformations from the basin b2 is shown (backbone atoms only). Gray conformations are from the NMR ensemble (free state) and the red conformation corresponds to the crystal structure (bound state). (B) Based on this basin, we could estimate the population of bound conformations in the structural ensembles of the free state of the Gab2503-524 sequences (WT and mutants) considered here. To see this figure in color, go online.
We then analyzed the influence of the basin b2 population on the ability of Grb2 to recognize and bind Gab2503-524. In particular, we mutated a set of residues in Gab2503-524 that are directly involved in the binding site of Grb2 SH3C (Fig. 1). ITC measurements provided direct evidence of the effects of these point mutations on the binding affinity. All mutations showed a decrease in the affinity for binding Grb2 SH3C, and a general trend that included an unfavorable entropy contribution and favorable enthalpy terms for the binding (Table 1). The strongest affinity impairment was measured for the mutations that affected the RxxK core motif, namely, R515A and K518A, for which no binding was detected. The other mutations tested targeted the proline residues in the binding site. Of these, P511A induced the strongest perturbation of the binding affinity (with a resulting Kd lower than the reliably quantifiable range), followed by P519A.
The δ2D (22,25) analysis showed that all of the mutants retain a degree of disorder with P511A and K518A having an increased coil content in RS1 and RS2, respectively, and P519A being less structured in both RS1 and RS2 (Fig. 4 A). Similarly, the RCI (26) suggests that all the peptides exhibit a similar disorder, with P519A being the most dynamical mutant (Fig. 4 B). An interesting result is associated with the analysis of PPII content in the various mutants. P→A mutations in RS1 affect mainly the PPII content in this region (Fig. 4 C). Of these, P511A shows the highest reduction in PPII structure, whereas in P510A the reduction of PPII is partially compensated for by a local increase in extended β content (Fig. 4 D). Another strong effect on the PPII content of RS1 is shown by R515A. Finally, K518A and P519A affect mainly the PPII in RS2, with P519A having the most significant perturbation, which is in part also transferred to RS1. No significant effects were detected for the helical residual structure (Fig. 4 E).
Figure 4.
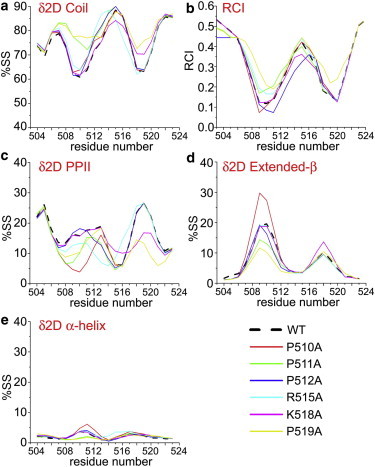
Effects of point mutations on the conformational properties of the free state of Gab2503-524 by analysis of CSs. (A) Populations of coil conformations from δ2D (22,25). (B) RCI (26). (C) Populations of PPII conformations from δ2D. (D) Populations of extended β conformations from δ2D. (E) Populations of helical conformations from δ2D. To see this figure in color, go online.
Overall, the residual structure in the mutants of Gab2503-524 shows that the partial impairment of binding affinity for Grb2 is correlated with a reduced amount of preorganized bound conformations in the free-state ensemble. This important finding is underlined by the analysis of the populations in basin b2 of the projected energy landscape (Fig. 3). If we exclude R515A and K518A, which in addition to the conformational perturbation of the backbone lack the key side chains for the RxxK-binding motif, the resulting trend of populations is consistent with the ranking obtained from ITC measurements (Table 1). Indeed, the ranking of populations in the region occupied by basin b2 is WT > P510A > P512A > P519A > P511A (Fig. 3 B). This remarkable correspondence indicates that one of the key factors governing the interaction propensities of IDPs is the population of preexisting bound conformations in the free-state ensemble. It is worth noting that other factors may also contribute to the decrease of affinity between Grb2 and Gab2503-524, including the loss of favorable native interactions in the protein-protein complex. For instance, these factors might contribute to the ∼15-fold loss of binding affinity associated with the P519A mutation (see Kd values in Table 1), which is not entirely accounted for by the decrease in the population of preexisting bound conformations in the energy landscape of Gab2503-524 (Fig. 3 B).
Discussion
It is now well established that a significant fraction of all proteins feature a partial or total degree of structural disorder (11,12,15,61). Disordered regions can influence intrinsic biological properties, such as the propensity to self-assemble (62) or allosteric modulation (63). It is also becoming increasingly evident that IDPs are actively employed in fundamental biological processes, including cellular signaling through disordered linkers (64) and protein translation or transcriptional regulation (65). Their remarkable ability to interact with multiple cellular partners is likely promoted by their inherent flexibility and energy landscapes encoding for multiple conformational minima, enabling shape adaptation during partner selection. Although the functional role of IDPs is attracting increasing interest, understanding the underlying structural and physical principles of their biological activity remains a difficult challenge. In this context, NMR spectroscopy is emerging as a major tool (17,18,22,66), and we illustrate this point very clearly by showing that it is possible to use new NMR approaches to address the functional determinants of the biological activity of IDPs.
One prominent representative of functional IDPs is Gab2, a platform protein involved in complex molecular signaling. Gab2 is a multisite docking molecule with a conserved, folded N-terminal domain that enables docking to the plasma membrane, and a large disordered region that mediates interactions with many signaling proteins. The long disordered region of Gab2 acts as a substrate region for multiple phosphorylations by tyrosine kinases. It also forms a multivalent docking station for SH2 and SH3 domains, which allows it to interact with a large number of proteins from different families (e.g., Crk/CRKL, Nck1, Grb2, PI3 kinase, and SHP2). Thus, as found for other IDPs, Gab2 behaves as a versatile hub for signaling pathway cross talk in several different cell types. It acts in many physiological processes, including growth of the bone marrow and cardiac function, as well as in pathological conditions such as leukemia and Alzheimer’s disease (67). Besides representing a convenient system in which to address the binding and recognition of IDPs, Gab2 is therefore a central node in cellular signaling of major biological relevance.
Using the information contained in the backbone CSs, the RCI (26) showed two regions of Gab2503-524, RS1 and RS2, with reduced conformational flexibility. The high occurrence of PPII and extended β structure in these regions, as revealed by δ2D (22,25), suggests a nonnegligible intrinsic tendency toward extended conformations. PPII conformations in RS1 are directly relevant to the conformation adopted in the complex with Grb2 SH3C (PDB code: 2vwf). Similarly, we propose that the marked residual content of PPII in RS2 has a role in nucleating an essential kink that conveys to the backbone an optimal conformation for a salt bridge made by residue R521. The intrinsic propensity to adopt residual structures converges with the evidence that the energy landscape of Gab2503-524 includes a basin of conformers with similar structural properties to the bound state. As shown by the mutant analysis of Gab2503-524, changes in the population of these conformers correlate with the impairment in the Grb2 binding affinity.
Overall, our analysis of transient conformations by NMR and statistical mechanics indicates that the presence of prearranged bound conformations in the heterogeneous structural ensemble of the disordered Gab2503-524 is an essential determinant of the binding affinity for its protein partner Grb2, as tested by mutational analysis and calorimetric measurements. Therefore, the results reported here suggest that the binding of Gab2503-524 requires a selection of conformations that appear to be intrinsically encoded in the energy landscape of this disordered state.
In conclusion, by using recently developed methods that exploit the information contained in NMR CSs, we investigated the conformational properties of disordered Gab2503-524, and found intrinsic conformations that influence the affinity for protein partner recognition and binding. Thus, the physical principles that emerge from this study add to our understanding of the recognition of IDPs by showing that preexisting bound conformations have a significant influence on the binding affinity. The elements reported in this work are likely to have very general implications for the biological behavior of IDPs in light of the fact that a large fraction of these proteins possess a specific propensity to form the PPII helix and adopt conformations that are more extended than the typical random-coil states.
Acknowledgments
We are grateful to the Tate group (Department of Chemistry, Imperial College London) for assisting with the peptide synthesis and purification.
This research was supported by the Centre for Structural Biology at Imperial College, Leverhulme Trust (A.De S.), Parkinson’s UK (G.F.), and FP7 Marie Curie Actions (C.C.).
Supporting Material
References
- 1.Boehr D.D., Nussinov R., Wright P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Simone A., Montalvao R.W., Vendruscolo M. Characterization of the interdomain motions in hen lysozyme using residual dipolar couplings as replica-averaged structural restraints in molecular dynamics simulations. Biochemistry. 2013;52:6480–6486. doi: 10.1021/bi4007513. [DOI] [PubMed] [Google Scholar]
- 3.Boehr D.D., Dyson H.J., Wright P.E. An NMR perspective on enzyme dynamics. Chem. Rev. 2006;106:3055–3079. doi: 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- 4.Masterson L.R., Mascioni A., Veglia G. Allosteric cooperativity in protein kinase A. Proc. Natl. Acad. Sci. USA. 2008;105:506–511. doi: 10.1073/pnas.0709214104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das R., Esposito V., Melacini G. cAMP activation of PKA defines an ancient signaling mechanism. Proc. Natl. Acad. Sci. USA. 2007;104:93–98. doi: 10.1073/pnas.0609033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tzeng S.R., Kalodimos C.G. Allosteric inhibition through suppression of transient conformational states. Nat. Chem. Biol. 2013;9:462–465. doi: 10.1038/nchembio.1250. [DOI] [PubMed] [Google Scholar]
- 7.Tzeng S.R., Kalodimos C.G. Protein activity regulation by conformational entropy. Nature. 2012;488:236–240. doi: 10.1038/nature11271. [DOI] [PubMed] [Google Scholar]
- 8.Selvaratnam R., Chowdhury S., Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. USA. 2011;108:6133–6138. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arai M., Ferreon J.C., Wright P.E. Quantitative analysis of multisite protein-ligand interactions by NMR: binding of intrinsically disordered p53 transactivation subdomains with the TAZ2 domain of CBP. J. Am. Chem. Soc. 2012;134:3792–3803. doi: 10.1021/ja209936u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tompa P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem. Sci. 2012;37:509–516. doi: 10.1016/j.tibs.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Uversky V.N. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 2013;22:693–724. doi: 10.1002/pro.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunker A.K., Obradovic Z., Brown C.J. Intrinsic protein disorder in complete genomes. Genome Inform. Ser. Workshop Genome Inform. 2000;11:161–171. [PubMed] [Google Scholar]
- 13.Dunker A.K., Silman I., Sussman J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008;18:756–764. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Uversky V.N. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem. Soc. Rev. 2011;40:1623–1634. doi: 10.1039/c0cs00057d. [DOI] [PubMed] [Google Scholar]
- 15.Tompa P. Unstructural biology coming of age. Curr. Opin. Struct. Biol. 2011;21:419–425. doi: 10.1016/j.sbi.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 16.Soranno A., Buchli B., Schuler B. Quantifying internal friction in unfolded and intrinsically disordered proteins with single-molecule spectroscopy. Proc. Natl. Acad. Sci. USA. 2012;109:17800–17806. doi: 10.1073/pnas.1117368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dyson H.J., Wright P.E. Unfolded proteins and protein folding studied by NMR. Chem. Rev. 2004;104:3607–3622. doi: 10.1021/cr030403s. [DOI] [PubMed] [Google Scholar]
- 18.Mittag T., Forman-Kay J.D. Atomic-level characterization of disordered protein ensembles. Curr. Opin. Struct. Biol. 2007;17:3–14. doi: 10.1016/j.sbi.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 19.Schneider R., Huang J.R., Blackledge M. Towards a robust description of intrinsic protein disorder using nuclear magnetic resonance spectroscopy. Mol. Biosyst. 2012;8:58–68. doi: 10.1039/c1mb05291h. [DOI] [PubMed] [Google Scholar]
- 20.Milojevic J., Esposito V., Melacini G. Understanding the molecular basis for the inhibition of the Alzheimer’s Aβ-peptide oligomerization by human serum albumin using saturation transfer difference and off-resonance relaxation NMR spectroscopy. J. Am. Chem. Soc. 2007;129:4282–4290. doi: 10.1021/ja067367+. [DOI] [PubMed] [Google Scholar]
- 21.De Simone A., Gustavsson M., Vendruscolo M. Structures of the excited states of phospholamban and shifts in their populations upon phosphorylation. Biochemistry. 2013;52:6684–6694. doi: 10.1021/bi400517b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Camilloni C., De Simone A., Vendruscolo M. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry. 2012;51:2224–2231. doi: 10.1021/bi3001825. [DOI] [PubMed] [Google Scholar]
- 23.Mittag T., Marsh J., Forman-Kay J.D. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure. 2010;18:494–506. doi: 10.1016/j.str.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jensen M.R., Markwick P.R.L., Blackledge M. Quantitative determination of the conformational properties of partially folded and intrinsically disordered proteins using NMR dipolar couplings. Structure. 2009;17:1169–1185. doi: 10.1016/j.str.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 25.De Simone A., Cavalli A., Vendruscolo M. Accurate random coil chemical shifts from an analysis of loop regions in native states of proteins. J. Am. Chem. Soc. 2009;131:16332–16333. doi: 10.1021/ja904937a. [DOI] [PubMed] [Google Scholar]
- 26.Berjanskii M.V., Wishart D.S. The RCI server: rapid and accurate calculation of protein flexibility using chemical shifts. Nucleic Acids Res. 2007;35(Web Server issue):W531–W537. doi: 10.1093/nar/gkm328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harkiolaki M., Tsirka T., Feller S.M. Distinct binding modes of two epitopes in Gab2 that interact with the SH3C domain of Grb2. Structure. 2009;17:809–822. doi: 10.1016/j.str.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 28.Lewitzky M., Kardinal C., Feller S.M. The C-terminal SH3 domain of the adapter protein Grb2 binds with high affinity to sequences in Gab1 and SLP-76 which lack the SH3-typical P-x-x-P core motif. Oncogene. 2001;20:1052–1062. doi: 10.1038/sj.onc.1204202. [DOI] [PubMed] [Google Scholar]
- 29.Lock L.S., Royal I., Park M. Identification of an atypical Grb2 carboxyl-terminal SH3 domain binding site in Gab docking proteins reveals Grb2-dependent and -independent recruitment of Gab1 to receptor tyrosine kinases. J. Biol. Chem. 2000;275:31536–31545. doi: 10.1074/jbc.M003597200. [DOI] [PubMed] [Google Scholar]
- 30.McDonald C.B., Seldeen K.L., Farooq A. Binding of the cSH3 domain of Grb2 adaptor to two distinct RXXK motifs within Gab1 docker employs differential mechanisms. J. Mol. Recognit. 2011;24:585–596. doi: 10.1002/jmr.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eulenfeld R., Schaper F. A new mechanism for the regulation of Gab1 recruitment to the plasma membrane. J. Cell Sci. 2009;122:55–64. doi: 10.1242/jcs.037226. [DOI] [PubMed] [Google Scholar]
- 32.Nishida K., Wang L., Hirano T. Requirement of Gab2 for mast cell development and KitL/c-Kit signaling. Blood. 2002;99:1866–1869. doi: 10.1182/blood.v99.5.1866. [DOI] [PubMed] [Google Scholar]
- 33.Nishida K., Yoshida Y., Hirano T. Gab-family adapter proteins act downstream of cytokine and growth factor receptors and T- and B-cell antigen receptors. Blood. 1999;93:1809–1816. [PubMed] [Google Scholar]
- 34.McDonald C.B., Bhat V., Farooq A. Bivalent binding drives the formation of the Grb2-Gab1 signaling complex in a noncooperative manner. FEBS J. 2012;279:2156–2173. doi: 10.1111/j.1742-4658.2012.08600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDonald C.B., Seldeen K.L., Farooq A. Assembly of the Sos1-Grb2-Gab1 ternary signaling complex is under allosteric control. Arch. Biochem. Biophys. 2010;494:216–225. doi: 10.1016/j.abb.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simister P.C., Feller S.M. Order and disorder in large multi-site docking proteins of the Gab family—implications for signalling complex formation and inhibitor design strategies. Mol. Biosyst. 2012;8:33–46. doi: 10.1039/c1mb05272a. [DOI] [PubMed] [Google Scholar]
- 37.Chan W.C., White P.D. Oxford University Press; Oxford, UK: 2000. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. [Google Scholar]
- 38.Pendergast A.M., Quilliam L.A., Cripe L.D., Bassing C.H., Dai Z., Li N., Batzer A., Rabun K.M., Der C.J., Schlessinger J. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75:175–185. [PubMed] [Google Scholar]
- 39.Wüthrich K. Protein structure determination in solution by nuclear magnetic resonance spectroscopy. Science. 1989;243:45–50. doi: 10.1126/science.2911719. [DOI] [PubMed] [Google Scholar]
- 40.Kohlhoff K.J., Robustelli P., Vendruscolo M. Fast and accurate predictions of protein NMR chemical shifts from interatomic distances. J. Am. Chem. Soc. 2009;131:13894–13895. doi: 10.1021/ja903772t. [DOI] [PubMed] [Google Scholar]
- 41.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular dynamics simulations. J. Chem. Theory Comput. 2008;4:435–437. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 42.Camilloni C., Robustelli P., Vendruscolo M. Characterization of the conformational equilibrium between the two major substates of RNase A using NMR chemical shifts. J. Am. Chem. Soc. 2012;134:3968–3971. doi: 10.1021/ja210951z. [DOI] [PubMed] [Google Scholar]
- 43.Camilloni C., Cavalli A., Vendruscolo M. Assessment of the use of NMR chemical shifts as replica-averaged structural restraints in molecular dynamics simulations to characterize the dynamics of proteins. J. Phys. Chem. B. 2013;117:1838–1843. doi: 10.1021/jp3106666. [DOI] [PubMed] [Google Scholar]
- 44.Best R.B., Mittal J. Protein simulations with an optimized water model: cooperative helix formation and temperature-induced unfolded state collapse. J. Phys. Chem. B. 2010;114:14916–14923. doi: 10.1021/jp108618d. [DOI] [PubMed] [Google Scholar]
- 45.Abascal J.L., Sanz E., Vega C. A potential model for the study of ices and amorphous water: TIP4P/Ice. J. Chem. Phys. 2005;122:234511. doi: 10.1063/1.1931662. [DOI] [PubMed] [Google Scholar]
- 46.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 47.Darden T., Perera L., Pedersen L. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure. 1999;7:R55–R60. doi: 10.1016/s0969-2126(99)80033-1. [DOI] [PubMed] [Google Scholar]
- 48.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 49.Gil S., Hošek T., Felli I.C. NMR spectroscopic studies of intrinsically disordered proteins at near-physiological conditions. Angew. Chem. Int. Ed. Engl. 2013;52:11808–11812. doi: 10.1002/anie.201304272. [DOI] [PubMed] [Google Scholar]
- 50.Camilloni C., Schaal D., De Simone A. Energy landscape of the prion protein helix 1 probed by metadynamics and NMR. Biophys. J. 2012;102:158–167. doi: 10.1016/j.bpj.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dyson H.J., Wright P.E. Defining solution conformations of small linear peptides. Annu. Rev. Biophys. Biophys. Chem. 1991;20:519–538. doi: 10.1146/annurev.bb.20.060191.002511. [DOI] [PubMed] [Google Scholar]
- 52.Dyson H.J., Wright P.E. Nuclear magnetic resonance methods for elucidation of structure and dynamics in disordered states. Methods Enzymol. 2001;339:258–270. doi: 10.1016/s0076-6879(01)39317-5. [DOI] [PubMed] [Google Scholar]
- 53.Maltsev A.S., Ying J., Bax A. Impact of N-terminal acetylation of α-synuclein on its random coil and lipid binding properties. Biochemistry. 2012;51:5004–5013. doi: 10.1021/bi300642h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voelz V.A., Jäger M., Pande V.S. Slow unfolded-state structuring in Acyl-CoA binding protein folding revealed by simulation and experiment. J. Am. Chem. Soc. 2012;134:12565–12577. doi: 10.1021/ja302528z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roche J., Ying J., Bax A. Impact of hydrostatic pressure on an intrinsically disordered protein: a high-pressure NMR study of α-synuclein. ChemBioChem. 2013;14:1754–1761. doi: 10.1002/cbic.201300244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ozenne V., Schneider R., Blackledge M. Mapping the potential energy landscape of intrinsically disordered proteins at amino acid resolution. J. Am. Chem. Soc. 2012;134:15138–15148. doi: 10.1021/ja306905s. [DOI] [PubMed] [Google Scholar]
- 57.Kang L., Moriarty G.M., Baum J. N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 2012;21:911–917. doi: 10.1002/pro.2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Libich D.S., Fawzi N.L., Clore G.M. Probing the transient dark state of substrate binding to GroEL by relaxation-based solution NMR. Proc. Natl. Acad. Sci. USA. 2013;110:11361–11366. doi: 10.1073/pnas.1305715110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen Y., Bax A. SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J. Biomol. NMR. 2010;48:13–22. doi: 10.1007/s10858-010-9433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Simone A., Dhulesia A., Dobson C.M. Experimental free energy surfaces reveal the mechanisms of maintenance of protein solubility. Proc. Natl. Acad. Sci. USA. 2011;108:21057–21062. doi: 10.1073/pnas.1112197108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tompa P., Kovacs D. Intrinsically disordered chaperones in plants and animals. Biochem. Cell Biol. 2010;88:167–174. doi: 10.1139/o09-163. [DOI] [PubMed] [Google Scholar]
- 62.De Simone A., Kitchen C., Frenkel D. Intrinsic disorder modulates protein self-assembly and aggregation. Proc. Natl. Acad. Sci. USA. 2012;109:6951–6956. doi: 10.1073/pnas.1118048109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferreon A.C., Ferreon J.C., Deniz A.A. Modulation of allostery by protein intrinsic disorder. Nature. 2013;498:390–394. doi: 10.1038/nature12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akimoto M., Selvaratnam R., Melacini G. Signaling through dynamic linkers as revealed by PKA. Proc. Natl. Acad. Sci. USA. 2013;110:14231–14236. doi: 10.1073/pnas.1312644110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 66.Jensen M.R., Houben K., Blackledge M. Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: application to the molecular recognition element of Sendai virus nucleoprotein. J. Am. Chem. Soc. 2008;130:8055–8061. doi: 10.1021/ja801332d. [DOI] [PubMed] [Google Scholar]
- 67.Reiman E.M., Webster J.A., Stephan D.A. GAB2 alleles modify Alzheimer’s risk in APOE epsilon4 carriers. Neuron. 2007;54:713–720. doi: 10.1016/j.neuron.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.