

Abstract
The validation of protein structures through functional assays has been the norm for many years. Functional assays perform this validation for water-soluble proteins very well, but they need to be performed in the same environment as that used for the structural analysis. This is difficult for membrane proteins that are often structurally characterized in detergent environments, although functional assays for these proteins are most frequently performed in lipid bilayers. Because the structure of membrane proteins is known to be sensitive to the membrane mimetic environment, such functional assays are appropriate for validating the protein construct, but not the membrane protein structure. Here, we compare oriented sample solid-state NMR spectral data of diacylglycerol kinase previously published with predictions of such data from recent structures of this protein. A solution NMR structure of diacylglycerol kinase has been obtained in detergent micelles and three crystal structures have been obtained in a monoolein cubic phase. All of the structures are trimeric with each monomer having three transmembrane and one amphipathic helices. However, the solution NMR structure shows typical perturbations induced by a micelle environment that is reflected in the predicted solid-state NMR resonances from the structural coordinates. The crystal structures show few such perturbations, especially for the wild-type structure and especially for the monomers that do not have significant crystal contacts. For these monomers the predicted and observed data are nearly identical. The thermostabilized constructs do show more perturbations, especially the A41C mutation that introduces a hydrophilic residue into what would be the middle of the lipid bilayer inducing additional hydrogen bonding between trimers. These results demonstrate a general technique for validating membrane protein structures with minimal data obtained from membrane proteins in liquid crystalline lipid bilayers by oriented sample solid-state NMR.
Introduction
There is a need to structurally validate helical membrane protein structures. Although many of these structures in the Protein Data Bank (PDB) appear to be native-like many others do not have such characteristics (5,6). It has been argued that the nonnative character in many of these structures is the result of poor modeling of the native environment for the structural characterization. Neither solution NMR nor x-ray crystallography can observe samples in native-like lipid bilayers. For both of these technologies detergents are extensively used to model the native membrane. To various extents the detergent environments lead to variable hydrophobic thicknesses, to weak hydrophobic environments, to increased water penetration into the transmembrane (TM) environment, to dampened lateral pressure profiles and, in the case of micelles, to a single hydrophilic surface. Indeed, as Anfinsen stated for proteins “that the native conformation is determined by the totality of interatomic interactions and hence by the amino acid sequence in a given environment,” unfortunately these last four words are frequently left off of this quote (7). It is, therefore, not surprising that detergent-based environments may not result in the same set of molecular interactions that stabilize the native membrane protein structure. For this reason we claim that helical structures, especially small helical structures characterized in detergent-based environments, should be structurally validated as described in this report for diacylglycerol kinase (DgkA).
DgkA is a helical TM protein that catalyzes the conversion of diacyl-glycerol and ATP to phosphatidic acid and ADP (8,9). Early on, three TM helices were identified for this protein of 121 amino acid residues (10) and Fourier transform infrared spectroscopy experiments estimated that 90% of the residues were in a helical conformation (11). Of importance, DgkA has been the subject of extensive studies associated with mutation-induced misfolding, which can lead to disease, and an understanding of the cellular control mechanisms for avoiding the presence of such misfolded proteins (12). The published DgkA structures are trimers, one monomer of which is highlighted for each structure in Fig. 1. The solution NMR structure and the x-ray structure of the wild-type (WT) protein both have an amphipathic helix and three TM helices plus additional hydrophilic helix on the cytoplasmic side accounting for 72% helicity in the solution NMR structure and 80% in the x-ray structure. The amphipathic helix is the N-terminus of the protein and presumably interacts with the membrane interfacial region.
Figure 1.
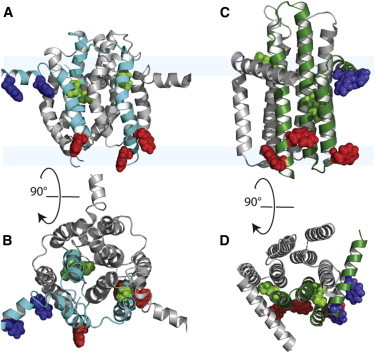
Structures of DgkA—cytoplasmic surface is at the top for the side views (A and C) and the end views (B and D) are from the cytoplasmic surface. (A and B) Side and end view of the solution NMR structure in dodecylphosphocholine micelles (PDB: 2KDC). (C and D) Side and end view of the x-ray diffraction WT structure in monoolein cubic phase (PDB: 3ZE4). TM helix tryptophan residues are in red, amphipathic helix tryptophan residues are in blue, and methionine residues are in green.
The recent crystal structures of DgkA (PDB: 3ZE3, 3ZE4, 3ZE5) obtained from lipidic cubic phase crystallization (13) represent very different structures from that obtained in a dodecylphosphatidylcholine (DPC) micelle environment (PDB: 2KDC) by solution NMR (Fig. 1) (14). The crystal structures include a WT structure (3ZE4) and two structures of thermally stabilized DgkA constructs, one with four single site mutations (3ZE5) and another with seven such mutations (3ZE3). Both the solution NMR and x-ray crystal structures are trimeric, with each monomer having three TM helices and an amphipathic helix. Helix 2 forms the core in the trimeric structure from both technologies. Although there are a few other similarities between the structures obtained in these different environments, the uniformity of the helical structures and the global packing in these structures is very different. The solution NMR structure is a domain-swapped structure in which the TM helices of the three monomers intermingle, but in the x-ray structures the three TM helices of each monomer form well-folded tertiary structures (15). The helices in the solution NMR structure are not well packed as indicated by large cavities between the helices, which would be filled with detergent or water in the sample used for structural characterization. In comparison the helices of the x-ray structures appear to be much more tightly packed. The helices in the solution NMR structure have an outward curvature resulting in lengthening the hydrogen bonds facing what would be the low dielectric of the fatty acyl environment. Furthermore, the high degree of curvature induces large crossing angles for the TM helices and a reduced helix-helix interface. In comparison, the helices of the x-ray structure are remarkably linear and uniform with small tilt angles and modest crossing angles with adjacent helices. These differences suggest that the DgkA structures should be validated with structural data obtained from a lipid bilayer environment. In fact, oriented sample solid-state NMR (OS ssNMR) spectra of DgkA in lipid bilayers had been published in 2007 (1) before the publication of these structures. Here, we will demonstrate the use of these data to evaluate the detergent-based structures.
Despite the structural differences, both works (13,14) performed functional assays in an attempt to ensure the biological relevance of the structure. For both studies the assays were performed in an environment intended to be very similar to that used for the structural characterization. However, the functional assays performed in a detergent micelle environment for the solution NMR structural characterization were not performed at the elevated temperature used for the spectroscopy. At a lower temperature for the assays the observed KM was significantly greater than in a native environment, indicating that the enzyme had somewhat less affinity for the substrate. Recently, these assays were repeated at the elevated temperature used for the spectroscopy and the protein was found to be nonfunctional (personal communication, C. Sanders, Department of Biochemistry and Center for Structural Biology, Vanderbilt University, Nashville, Tennessee; 2014), whereas the functional assays performed in a monoolein cubic phase environment as for x-ray crystallography showed that the protein was fully functional. Of importance, these functional assays only validate the structure when they are performed in the same environment as that of the sample preparation used for structural characterization. Such assays for membrane proteins involved in transport are very difficult, if not impossible to perform in detergent environments. However, functional assays performed in lipid bilayers can still be useful if the protein sequence has been modified, as is frequently done to thermally stabilize the structure or to enhance the crystal contacts via inserting additional residues in loops between helices or by complexing the protein with an antibody. The functional assays can still be useful for validating the construct as functional, but such assays do not validate the structure characterized in a different environment. Here, the enzymatic assays performed for DgkA in monoolein cubic phase provide strong support for a native-like structure.
Recent literature has clearly shown that helical membrane protein structure can be influenced by the membrane mimetic used for structural analysis (5,6,16,17). For membrane proteins, especially for relatively small membrane proteins, such as DgkA, the influence of the environment can be very significant, having a relatively large surface area for interactions with the membrane protein versus the relatively small internal structural volume for interhelical interactions. Note that this balance shifts as the number of TM helices increases. The TM domains of small helical proteins have a very high hydrophobic content, even for those residues facing the interior of the protein (16). Consequently, there are few or only weak electrostatic interactions that stabilize the tertiary structure of the TM domains of these small proteins, unless there are hydrophilic ligands or cofactors bound in the TM region to enhance the structural stability (e.g., electron transport or light harvesting complexes). With few strong interactions between helices and the low dielectric of the environment, the helices have a tendency to be more uniform and more linear in TM domains than in water-soluble proteins (18,19). This is the result of strengthened intrahelical hydrogen bonds in the low dielectric environment compared to water-soluble proteins, despite the frequent high content of helix breaking Gly and Pro residues (20). It is therefore important to analyze each protein structure to see if the helices are well packed, i.e., few cavities between the helices except where such a cavity might be needed for functional activities. Furthermore, to facilitate tertiary and quaternary structural stability most helical TM proteins have glycine or alanine motifs (small residues on a single face of a helix) that facilitate the close packing of helical pairs (21,22). Such motifs only function if they have substantial van der Waals contact with a neighboring helix. Helix uniformity and the packing of glycine/alanine motifs represent two parameters that can be used to help evaluate helical TM proteins.
An aqueous solution of detergent micelles provides a hydrophobic domain, a bulk aqueous environment, and an interfacial region. However, as a mimetic for membrane proteins they have several shortcomings (6). In the presence of a protein the hydrophobic dimension is not fixed but can expand or contract for a lowest energy state of the complex, which may result in a nonnative protein structure. Many examples of micelle solubilized proteins have shown that the TM helices can undergo partial hydrogen/deuterium exchange even at the center of the TM helix and hence the interstices of the micelle environment do not have the same low dielectric environment that a bilayer has (23,24). The single surface of the micelle implies that any portion of a TM helix can reach the hydrophilic surface with only a modest displacement, such as the outward curvature of helices seen in multiple solution NMR structures (23,25,26). The lengthened hydrogen bonds on what would be the fatty acyl-facing surface of the helix expose backbone hydrophilic atoms in the micelle to a more hydrophilic environment than would be available in a lipid bilayer or a native membrane.
Few structures have been characterized by x-ray diffraction of proteins crystallized from a lipidic cubic phase. For the structure of DgkA, this phase was generated with monololein, a detergent forming a Pn3m phase with two separate continuous, but highly curved surfaces (27,28). The two hydrophilic surfaces and highly hydrophobic interstices suggest that this may be a better membrane mimetic for membrane protein crystallization than detergent micelles. In particular, this cubic phase may be useful for the crystallization of small helical membrane proteins with their enhanced sensitivity for the environment. However, the crystallization of gramicidin A (gA) from the lipidic cubic phase resulted in a nonnative structure nearly identical to a structure crystallized from organic solvents (29,30). Indole side chains are well known to interact with the bilayer interface and for gA in a lipid bilayer the only way these interactions are possible is through a single stranded amino terminus to amino terminus dimer (31,32). In the lipidic cubic phase structure the gA indoles in the middle of the structure appear to be stabilized by the polyethylene glycol precipitant in the middle of what would be the lipid bilayer (33). To further investigate the effects that these environments have on helical membrane protein structure we will compare the DgkA structures obtained from detergent micelles and lipidic cubic phase membrane mimetics with data obtained by ssNMR in liquid crystalline lipid bilayers.
To study membrane proteins in lipid bilayers the overall correlation time for the tumbling of proteoliposomes is too long for obtaining high resolution solution NMR spectra useful for structural characterization. Alternatively, a set of tools that are known as ssNMR methods are used to study these proteins in frozen or liquid crystalline lipid bilayer environments. Using magic angle spinning (MAS, a technique that spins the samples at tens of kHz at an angle of 54.7° with respect to the magnetic field axis) it is possible to obtain isotropic chemical shift spectra and distance restraints that can be used in much the same way that nuclear Overhauser effect restraints from solution NMR spectroscopy are used to help define protein structure. Furthermore, like solution NMR, the isotropic chemical shifts of the polypeptide backbone obtained from MAS ssNMR can be used to restrain the torsion angles in the polypeptide backbone, another example of relative restraints. Identifying these restraints requires sequence-specific resonance assignments for most sites in the protein. For multiple reasons these assignments have been very difficult to obtain, preventing the widespread determination of helical membrane protein structures using MAS techniques alone. However, progress is being made and methodologies are on the horizon that could result in better resolution possibly leading to the ability to routinely determine membrane protein structures solely using MAS techniques (34–39).
Here, we will discuss the use of orientational restraints derived from another ssNMR technique, OS ssNMR, to validate detergent-based structures. This is a technique that has been shown to be particularly appropriate for characterizing such small helical membrane proteins as DgkA (4,38,40–42). The observation of anisotropic (orientation dependent) nuclear spin interactions from uniformly oriented lipid bilayer preparations of a protein provides high-resolution orientational restraints that characterize the helical structure very well and dramatically reduce the dependence on distance restraints to define the tertiary and quaternary structure of a helical membrane protein (2,39,43). Furthermore, recent developments in the sample preparation for OS ssNMR of membrane proteins (44,45) and the application to larger protein systems than ever studied before (46) point to the general use of the technique. PISEMA spectra from which 1H -15N dipolar and anisotropic 15N chemical shift restraints are obtained for amino acid-specific-labeled DgkA were published in 2007 (1) before the publication of any DgkA structure. We will discuss this data in light of the structures of this protein that have been published in recent years.
Orientational Restraints are Absolute Restraints
OS ssNMR uses anisotropic dipolar, quadrupolar, or chemical shifts. These observables lead to a set of orientational restraints that restrict atomic sites within the protein with respect to the magnetic field axis. This axis is fixed in the laboratory frame of reference as is the orientation frame for the sample. Here, the bilayer normal is fixed parallel to the magnetic field axis. Such absolute restraints where a protein site is defined relative to a laboratory fixed axis frame are fundamentally different from relative restraints. These latter restraints are, for example, the short range distances measured between atoms in the protein or isotropic chemical shifts used to restrain backbone torsion angles, again restraining one position in the protein relative to another.
To compare these restraints consider a set of relative restraints, each with an appropriate error bar, throughout an isolated helix, i.e., we do not consider the helix-helix interactions for this illustration (Fig. 2). For comparison, consider a set of orientational restraints with an appropriate error bar throughout a second isolated helix. Following restrained molecular dynamics on each helix the three lowest energy structures are superimposed using the six N-terminal residues for each structure. The two sets of structures illustrate an important difference between these two classes of restraints. Long-range structure perturbations, such as helix bending can result from the use of relative restraints, because the errors from restraints at the N-terminus of the helix add to those at the C-terminus, i.e., the restraints are not independent. However, the errors from the orientational restraints do not add, because they are each referenced to the laboratory frame of reference and are independent of each other. In characterizing each peptide plane of the helix, not only is the secondary structure characterized by these restraints, but its orientation relative to the magnetic field and the bilayer normal is defined simultaneously. This restraint to the laboratory frame means that there is no summation of errors across a molecular structure that leads to the perturbations observed when relative restraints are used to define the molecular structure.
Figure 2.
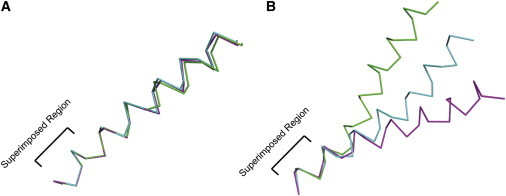
Comparison of helical structures restrained by absolute (orientational) restraints (A) and relative (distance) restraints (B). Calculations were performed in XPLOR-NIH. An extended conformation was equilibrated at 3500 K for 20 ps using torsion angle restraints (φ = −60°, ψ = −45°, ±30°) followed by slow cooling simulated annealing from 3500 to 100 K. Both calculations used torsion angle, helical hydrogen bond, bond angle, improper dihedral, and van der Waals restraints for simulated annealing. The absolute calculation was based on an ideal helix tilted at 25° that was also restrained by 20 15N chemical shift anisotropy restraints using a generous error bar of ±10 ppm and tensor element values of δ11 = 57.3, δ22 = 81.2, δ33 = 227.8ppm (2), as well as 20 1H-15N dipolar coupling restraints with an error bar of ±0.5 kHz and dipolar magnitude of 10.375 kHz (2,3). The relative calculation was also based on the same ideal helix and was also restrained by 20 nitrogen (i, i + 4) and 20 α-carbon (i, i + 4) distance restraints (5Å cutoff). All restraints were modeled as square-well potentials and the scaling of the force constants for the structure calculation was the same as that used for the Influenza A M2 monomer (4). Three representative, lowest energy, structures are shown for each calculation. Superposition of the structures was calculated using the backbone atoms of residues 1–6 of each helix.
The error bars for the orientational restraints are dependent on a variety of factors including how uniformly the sample is aligned, the linewidths of the resonances in the oriented samples compared to the magnitude of the anisotropic spin interaction, and the specific orientation of the tensor for a given molecular site with respect to the magnetic field. We have shown using 2H quadrupolar resonances having a linewidth of 1.2 kHz, an orientational error of as little as ±0.2° (47). This is a motionally averaged value. It is not that a particular site is held rigidly to within ±0.2°, but that the distribution of orientations over our entire sample has a half-width distribution of ±0.2°. Typically our aligned samples have a half-width distribution of <±1.0°. At first glance, one would think that positioning the sample in the radiofrequency coil would have a greater error bar than this half-width distribution. Indeed, the glass slides that we use for bilayer alignments are often perceptibly misaligned (by several degrees) with respect to the sample tube. However, such misalignment, has routinely been observed not to influence the frequency of the observed resonances, because there is significant magnetic alignment of the protein in lipid bilayers. The diamagnetic susceptibility of the carbonyl groups in the helices dominates that of the methylene groups in the lipids. The helix carbonyls are aligned relatively close to the helix axis and with rotation of the protein about the bilayer normal the protein susceptibility tends to align the bilayer normal parallel with the external magnetic field. This magnetic alignment of the bilayer normal minimizes the width of the resonance frequencies. In addition to the overall alignment, the orientational error associated with each restraint is dependent on the tensor orientation with respect to the magnetic field and the bilayer normal via a P2 cos θ dependence. For the 1H-15N bond, with its ν|| axis aligned nearly parallel to the bilayer normal, the dipolar coupling dependence is not very sensitive to the angle θ. But if the angle is 10° or 15° the dipolar coupling sensitivity to this angle becomes greatly enhanced. Fortunately, we use not only this dipolar interaction, but the anisotropic 15N chemical shift and these tensors are not collinear and are complementary. Thus, the combination of these orientational restraints represents an excellent tool for accurately orienting each peptide plane with respect to the bilayer normal.
Structural Validation
High-resolution OS ssNMR spectra (PISEMA spectra) were published for DgkA in 2007 (Fig. 3) (1). Three samples had been prepared; one with uniform 15N-labeling and two with amino acid-specific 15N-labeling. The spectrum of the uniformly labeled sample displayed severely overlapped resonances, but strongly suggested that the TM helices were restricted to tilt angles of <20°. For helix 3 a tilt angle estimate of 12 ± 3° was published. For DgkA there are five tryptophan residues corresponding to five well-resolved backbone amide resonances in the OS spectrum and three methionine residues that were not resolved, but span a narrow range of anisotropic chemical shifts and dipolar couplings. Note that a single set of resonances for each amino acid residue was observed indicating that the structure has threefold symmetry about the bilayer normal. If each monomer in the trimer had a slightly different conformation, three resonances would be observed for each residue (see below and Fig. 3). Furthermore, because the helical backbone dynamics are similar from one membrane protein to another it is possible to predict the observed OS ssNMR spectral frequencies accurately from the coordinates of the DgkA structures using typical values of the 1H-15N dipolar interaction (10.375 kHz) and 15N chemical shift anisotropy (δ11 = 227.8, δ22 = 81.2, δ33 = 57.3 ppm) (2). Global rotation of the protein about the bilayer normal does occur as mentioned previously, but does not average the anisotropic resonance frequencies in OS ssNMR when the bilayer normal is aligned parallel to the magnetic field.
Figure 3.
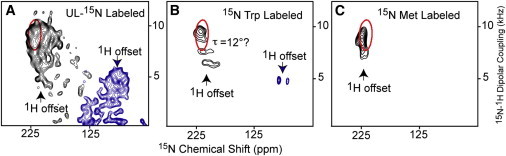
PISEMA spectra from DgkA in liquid crystalline lipid bilayers of dimyristoyl-phosphatidylcholine and dimyristoyl phosphatidylglycerol in a 4:1 molar ratio. The molar ratio of lipid/protein was ∼200:1. (A) Spectra of uniform 15N-labeled protein. (B) Spectra of 15N tryptophan-labeled protein. (C) Spectra of 15N methionine-labeled protein. Because of the sensitivity of this experiment to 1H offset, to record the full spectrum, the 1H offset was set at two different frequencies for the UL 15N and 15N tryptophan-labeled samples (1). For these samples no functional assays were performed.
The methionine residues are in the TM helices at positions 63 and 66 in helix 2, whereas residue 96 is in helix 3 near the cytoplasmic interfacial region, but still well within the helix based on both the solution NMR and crystal structures (Fig. 1, A and C). To initiate the prediction of the anisotropic 15N chemical shifts and the 1H-15N dipolar interactions, the structures were oriented relative to the bilayer normal. Based on the threefold symmetry axis for the solution NMR structure (PDB: 2KDC) this could be done accurately resulting in identical values for the anisotropic spin interactions from each monomer in the trimer. The predictions from the solution structure for residues 63 and 66, which reside in the core of the structure, are consistent with the experimental data suggesting that this part of the structure is similar to that of the native protein in a membrane environment (Fig. 4 A). However, the prediction for M96 of helix 3, is far removed from the observed frequencies (Fig. 4 A). The dipolar interaction for this latter residue is altered by >5 kHz and the anisotropic chemical shift by 35 ppm suggesting a minimum of a 25° change in the peptide plane orientation relative to that observed in lipid bilayers.
Figure 4.
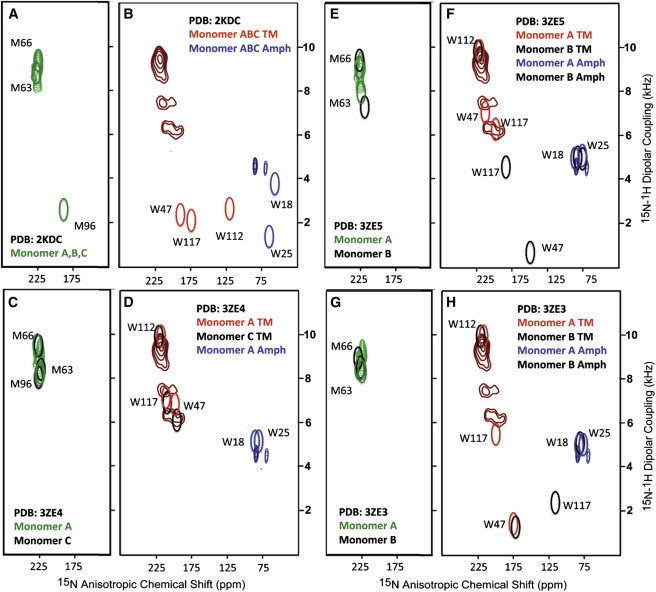
Comparisons of predicted resonance frequencies from the DgkA structures with the 15N tryptophan and methionine-labeled DgkA experimental data shown in Fig. 3. Methionine resonance contours are green and TM tryptophan resonances are red and amphipathic helix tryptophan resonances are blue. (A and B) Comparison with the solution NMR structure (PDB: 2KDC). M63 and M66 fit well with the experimental data and W18 is not too far from one of the amphipathic helix experimental resonances, but the other resonances are not in agreement. (C and D) Comparison with the WT DgkA x-ray structure (PDB: 3ZE4). The A (green, red, blue) and C (black) monomers were used for the predictions. The amphipathic helix of monomer C did not diffract well enough for a structural characterization. (E and F) Comparison with the thermally stabilized (4 mutations) DgkA x-ray structure (PDB 3ZE5) using monomers A (green, red, blue) and B (black). One of the mutations is M96L and therefore this resonance is not predicted. (F and G) Comparison with the thermally stabilized (7 mutations) DgkA structure (PDB 3ZE3) using monomers A (green, red, blue) and B (black). Two thermal stabilization mutations affect this spectrum M96L as in 3ZE5 and A41C. Predictions were based on proteins with their symmetry axis aligned parallel to the magnetic field and the chemical shift and dipolar interactions defined in the Fig. 2 legend.
The predictions from the crystal structures are more complicated because the structures are only pseudo-threefold symmetric, due to crystal contacts between trimers in the crystal lattice, thermostabilizing mutations, and a dimer of trimers in the crystal lattice for 3ZE3. For these structures, select residues were used to define a membrane normal vector. Near the periplasmic side of the protein, L57 residues line the center of the DgkA structures, whereas L70 lines the center of the protein near the cytoplasmic side of the protein. These residues are at least one helical turn into the lipid bilayer. The center of mass for the nitrogen atoms was used to define endpoints for the membrane normal vector. This vector was used to predict the NMR observables. For both 3ze3 and 3ze5 M96 is mutated to leucine and therefore the M96 resonance is missing from the predictions for these structures, but for M96 (helix 3) in the WT (3ZE4) structure it is consistent with the experimental methionine resonance envelope (see Fig. 4 C). The M63 and M66 residues on helix 2 are near the center of the structure and their calculated frequencies do not shift substantially between the monomers or between the three crystal structures—again all of these predictions are consistent with the ssNMR methionine resonance envelope (see Fig. 4, C, E, and G). In the spectral region near the observed methionine resonances a change of 10 ppm in chemical shift could suggest a change in orientation of the peptide plane of as much as 7° and a change in the dipolar interaction of 1 kHz could be construed to be as much as a 10° change in the orientation of the N-H vector. For these Met residues in the WT (3ZE4) structure the deviations from experimental data are significantly less than this. Indeed the fit is remarkable.
Although the methionine residues report on TM helix 2 and 3, the tryptophan residues sample TM helices 1 and 3, as well as the amphipathic helix. Two of the tryptophan residues are in the N-terminal amphipathic helix at positions 18 and 25, and three are in the TM helices near the periplasmic bilayer interface at position 47 in TM helix 1 and at positions 112 and 117 in TM helix 3 (Fig. 1). In others words, between the methionine and tryptophan labels we have reporter groups from all of the helices in DgkA covering both the periplasmic and cytoplasmic interfacial regions. The predictions from the solution NMR structure for tryptophan residues in the amphipathic helix have somewhat similar chemical shifts to the observed resonances (potentially within 20 ppm depending on assignment). The dipolar interactions are substantially different especially for W25, which suggests a change of at least 20° in N-H orientation (Fig. 4 B). The amphipathic helix in the solution NMR structure has a substantial break between residues 15 and 17 and the location of the charged residues in this domain suggest that rotational orientation is also non native-like (Fig. 5 A). This is not surprising, because the micelle provides a highly curved interaction surface for the amphipathic helices. The predictions for the amphipathic helix from the crystal structures are quite consistent with the observed data for the two monomers in each trimer for which these helices were observed in the crystal structures (Fig. 4, D, F, and H). It appears that the crystal contacts may have disrupted the stability of one out of three amphipathic helices in these structures preventing observation of the electron density for them. The good agreement between the observed frequencies from lipid bilayers and the predictions for the amphipathic helix sites where structural data is available suggest that the rotational orientation of the amphipathic helix in these structures is similar to that in a lipid bilayer. However, the predictions from all of the W18 and W25 structures consistently suggest a somewhat greater dipolar coupling by ∼0.5 kHz indicating that in lipid bilayers the helical axes may have an increased tilt with respect to the bilayer surface than in the x-ray structures.
Figure 5.
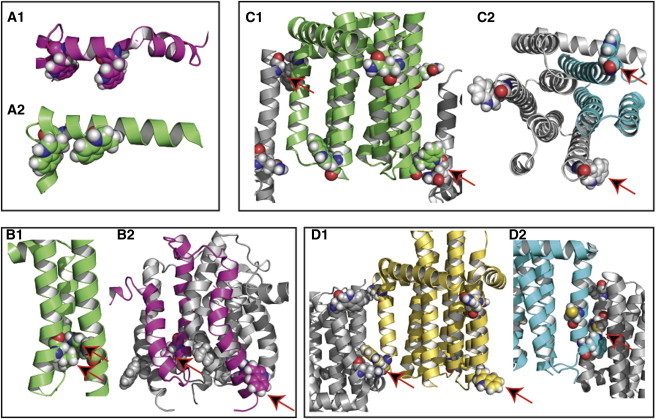
Highlights of structural perturbations involving the tryptophan residues: Panel A – W18, 25; panel B – W112; panel C – W117; panel D – W47. The carbons of the structures are color coded: 2KDC – magenta and gray; 3ZE4 – green and gray; 3ZE5 – yellow and gray; 3ZE3 – cyan and gray. The residues of interest are shown as van der Waals spheres. (A1 and A2) Comparison of the W18 and W25 in the solution NMR (2KDC) and the WT x-ray structure (3ZE4)—note in the later structure that the indole N-H groups are partially oriented toward what would be the bilayer interfacial region. (B1 and B2) Another comparison between 2KDC and 3ZE4. The x-ray structure shows an interhelical hydrogen bond between W112 indole and the carbonyl of L58. In 2KDC these residues are far apart. (C1 and C2) W117 indole of 3ZE4 forms a hydrogen bond with Q33 in a neighboring trimer without significantly affecting the backbone amide conformation. However, in 3ZE3 one of the helices is shortened by a turn and the W117 backbone orientation is nonhelical. (D1 and D2) In 3ZE5 Q33 hydrogen bond to the amide of W47 between trimers distorts the structure in comparison to the other W47 residues. In 3ZE3 W47 indole hydrogen bonds to C41, one of the temperature stabilizing mutations, in a neighboring trimer, distorting the helical structure.
The predictions for the Trp amide resonances in the TM helices from the solution NMR structure display no correspondence with the observed resonances (Fig. 4 B). The orientation of the peptide planes for these sites near the periplasmic surface, bear no resemblance to the structure in lipid bilayers. The dipolar interactions are all <3 kHz in 2KDC, whereas those observed in lipid bilayers range from 6.5 to 10 kHz and the W112 prediction is 100 ppm removed from the observed resonance. The predictions from the crystal structures show considerable agreement with the observed resonances for these interfacial tryptophan residues. In particular, W112 in all monomers of the three crystal structures results in predictions that agree precisely with one of the observed tryptophan resonances. From the crystal structures this indole in helix 3 forms a hydrogen bond with the backbone carbonyl of L58 in helix 2 of the same monomer, a relatively rare interhelical hydrogen bond (Fig. 5 B). This tertiary interaction is conserved within monomers of the various DgkA constructs and results in uniform predictions for this site, consistent with the observations in liquid crystalline lipid bilayers. The lack of agreement between the predictions from the solution NMR structure and the OS ssNMR data suggest that not only the local structure but potentially the tertiary structure may be different because it appears that this hydrogen bond is not present in the detergent micelle preparation.
The remaining two tryptophan residues, 47 and 117 are on the exterior of the trimeric structure. Located near the periplasmic end of helix 3, W117 has more variation than W112 in the predicted values of the anisotropic chemical shift and dipolar interactions from the crystal structures. Each of the W117 indole N-H bonds is oriented toward what would be the lipid interface to form the well-known interaction between indole and hydrophilic sites in the lipid interface. However, monomer A in 3ZE4 has the W117 indole N-H hydrogen bonded to the Q33 backbone carbonyl oxygen in an intertrimer crystal contact (Fig. 5 C1). Another crystal contact hydrogen bond involving these two residues occurs between the Q33 side-chain amide to the W117 carbonyl oxygen. However, these interactions do not significantly perturb the predicted amide resonance frequencies for the monomer A W117 because it has the same predicted resonance frequencies as W117 of monomers B and C in 3ZE4 that do not participate in such crystal contacts (Fig. 4 C). In part, this may be because the W117 carbonyl is not in the same peptide plane as the observed 15N site. It may also be that this interaction fortuitously stabilizes the indole in an appropriate orientation for interacting with the lipid bilayer. Similarly, the W117 from the 3ZE5 monomer A displays the same intertrimer interactions with insignificant perturbation of its predicted frequencies (Fig. 4 F). Hence, it is interesting to note that such hydrogen bonding between lattice sites does not necessarily result in structural perturbations. This particular interaction does not appear to take place in the interdimer (of trimers) crystal contacts for 3ZE3, but monomer C W117 has such an intertrimer interaction and again, although it is not shown in Fig. 4, it has the same predicted NMR resonance frequencies as the experimentally observed resonance frequencies. However, there are some differences between the x-ray predictions and ssNMR data. For example, in monomer B of 3ZE3 helix 3 terminates at residue 116, although in the other monomers it continues for another turn of the helix. This results in a nonhelical W117 resonance for monomer B (Fig. 4 H and Fig. 5 C2).
Trp-47 is at the periplasmic end of helix 1 and in most of the monomers of the three crystal structures this indole N-H bond is oriented toward the bilayer interface. However, for monomer B in the 3ZE5 structure the W47 carbonyl forms an intertrimer hydrogen bond with the amide side chain of Q33 (Fig. 5 D1). The result is a distorted orientation of the indole and a termination of the helical residues at A45 resulting in nonhelical resonance frequencies for W47. Most significantly, the indole N-H bond of W47 in monomer B of 3ZE3 intercalates between helices 1 and 3 of the neighboring trimer and also hydrogen bonds with C41 of the same neighboring trimer (Fig. 5 D2). Because of this one-sided, asymmetric, interaction the two structures have significant structural differences. A41C is one of the thermostabilizing mutations that result in a significant change in helix packing and a dramatic change in the predicted anisotropic spin interactions. This is a result of integrating an additional hydrophilic residue into the TM domain of the protein.
In the 2007 publication (1) of the experimental data the tilt angle for helix 3 was estimated to be 12 ± 3°. Even without resolved resonances and all of the assignments, the OS ssNMR data was used quantitatively to define a helix tilt. However, helix 3 of the solution NMR structure is a highly curved helix with segments having tilt angles upward of 40°, although the tilt angle for this helix is ∼9° in the 3ZE4 crystal structure. As seen in Fig. 4 the OS ssNMR experimental data fit with the predictions for the 3ZE4 structure very well, whereas the fit with the 2KDC predictions was much worse. Moreover, the analysis of the helical tilts also agrees with the assessment from the individual resonances.
An Assessment of the DGKA Structures and their Environment
As mentioned earlier, the structures are trimers; they have helix 2 at the core of the oligomer with helices 1 and 3 on the exterior along with the amphipathic helix. Moreover, the residues that make up the TM helices are very similar and the overall helical content is similar. There are, however, numerous features that are different. The amphipathic helix in the solution NMR structure was not well restrained by the interfacial region of the detergent micelle and, consequently, neither was the position of the helical segments nor their rotational orientation appropriate for an interfacial interaction. Although the crystal structures present a well-oriented amphipathic helix for two of the three DgkA monomers, it appears that crystal contacts between trimers in the crystal lattice disrupt one of these helices such that its electron density is not characterized. The crystal lattice is somewhat bilayer-like and the detergents that diffract well are packed in a bilayer-like array that may be responsible for the appropriate positioning and orientation of those amphipathic helices that are observed. There are, however, significant crystal contacts through an extensive van der Waals surface and hydrogen bonding between W117 and Q33 in a neighboring trimer. In 3ZE3 hydrogen bonding of the W47 indole to C41 of a neighboring trimer was observed. Among the three DgkA constructs that were crystallized the superposition of helix 1 shows the greatest structural dispersion (13). Even though some crystal contacts did not appear to disturb the structures, other contacts result in significant resonance perturbations and in structural perturbations. The most significant perturbations appear to be the result of introducing what some may consider a conservative thermostabilizing mutation, A41C. However, such mutations that introduce hydrogen bonding capacity into the middle of a TM helix near the interface with the fatty acyl environment should not be considered a conservative mutation for membrane proteins.
The helices of the solution NMR structure are domain swapped in that helix 1 and 3 interact primarily with helices from different monomers. There appear to be very few examples of such domain swapping in the PDB, although a recent solution NMR structure of the p7 protein from hepatitis C virus (48) is another example, but it too appears to have nonnative-like characteristics based on the criteria presented by Zhou and Cross (5) and discussed briefly here. Another difference is that the helices are much more uniform for the crystal structure than for the solution NMR structure. In other words, the torsion angles and the helical tilts are both more uniform in the crystal structure than in the solution NMR structure, where the helices have an outward curvature. The net effect is a cylindrical x-ray structure and a ball-shaped solution NMR structure. For the solution NMR structure this suggests multiple issues. The nonuniformity of the torsion angles suggests that the membrane mimetic environment is not as hydrophobic and does not have as low a dielectric constant as that of a native membrane. The curvature causes a lengthening of the hydrogen bonds on the helical surface facing the very low dielectric of what would be the fatty acyl environment, while shortening the hydrogen bonds on the somewhat more hydrophilic interior of the TM domain. This curvature also results in fenestrations into the protein interior and a lack of interaction between the helices in the bilayer interstices reducing the stability of the protein. Indeed, helical TM proteins go to great lengths to strengthen the interactions between helices by tolerating glycine and proline residues in the TM helices (20). Here, there are not many glycine residues, but there are two alanine motifs in the TM helices of each DgkA monomer. Neither the motif in helix 1 (Alanine 37, 41, 45) nor the motif in helix 3 (Alanine 100, 104, 108) facilitate close contacts in the solution NMR structure. However, both of these motifs facilitate close contacts in the crystal structure. In addition, G35 and S61 do have van der Waals interactions with neighboring helices in the crystal structure, but neither of these residues form close contacts with neighboring helices in the solution NMR structure.
As stated previously, these two structures are obtained in detergent environments; the solution structure in DPC detergent micelles and the crystal structure in a monoolein cubic phase. Based both on criteria for a native-like structure as discussed here and on the predictions of the OS ssNMR data, the solution NMR structure is clearly nonnative-like. The characterization of the DgkA structure by solution NMR was a very significant challenge for the 40 kDa mass of this trimer embedded in a detergent complex. The slow correlation times resulted in broad resonances and the dynamics resulted in further challenges. Without side-chain resonance assignments it was not possible to determine interhelical distance restraints from nuclear Overhauser effect measurements. The disulfide bond restraints and the paramagnetic relaxation restraints used for the solution NMR structure are both highly susceptible to the influence of dynamics. The residual dipolar couplings, the solution NMR form of absolute restraints, have the disadvantage that the alignment tensor is dependent on the alignment media, unlike the OS ssNMR restraints where the alignment frame is fixed in the laboratory frame of reference. Typically, multiple alignments are used in solution NMR, so that these restraints can be accurately interpreted, but these were not available for DgkA. As a result, from the entire protein, only in a fragment of helix 1 displayed the typical sinusoidal oscillation of the observed couplings. Given these issues, it is possible that the data was overinterpreted and the structure under restrained.
Although the interpretation of the data may have been a problem, it is also clear that the detergent micelle provides a very different environment from that of lipid bilayers or native membranes. Consequently, this membrane mimetic environment is also responsible for many of the observed problems. For instance, the outward curvature of the helices has been seen multiple times in detergent micelles and only in detergent micelles. The irregularity of the helices, which should have been well restrained by the solution NMR backbone restraints, is inconsistent with the low dielectric and hydrophobic environment of the lipid bilayer. The functional assays in the same environment, but at a lower temperature than that used for the spectroscopy demonstrated catalytic activity, but with a higher KM suggesting that the structure is either significantly distorted or excessively dynamic (49). The recently observed failure of the functional assay at the higher NMR sample temperature now also suggests a nonnative structure. The crystal structure also showed some problems, especially with the thermostabilizing mutations, but far fewer than the solution structure. By focusing on the monomers within the trimer that do not have significant crystal contacts a structure is observed that predicts the OS ssNMR resonance frequencies extremely well, thereby validating this structure as a native-like structure.
In addition, it should be noted that there is an increasing number of solid-state NMR structures of relatively small helical membrane proteins some of which have been compared to solution NMR structures and x-ray crystal structures. The pentameric phospholamban solution NMR structure obtained in detergent micelles (50) has been shown to be nonnative-like by OS ssNMR data in lipid bilayers and by high resolution structure (51). The WT M2 proton channel solution NMR structure in detergent micelles (52) and the detergent based crystal structure (53) have both been identified as nonnative-like structures based on OS ssNMR and MAS ssNMR data (4,54,55). Other ssNMR structures have been obtained (fd coat (57), MgtC/MgtR (58), MerF (59), CXCR1 (38); Cyt b5 (41)) demonstrating that multiple labs are producing native-like membrane protein structures in lipid bilayer environments.
Conclusions
The DgkA structure obtained from detergent micelles is a severely distorted structure. This solution NMR characterization was a heroic effort for such a large membrane protein structure. The sample for structural study was recently shown to be nonfunctional and the result was a structure that reflected many of the nonnative environmental effects of a detergent micelle rather than that of a lipid bilayer or native membrane. The WT DgkA structure obtained from x-ray diffraction is a triumph for lipidic cubic phase sample preparation. The agreement between the OS ssNMR data, which has typical orientational error bars of 1–2° is predicted with excellent accuracy for those monomers not involved in crystal contacts. Even then, the crystal contacts did not always lead to significant distortions. However, some temperature stabilizing mutations did induce significant local distortions that caused dramatic deviations for the predicted resonances. In particular, the A41C mutation introduced a hydrophilic residue in the TM domain and caused significant changes in helix packing as a result of a hydrogen bond formation between trimers. Of importance, this is not a blanket recommendation for lipidic cubic phase, as we have seen the structure of gA was not native-like when crystallized from such an environment, but it raises hope that this approach may be more generally useful than more traditional detergent environments for crystallization, especially for small helical membrane proteins.
In the past decade functional assays for water-soluble proteins have provided validation for these structures. Indeed, such validation has become the norm for water-soluble proteins. However, functional assays do not provide structural validation unless they are performed in the same environment as the structural characterization. For membrane proteins this is a problem, because functional assays can rarely be achieved in the environment used for structural analysis. It is clear that validation of membrane protein structures is even more important than it is for water-soluble proteins, because the mimetic environment used for structural characterization can substantially influence the structure (5,6). Structural validation of membrane proteins needs to be established as the norm to avoid misleading the broader scientific community where the structural information is used to develop functional models. The classic example is the paddle model for voltage sensing, which was based on a structure that is now well recognized as a highly distorted structure. Even a decade after the publication of the KvAP structure (60) this distorted structure and its implications are still influencing the discussion of voltage regulation. Although it was not possible then to obtain data on KvAP in lipid bilayers, today it is possible and should be obtained. Validation of membrane proteins is critically important so that scientific communities are not misled for a decade or more. An approach for validating membrane protein structures is by collecting OS ssNMR structural data in lipid bilayer environments from a minimal number of isotopically labeled samples.
Acknowledgments
This work was supported, in part, by the National Institutes of Health (NIH) grants AI074805 and AI023007. The NMR experiments were performed at the National High Magnetic Field Laboratory supported by a National Science Foundation cooperative agreement (DMR-1157490) with the State of Florida.
References
- 1.Li C., Gao P., Cross T.A. Uniformly aligned full-length membrane proteins in liquid crystalline bilayers for structural characterization. J. Am. Chem. Soc. 2007;129:5304–5305. doi: 10.1021/ja068402f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang J., Denny J., Cross T.A. Imaging membrane protein helical wheels. J. Magn. Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 3.Denny J.K., Wang J., Quine J.R. PISEMA powder patterns and PISA wheels. J. Magn. Reson. 2001;152:217–226. doi: 10.1006/jmre.2001.2405. [DOI] [PubMed] [Google Scholar]
- 4.Sharma M., Yi M., Cross T.A. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou H.X., Cross T.A. Influences of membrane mimetic environments on membrane protein structures. Annu. Rev. Biophys. 2013;42:361–392. doi: 10.1146/annurev-biophys-083012-130326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cross T.A., Murray D.T., Watts A. Helical membrane protein conformations and their environment. Eur. Biophys. J. 2013;42:731–755. doi: 10.1007/s00249-013-0925-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anfinsen C.B. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 8.Loomis C.R., Walsh J.P., Bell R.M. sn-1,2-Diacylglycerol kinase of Escherichia coli. Purification, reconstitution, and partial amino- and carboxyl-terminal analysis. J. Biol. Chem. 1985;260:4091–4097. [PubMed] [Google Scholar]
- 9.Walsh J.P., Fahrner L., Bell R.M. sn-1,2-diacylglycerol kinase of Escherichia coli. Diacylglycerol analogues define specificity and mechanism. J. Biol. Chem. 1990;265:4374–4381. [PubMed] [Google Scholar]
- 10.Smith R.L., O’Toole J.F., Sanders C.R., 2nd Membrane topology of Escherichia coli diacylglycerol kinase. J. Bacteriol. 1994;176:5459–5465. doi: 10.1128/jb.176.17.5459-5465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanders C.R., 2nd, Czerski L., Smith S.O. Escherichia coli diacylglycerol kinase is an alpha-helical polytopic membrane protein and can spontaneously insert into preformed lipid vesicles. Biochemistry. 1996;35:8610–8618. doi: 10.1021/bi9604892. [DOI] [PubMed] [Google Scholar]
- 12.Nagy J.K., Sanders C.R. Destabilizing mutations promote membrane protein misfolding. Biochemistry. 2004;43:19–25. doi: 10.1021/bi035918s. [DOI] [PubMed] [Google Scholar]
- 13.Li D., Lyons J.A., Caffrey M. Crystal structure of the integral membrane diacylglycerol kinase. Nature. 2013;497:521–524. doi: 10.1038/nature12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Horn W.D., Kim H.J., Sanders C.R. Solution nuclear magnetic resonance structure of membrane-integral diacylglycerol kinase. Science. 2009;324:1726–1729. doi: 10.1126/science.1171716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng J., Jia Z. Structural biology: tiny enzyme uses context to succeed. Nature. 2013;497:445–446. doi: 10.1038/nature12245. [DOI] [PubMed] [Google Scholar]
- 16.Zhou H.X., Cross T.A. Modeling the membrane environment has implications for membrane protein structure and function: influenza A M2 protein. Protein Sci. 2013;22:381–394. doi: 10.1002/pro.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cross T.A., Sharma M., Zhou H.X. Influence of solubilizing environments on membrane protein structures. Trends Biochem. Sci. 2011;36:117–125. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim S., Cross T.A. Uniformity, ideality, and hydrogen bonds in transmembrane alpha-helices. Biophys. J. 2002;83:2084–2095. doi: 10.1016/S0006-3495(02)73969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Page R.C., Kim S., Cross T.A. Transmembrane helix uniformity examined by spectral mapping of torsion angles. Structure. 2008;16:787–797. doi: 10.1016/j.str.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong H., Sharma M., Cross T.A. Glycines: role in α-helical membrane protein structures and a potential indicator of native conformation. Biochemistry. 2012;51:4779–4789. doi: 10.1021/bi300090x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Javadpour M.M., Eilers M., Smith S.O. Helix packing in polytopic membrane proteins: role of glycine in transmembrane helix association. Biophys. J. 1999;77:1609–1618. doi: 10.1016/S0006-3495(99)77009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russ W.P., Engelman D.M. The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 23.Maslennikov I., Klammt C., Choe S. Membrane domain structures of three classes of histidine kinase receptors by cell-free expression and rapid NMR analysis. Proc. Natl. Acad. Sci. USA. 2010;107:10902–10907. doi: 10.1073/pnas.1001656107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nymeyer H., Zhou H.X. A method to determine dielectric constants in nonhomogeneous systems: application to biological membranes. Biophys. J. 2008;94:1185–1193. doi: 10.1529/biophysj.107.117770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klammt C., Maslennikov I., Choe S. Facile backbone structure determination of human membrane proteins by NMR spectroscopy. Nat. Methods. 2012;9:834–839. doi: 10.1038/nmeth.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berardi M.J., Shih W.M., Chou J.J. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature. 2011;476:109–113. doi: 10.1038/nature10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caffrey M., Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caffrey M. Crystallizing membrane proteins for structure determination: use of lipidic mesophases. Nat. Protoc. 2009;38:29–51. doi: 10.1146/annurev.biophys.050708.133655. [DOI] [PubMed] [Google Scholar]
- 29.Höfer N., Aragão D., Caffrey M. Crystallizing transmembrane peptides in lipidic mesophases. Biophys. J. 2010;99:L23–L25. doi: 10.1016/j.bpj.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burkhart B.M., Gassman R.M., Duax W.L. Heterodimer formation and crystal nucleation of gramicidin D. Biophys. J. 1998;75:2135–2146. doi: 10.1016/S0006-3495(98)77656-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ketchem R.R., Roux B., Cross T.A. High-resolution polypeptide structure in a lamellar phase lipid environment from solid state NMR derived orientational constraints. Structure. 1997;5:1655–1669. doi: 10.1016/s0969-2126(97)00312-2. [DOI] [PubMed] [Google Scholar]
- 32.Ketchem R.R., Hu W., Cross T.A. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science. 1993;261:1457–1460. doi: 10.1126/science.7690158. [DOI] [PubMed] [Google Scholar]
- 33.Separovic F., Killian J.A., Cross T.A. Modeling the membrane environment for membrane proteins. Biophys. J. 2011;100:2073–2074. doi: 10.1016/j.bpj.2011.02.058. author reply 2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhate M.P., Wylie B.J., McDermott A.E. Preparation of uniformly isotope labeled KcsA for solid state NMR: expression, purification, reconstitution into liposomes and functional assay. Protein Expr. Purif. 2013;91:119–124. doi: 10.1016/j.pep.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhate M.P., McDermott A.E. Protonation state of E71 in KcsA and its role for channel collapse and inactivation. Proc. Natl. Acad. Sci. USA. 2012;109:15265–15270. doi: 10.1073/pnas.1211900109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Cruijsen E.A., Nand D., Baldus M. Importance of lipid-pore loop interface for potassium channel structure and function. Proc. Natl. Acad. Sci. USA. 2013;110:13008–13013. doi: 10.1073/pnas.1305563110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weingarth M., Prokofyev A., Baldus M. Structural determinants of specific lipid binding to potassium channels. J. Am. Chem. Soc. 2013;135:3983–3988. doi: 10.1021/ja3119114. [DOI] [PubMed] [Google Scholar]
- 38.Park S.H., Das B.B., Opella S.J. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–783. doi: 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murray D.T., Das N., Cross T.A. Solid state NMR strategy for characterizing native membrane protein structures. Acc. Chem. Res. 2013;46:2172–2181. doi: 10.1021/ar3003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park S.H., Mrse A.A., Opella S.J. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein “u” (Vpu) from HIV-1. J. Mol. Biol. 2003;333:409–424. doi: 10.1016/j.jmb.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 41.Ahuja S., Jahr N., Ramamoorthy A. A model of the membrane-bound cytochrome b5-cytochrome P450 complex from NMR and mutagenesis data. J. Biol. Chem. 2013;288:22080–22095. doi: 10.1074/jbc.M112.448225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verardi R., Shi L., Veglia G. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc. Natl. Acad. Sci. USA. 2011;108:9101–9106. doi: 10.1073/pnas.1016535108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marassi F.M., Opella S.J. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Das N., Murray D.T., Cross T.A. Lipid bilayer preparations of membrane proteins for oriented and magic-angle spinning solid-state NMR samples. Nat. Protoc. 2013;8:2256–2270. doi: 10.1038/nprot.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Angelis A.A., Opella S.J. Bicelle samples for solid-state NMR of membrane proteins. Nat. Protoc. 2007;2:2332–2338. doi: 10.1038/nprot.2007.329. [DOI] [PubMed] [Google Scholar]
- 46.Murray D.T., Hung I., Cross T.A. Assignment of oriented sample NMR resonances from a three transmembrane helix protein. J. Magn. Reson. 2014;240C:34–44. doi: 10.1016/j.jmr.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cross T.A., Ketchem R.R., North C.L. Structure and dynamics of a membrane bound polypeptide. Bull. Magn. Reson. 1992;14:96–101. [Google Scholar]
- 48.OuYang B., Xie S., Chou J.J. Unusual architecture of the p7 channel from hepatitis C virus. Nature. 2013;498:521–525. doi: 10.1038/nature12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Horn W.D., Sanders C.R. Prokaryotic diacylglycerol kinase and undecaprenol kinase. Annu. Rev. Biophys. 2012;41:81–101. doi: 10.1146/annurev-biophys-050511-102330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oxenoid K., Chou J.J. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc. Natl. Acad. Sci. USA. 2005;102:10870–10875. doi: 10.1073/pnas.0504920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Traaseth N.J., Shi L., Veglia G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc. Natl. Acad. Sci. USA. 2009;106:10165–10170. doi: 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schnell J.R., Chou J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stouffer A.L., Acharya R., DeGrado W.F. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Can T.V., Sharma M., Cross T.A. Magic angle spinning and oriented sample solid-state NMR structural restraints combine for influenza a M2 protein functional insights. J. Am. Chem. Soc. 2012;134:9022–9025. doi: 10.1021/ja3004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miao Y., Qin H., Cross T.A. M2 proton channel structural validation from full-length protein samples in synthetic bilayers and E. coli membranes. Angew. Chem. Int. Ed. Engl. 2012;51:8383–8386. doi: 10.1002/anie.201204666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reference deleted in proof.
- 57.Marassi F.M., Opella S.J. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci. 2003;12:403–411. doi: 10.1110/ps.0211503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jean-Francois F.L., Dai J., Cross T.A. Binding of MgtR, a Salmonella transmembrane regulatory peptide, to MgtC, a Mycobacterium tuberculosis virulence factor: a structural study. J. Mol. Biol. 2014;426:436–446. doi: 10.1016/j.jmb.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Das B.B., Nothnagel H.J., Opella S.J. Structure determination of a membrane protein in proteoliposomes. J. Am. Chem. Soc. 2012;134:2047–2056. doi: 10.1021/ja209464f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang Y., Lee A., MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]