

Abstract
The neuropathology underlying dementia syndromes in older populations is complex. The contributions of Alzheimer’s and Lewy body pathology are well appreciated. Recent studies with brain autopsies have highlighted the high prevalence of vascular disease as an independent, but often co-morbid contributor to dementia. The Adult Changes in Thought Study is a community-based, longitudinal study of brain aging and cognitive decline which has recently confirmed cerebral microinfarcts as a strong correlate of cognitive impairment and dementia. This study examines correlations between clinical characteristics including extensive, longitudinal medication histories, and longitudinal cognitive testing against structural and biochemical features of disease. Keywords: Aging, community-based, microinfarct, longitudinal, neuropathology.
Keywords: Aging, community-based, microinfarct, longitudinal, neuropathology
INTRODUCTION
Dementia is a syndrome, a clinically-defined entity with multiple causes. The molecular mechanisms of the diseases that cause cognitive impairment and dementia in the elderly are only partially understood, but may be classified by pathologic criteria (Fig. 1). While there are many causes of dementia, results from community- or population-based studies with autopsy evaluations lead to the conclusion that diseases characterized by accumulation of misfolded proteins and amyloid, most commonly Alzheimer’s disease (AD) and Lewy body disease, as well as vascular brain injury (VBI) are the two most prevalent disease processes contributing to dementia [1–3].
Figure 1.
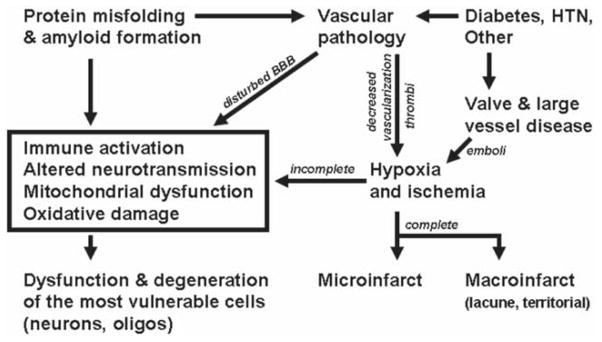
Proposed mechanisms underlying age related cognitive decline and dementia.
In a seminal paper in 1976, Dr. Robert Katzman applied a chronic disease model to AD and proposed the existence of latency where some structural damage accrues but there are no functional or behavioral deficits, followed by a prodrome during which there is more structural damage with mild functional and behavioral changes occur, and ultimately by dementia that is characterized by substantial irreversible damage and severe behavioral abnormalities (Table 1) [4]. While such a model likely does not reflect the full complexity of AD, clinical, neuroimaging, and pathologic data strongly support this sequence of disease progression that almost certainly forms a continuum beginning many years prior to clinical diagnosis of dementia. Indeed, the chronic disease model for AD has been codified with well-established clinical criteria for dementia and prodromal stages. Intense investigation is now being focused on developing neuroimaging or clinical laboratory tests to identify latent AD since this is likely where future mechanism-based therapeutic interventions will be most effective. This will be a serious public health challenge, akin to mammograms or prostate specific antigen measurements, which will need to be implemented at the level of primary care since, by definition, individuals with latent disease do not present with cognitive complaints.
Table 1.
Chronic disease model for common causes of dementia
| Stage of Disease | None | Latency | Prodrome | Dementia |
|---|---|---|---|---|
| Clinical presentation | Age appropriate | Age appropriate | Deficit insufficient for diagnosis of dementia | Meets criteria for dementia |
| Pathological findings | None | Mild | Moderate | Severe |
Vascular cognitive impairment (VCI) is the clinical manifestation of the many different forms of VBI that can produce cognitive impairment [5]. Although VBI from large vessel disease can lead to cognitive impairment and dementia, the entities of multi-infarct dementia or strategic infarcts appear to be relatively less common causes of dementia than VBI from small vessel disease (VBI-SVD) [1–3]. Difficulties immediately ensue from these deceptively simple categorizations since risk factors and pathogenic mechanisms for large vessel disease and SVD overlap, and there is no precise definition of large versus small vessels. Nevertheless, cognitive impairment from VBI-SVD commonly presents clinically in such a way that is difficult for experienced physicians to distinguish from AD and related protein misfolding diseases [6–8]. Others already have proposed categorizations for prodrome and dementia stages of disease from VCI-SVD that parallel those of AD and Lewy body disease [9]. In many respects, identification of latent VBI-SVD is far ahead of AD since identification of contributing factors already occurs at the level of primary care: detection and management of abnormalities in blood pressure, lipids, or glucose metabolism.
Despite this consideration of AD with or without Lewy body disease and VBI as separate disease processes, they are commonly co-morbid. Is this the coincidence of two common diseases in the elderly or something more? Indeed, much has been proposed on the convergence of congophilic angiopathy of AD and VBI-SVD [10]. However, this topic has been difficult to address definitively because of limits to clinical investigation and inadequate experimental models. Indeed, it remains unclear the extent to which AD and VBI-SVD intersect mechanistically. Autopsy remains the gold standard for classifying these commonly co-morbid conditions and remains a powerful tool for gaining insight into the complexity of diseases that can lead to dementia.
ADULT CHANGES IN THOUGHT (ACT) STUDY
ACT is one of the few community- or population-based studies of brain aging and dementia that have autopsy as an endpoint. ACT is an ongoing, longitudinal, community-based observational study of brain aging and incident dementia. Participants are individuals 65 years of age or older who were randomly sampled from Group Health Cooperative, a large, multicenter health management organization in King County, Washington, whose membership is reflective of the population of the region [11]. Approximately one quarter of the cohort who die undergo brain autopsy and receive an extensive neuropathologic evaluation. Of those autopsied, approximately one third meet criteria for dementia, half test within the normal range on serial cognitive screening performed every two years, and the remainder fall below the normal range on cognitive testing, but do not meet criteria for dementia. This allows neuropathologic correlation in a representative US population of individuals with none versus all levels of dementia-related illness from latency to clinically overt disease. This and similar population- or community-based studies of brain aging with autopsy endpoints offer a unique window into the neuropathologic changes underlying dementing illnesses in a manner that is directly relevant to living populations.
MICROINFARCTS
Perhaps the most important neuropathologic finding in ACT to date has been the correlation between the presence of a relatively small number of cerebral microinfarcts (CMI), i.e., microscopically identified cerebral infarcts, and cognitive decline or dementia. There have been no widely accepted neuropathologic guidelines for the diagnosis of VCI [12]. Previously, the Honolulu-Asia Aging Study (HAAS) used an epidemiologic approach to evaluate neuropathologic findings as risk factors for a clinical diagnosis of dementia. They identified AD, hippocampal sclerosis, cortical Lewy bodies, and > 2 remote microinfarcts in cortical gray and underlying white matter or in deep nuclei as independent pathologic processes underlying dementia in their cohort [2]. HAAS investigators considered a number of measures of cerebrovascular disease in their study including both acute and remote large infarcts, lacunes, microinfarcts, hemorrhages, amyloid angiopathy, and aneurysms. One of the tools used in HAAS to assess cognitive status is a 100-point questionnaire developed for use in Japanese and English called the Cognitive Assessment Screening Instrument (CASI). Using stepwise regressions, HAAS investigators identified that once remote cortical and deep microinfarcts were taken into account, none of the other cerebrovascular variables added significantly to the variance in the final cognitive function test scores; i.e., only microinfarct number was independently associated with poor CASI test scores [2]. These results suggested that microinfarct burden may have utility as a pathologic index of dementing illness from VBI-SVD in older Japanese-American men.
ACT uses CASI both as a cognitive testing instrument and as a trigger for dementia evaluation in a manner similar to that of HAAS [11]. In ACT, the same published protocol for the evaluation and counting of cortical and deep cerebral microinfarcts as used in HAAS were performed [2]. In addition, quantified neuropathologic changes based on published criteria for the evaluation of AD and Lewy body disease were performed [13–18]. Based on a sample of 221 autopsies, the pathologic correlates of dementia in our cohort were: a) cortical neuritic plaque number as assessed by the CERAD score; b) neurofibrillary tangle distribution by Braak stage; c)>2 cerebral microinfarcts; d) the presence of neocortical Lewy bodies; and e) amyloid angiopathy. When these neuropathologic indices were included jointly in a log-linear model, only Braak stage, > 2 cerebral microinfarcts, and the presence of neocortical Lewy bodies were statistically significant as correlates of dementia status [19]. Further, having at least one cystic infarct appeared to be associated with risk of dementia, although this did not attain statistical significance (p-value 0.107). We conclude that these findings validate the use of cerebral microinfarct number as an index of VBI-SVD sufficient to cause VCI in a typical urban and suburban US population. We do not suggest that these findings serve to promote the primacy of microinfarcts over other neuropathologic changes of VBI. Indeed the various neuropathologic indices of VBI tend to be correlated with one another and these statistical associations could be cohort dependent. Nevertheless, we believe that having tested a priori the hypothesis that cerebral microinfarct burden would correlate both with CASI score and dementia status offers reasonable evidence for the utility of this metric. The specific disease relevance of cerebral microinfarcts compared to other measures of VBI has yet to be established in humans or potential animal models.
COMORBIDITY
Numerous studies have identified common age-related conditions as contributing to cognitive decline and dementia [7,20–22]. As described above, in the ACT cohort we have identified three common neuropathologic correlates of dementia: Braak stage for neurofibrillary tangles, neocortical Lewy body disease, and cerebral microinfarct burden. Of those in ACT who died and came to autopsy, Table 2 summarizes the proportion who had sufficient pathologic changes within each neuropathologic category for the classification of dementia without consideration of other neuropathologic co-morbidity.
Table 2.
Proportion of ACT partipants that underwent autopsy and meet individual neuropathologic criteria associated with dementia
| Neuropathologic process | Percent of ACT meeting criteria n = 247* |
|---|---|
| CMI | 21 |
| NLB | 6 |
| AD | 34 |
| Any CMI | 45 |
10 cases were excluded due to lack of neuropathologic data.
Abbreviations: CMI = > 2 cerebral microinfarcts, NLB = presence of neocortical Lewy bodies by immunohistochemistry, AD = Braak stage V and VI for neurofibrillary tangles.
There is the possibility that these three neuropathologic processes may not be independent of one another. Indeed the pathologic changes of AD have been associated with various vascular changes or Lewy bodies [23]. This has led to speculation that the diseases may be related. We do not observe these associations in our study. Table 3 compares the proportion of ACT participants who have come to autopsy with mixed neuropathologic changes against the proportion that would be expected by multiplication of the proportions from Table 2, that is to say, by chance alone. The observed rates of pathological comorbidity closely approximate the rates expected by chance; i.e., they are independently distributed throughout this population. We tested the stability of this finding with sensitivity analyses. The finding of similar proportions to those expected by chance alone was robust to altering the thresholds used to define normal versus abnormal (i.e., Braak stage intermediate vs. high, or CMI > 2 versus any CMI). Similarly, our findings were similar when we limited the study cohort to those who were clinically demented during life or those who were not demented during life. This apparent lack of association is similarly observed in the HAAS cohort (personal communication with Dr. Lon White).
Table 3.
Expected for chance versus observed rates of mixed neuropathologic causes for dementia in ACT.
| Neuropathologic processes | Expected proportion (%) by chance alone | Observed proportion (%) N = 247* |
|---|---|---|
| CMI + AD | 7 | 8 |
| CMI + NLB | 1 | 0.4 |
| AD + NLB | 2 | 2 |
| CMI + AD + NLB | 0.4 | 0.4 |
| Any CMI + AD | 15 | 15 |
10 cases were excluded due to lack of neuropathologic data.
Expected proportions assume independence of all neuropathologic variables.
Abbreviations: CMI = presence of >2 cerebral microinfarcts, NLB = presence of cortical Lewy bodies by immunohistochemistry, AD = Braak stage V and VI for neurofibrillary tangles.
Although these common processes are independently distributed, they might not be clinically independent in that they may have synergistic effects on cognitive outcomes. In Table 4, we compare the proportion of the autopsy cohort who were clinically demented in life as categorized by their postmortem neuropathologic findings. Here individuals lacking a premortem diagnosis of dementia, but who had their most recent CASI evaluation more than 2 years preceding death, were excluded since their cognitive status was uncertain. Individuals were categorized by the presence of cerebral pathology (Braak stage intermediate and high, any CMI, or the presence of cortical Lewy bodies) alone or in combination. Although the proportion of individuals with dementia was relatively low in those with a single pathologic process, this rate doubles in those with combined pathology. Although these neuropathologic processes appear to be independently distributed in our cohort, these data suggest an additive effect of pathology in producing the dementia syndrome during life.
Table 4.
Proportion of ACT who underwent autopsy having dementia in life categorized by single or multiple pathologies.
| Neuropathologic process | % of Autopsies | Demented/Total* | % Demented |
|---|---|---|---|
| No significant cerebral pathology | 25 | 8/62 | 13 |
| Single pathology | 42 | 40/102 | 39 |
| CMI | 16 | 12/39 | 31 |
| NLB | 1 | 1/3 | 33 |
| AD | 24 | 27/60 | 45 |
| Dual Pathology | 27 | 49/67 | 73 |
| NLB + CMI | 2 | 3/5 | 60 |
| AD + NLB | 2 | 3/4 | 75 |
| AD + CMI | 18 | 43/58 | 74 |
| AD + NLB + CMI | 6 | 10/14 | 71 |
12 cases were excluded for missing dementia diagnosis or neuropathologic data.
CMI = presence of any cerebral microinfarcts, NLB = presence of neocortical Lewy bodies by immunohistochemistry, AD = Braak stage III through VI for neurofibrillary tangles.
DETERGENT INSOLUBLE PROTEIN
Accumulation of detergent-insoluble (DI) and abnormally aggregated proteins is a common feature of neurodegenerative diseases thought to be caused by protein misfolding. We investigated the correlation between the concentrations of these proteins in cerebral cortex from rapidly obtained frozen brain from ACT subjects and cognitive performance. Protein was extracted from frozen cerebral cortex using detergents of increasing strength followed by formic acid extraction for DI protein and measured by ELISA. Within the ACT sample, elevated concentrations of 17 proteins and protein variants correlated with each other within 3 clusters: amyloid (A) β, tau, and α-synuclein [24]. Elevated protein concentrations from each of the clusters independently correlated with cognitive performance, however, only elevated concentrations of DI α-synuclein and DI Aβ42 in prefrontal cortex and detergent soluble Aβ peptides in temporal cortex were correlated with cognitive performance when assessed by multivariate analysis [24]. Further, increases in DI Aβ peptides concentration are associated with increasing age of participants throughout cortex in both demented and nondemented individuals while DI tau species are seen primarily in individuals who were demented during life. These biochemical findings offer an objective, quantitative approach to protein misfolding diseases in human brain that are not subject to the same inter-observer variability as neuropathologic data. Further, correlating cognitive performance with these abnormal protein accumulations in a large series of consecutive autopsies from a population-based sample illuminates their potential functional significance. Finally, we identified a possible regional significance of abnormal Aβ and α-synuclein between the frontal and temporal cortex.
OXIDATIVE DAMAGE
Oxidative damage has been associated with neurodegeneration from a variety of causes [25]. Disappointingly, antioxidant supplement usage has not been shown clearly to decrease the risk for neurodegenerative disease. Recognizing that the two most common causes of dementing illness in the ACT autopsy cohort are AD as characterized by high Braak stage for neurofibrillary tangles and cerebral microinfarct burden, we investigated the pattern of oxidative damage to different tissue elements and between cerebrum and cerebellum. We extracted lipid-soluble compounds from frozen gray matter and measured eicosanoid products of oxidative damage to lipid membranes (F2-isoprostanes, measures of free radical damage to arachidonic acid that is evenly distributed throughout cells in brain, and F4-neuroprostanes, measures of free radical damage to docosohexaenoic acid that is highly concentrated in neuronal membranes) by gas chromatography/mass spectrometry using stable isotope dilution assays [26]. Further, since tobacco smoking has been associated with systemic oxidative damage in humans, we stratified participants by current smoking status. As expected we found increased free radical damage to cerebral cortical neuronal membranes in AD cases and in current smokers. Oxidative damage to neurons as well as other tissue elements was present in those with VBISVD as assessed by having at least two cerebral microinfarcts. The most severe levels of oxidative damage were identified in those individuals with both high Braak stage for NFTs and at least two cerebral microinfarcts. We are able to identify the expected pattern of oxidative damage in both AD and current smokers and to refine this characterization by identifying the target of this damage to cortical neurons. The characterization of oxidative damage in the setting of VBI-SVD is novel, although broadly consistent with other types of ischemic brain injury. These findings highlight the significance of increased oxidative damage in the disease processes that underlie AD, VBI-SVD, and smoking.
DIABETES
The association between diabetes mellitus (DM) and increased risk for dementia during aging is well known, however multiple mechanisms have been proposed [27]. Prior clinical studies have been inconsistent in reporting the associations between DM and causes of clinical dementia [28]. Previous autopsy studies have been illuminating, but highlight discrepancies in the relationships between DM and AD or VBI [29–33]. Autopsy cases from ACT were categorized into four groups based on premortem dementia status and diabetes diagnosis based on information obtained from the participant’ extensive medical record [34]. Analysis was performed on neuropathologic data on all consecutive autopsies for which there was information on DM status (n = 196). Biochemical analysis was performed on a subset of these who had rapidly frozen brain tissue (n = 57). In those individuals without dementia, neither neuropathologic or biochemical values differed significantly by DM status. However, in those with dementia, those without DM had greater biochemical measures of Aβ and increased evidence of oxidative damage to cell membranes as identified by F2-isoprostanes in cerebral cortex. Those demented individuals with DM had greater numbers of CMIs and increased biochemical measures of neuroinflammation as identified by increased interleukin-6 concentration in cortex. Of specific interest, when individuals with both diabetes and dementia were divided based on whether they had received any medical diabetic therapy, two additional patterns emerged. CMI number was greatest in the striatum, thalamus, and deep white matter of treated diabetics with dementia compared with all other groups. These different patterns of cerebral injury in patient with dementia depending on DM status could have etiologic or therapeutic implications.
DRUG EXPOSURES
A key strength of ACT is the availability of data from a prescription database that is decades long. Group Health Cooperative (GHC) of Puget Sound pharmacy adopted a computerized medical record for prescription medications in 1977. Interviews with GHC subjects have shown that 97.5% of GHC enrollees purchase all or almost all of their medications from a GHC pharmacy. We are unaware of any other population-based study that combines high quality, long-term drug exposure data with autopsies.
Statins
Treatment with 3-hydroxy-3-methylglutaryl-coenzyme- A reductase inhibitors (“statins”) has been associated in some epidemiologic studies with decreased risk of cognitive impairment [35], all-cause dementia [36,37], and AD [38]. We investigated the association between AD neuropathology and lifetime exposure to statins. Statin users were defined as those who had received at least three prescriptions (≥ 15 pills/prescription) of statins available on the GHC formulary (simvistatin, lovastatin, pravastatin, or atorvastatin) in the first 110 autopsies. Average age at death was 81 years. One-third of this cohort met our criteria for statin users. Compared to non-statin users, statin users were significantly more likely to be male, had higher rates of cardiovascular disease and diabetes mellitus and had somewhat higher rates of lifetime pack years of smoking and lower CASI scores. Statin users showed nonsignificant trends toward having grossly visible cystic infarcts (39 vs. 24%, p = 0.20), CMI (56 vs. 38%, p = 0.13), moderate to severe atherosclerosis (47 vs. 28%, p = 0.09), and lower brain weight (p = 0.20). After controlling for age at death, gender, CASI score at entry, brain weight, and CMI, statin users had a significantly reduced risk for AD neuropathology (Braak stage ≥ IV and CERAD rating ≥ moderate; Odds Ratio (OR) 0.21, 95% CI [0.05, 0.88]). Much of this association appeared to derive from a 2-fold decrease in the risk of elevated Braak stage (OR 0.44, 95% CI [0.20, 0.95]) while the risk for higher CERAD ratings did not differ from the null. A major potential bias in this analysis is confounding by indication. Subjects with vascular risk factors are more likely to be prescribed statins. As vascular disease and diabetes have been associated with increased risk of AD [27], this could result in increased burden of AD pathology in statin users and offset effect of statins. We attempted to adjust for this potential bias by including CMI as covariate in the logistic regression revealing a significant association between statin use and Braak stage. Unlike in ACT, a subsequent study in Catholic clergy found no association [39]. These were some of our earliest analyses and may not have adequately controlled for issues of selection bias including survivor effects. Our subsequent work has evolved to utilize statistical methods to address these issues and is described below in the section on limitations.
Antioxidants
Previously we have shown that the use of antioxidant vitamin supplements (vitamin C, vitamin E, or both) was not associated with a lower risk for developing clinical dementia over 5.5 years follow-up in ACT [40]. Utilizing the same quantitative markers of in vivo oxidative damage as described above, we examined the association between oxidative damage and self-reported use of vitamins C and E, and multivitamins alone or in combination in the whole cohort, those with AD, those with AD and CMIs, and in current smokers. We are unable in any of these groups to detect any pharmacologic benefit from the community use of antioxidant supplements [41]. These findings lead to several intriguing hypotheses. One is that the dose of antioxidant vitamins used in the community was too low to produce a measurable impact on oxidative damage. A second is that the antioxidant vitamins used in the community do not have an impact on oxidative damage in the brain. Further evaluations in other settings will be necessary to disentangle these hypotheses.
GENERAL LIMITATIONS OF THE ACT AUTOPSY COHORT
Autopsy studies are potentially influenced by the mechanism of selection for autopsy, such that those who come to autopsy, may not be representative of the ACT cohort as a whole. We have previously demonstrated that those who died and had autopsy are older and more likely to be demented during life than the cohort as a whole. We have also previously demonstrated that the autopsy cohort does not differ with respect to several potential confounders including gender, education, apoE allele, or marital status [19]. In our more recent analyses, we have utilized inverse probability weighting to improve the generalizability and interpretability of our findings to the ACT cohort as a whole [19,42].
A second limitation inherent in autopsy-based studies of dementia is sample size. For example, although several positive findings were illuminated by our investigation of statin use and risk of dementia, this was one of our earliest studies and the number of autopsies from individuals exposed to statins was relatively low. Power concerns meant that only the most common neuropathologic findings could be addressed. Future analysis of this cohort would benefit from analyzing additional pathologic measurements including a more robust characterization of relationships between dementia and potential risk factors.
SUMMARY
Investigation of autopsies from population-based studies like ACT provides an important perspective on the molecular, structural, and neuropathologic correlates of cognitive impairment and dementia. While these observational studies have obvious limitations compared to experimental studies in animals, their main advantage is that they provide much needed insight into brain aging and dementia as it exists in the elderly people rather than the approximations in animal models. A special strength of ACT is its extensive pharmacy database. Future studies will focus on the associations between the burden of structural and biochemical features of disease and exposure to commonly used drugs.
References
- 1.Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD, Craft S, Leverenz JB, Montine TJ. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62:406–413. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 2.White L, Petrovitch H, Hardman J, Nelson J, Davis D, Ross G, Masaki K, Launer L, Markesbery W. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann N Y Acad Sci. 2002;977:9–23. doi: 10.1111/j.1749-6632.2002.tb04794.x. [DOI] [PubMed] [Google Scholar]
- 3.Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS. Neuropathology of older persons without cognitive impairment from two communitybased studies. Neurology. 2006;66:1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 4.Katzman R. Editorial: The prevalence and malignancy of Alzheimer disease. Amajor killer. Arch Neurol. 1976;33:217–218. doi: 10.1001/archneur.1976.00500040001001. [DOI] [PubMed] [Google Scholar]
- 5.Selnes OA, Vinters HV. Vascular cognitive impairment. Nat Clin Pract Neurol. 2006;2:538–547. doi: 10.1038/ncpneuro0294. [DOI] [PubMed] [Google Scholar]
- 6.Knopman DS, Parisi JE, Boeve BF, Cha RH, Apaydin H, Salviati A, Edland SD, Rocca WA. Vascular dementia in a population-based autopsy study. Arch Neurol. 2003;60:569–575. doi: 10.1001/archneur.60.4.569. [DOI] [PubMed] [Google Scholar]
- 7.Chui HC, Zarow C, Mack WJ, Ellis WG, Zheng L, Jagust WJ, Mungas D, Reed BR, Kramer JH, Decarli CC, Weiner MW, Vinters HV. Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Ann Neurol. 2006;60:677–687. doi: 10.1002/ana.21009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu C, Chute DJ, Farag ES, Garakian J, Cummings JL, Vinters HV. Comorbidity in dementia: an autopsy study. Arch Pathol Lab Med. 2004;128:32–38. doi: 10.5858/2004-128-32-CID. [DOI] [PubMed] [Google Scholar]
- 9.Hachinski V, Iadecola C, Petersen RC, Breteler MM, Nyenhuis DL, Black SE, Powers WJ, DeCarli C, Merino JG, Kalaria RN, Vinters HV, Holtzman DM, Rosenberg GA, Dichgans M, Marler JR, Leblanc GG. National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke. 2006;37:2220–2241. doi: 10.1161/01.STR.0000237236.88823.47. [DOI] [PubMed] [Google Scholar]
- 10.Bailey TL, Rivara CB, Rocher AB, Hof PR. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res. 2004;26:573–578. doi: 10.1179/016164104225016272. [DOI] [PubMed] [Google Scholar]
- 11.Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD, van Belle G, Jolley L, Larson EB. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59:1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 12.Jellinger KA. The pathology of vascular dementia: a critical update. J Alzheimers Dis. 2008;14:107–123. doi: 10.3233/jad-2008-14110. [DOI] [PubMed] [Google Scholar]
- 13.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 14.Mirra SS. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: a commentary. Neurobiol Aging. 1997;18:S91– 94. doi: 10.1016/s0197-4580(97)00058-4. [DOI] [PubMed] [Google Scholar]
- 15.Braak E, Griffing K, Arai K, Bohl J, Bratzke H, Braak H. Neuropathology of Alzheimer’s disease: what is new since A. Alzheimer? Eur Arch Psychiatry Clin Neurosci. 1999;249(Suppl 3):14–22. doi: 10.1007/pl00014168. [DOI] [PubMed] [Google Scholar]
- 16.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg SM, Vonsattel JP. Diagnosis of cerebral amyloid angiopathy. Sensitivity and specificity of cortical biopsy. Stroke. 1997;28:1418–1422. doi: 10.1161/01.str.28.7.1418. [DOI] [PubMed] [Google Scholar]
- 18.Leverenz J, McKeith I. Dementia with Lewy bodies. Med Clin North Am. 2002;86:519–535. doi: 10.1016/s0025-7125(02)00012-3. [DOI] [PubMed] [Google Scholar]
- 19.Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD, Craft S, Leverenz JB, Montine TJ. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62:406–413. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 20.Fernando MS, Ince PG. Vascular pathologies and cognition in a population-based cohort of elderly people. J Neurol Sci. 2004;226:13–17. doi: 10.1016/j.jns.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Jellinger KA, Attems J. Neuropathological evaluation of mixed dementia. J Neurol Sci. 2007;257:80–87. doi: 10.1016/j.jns.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 22.Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Davis DG, Poduska JW, Patel E, Mendiondo MS, Markesbery WR. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol. 2008 doi: 10.1111/j.1750-3639.2008.00244.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katzman R, Galasko D, Saitoh T, Thal LJ, Hansen L. Genetic evidence that the Lewy body variant is indeed a phenotypic variant of Alzheimer’s disease. Brain Cogn. 1995;28:259– 265. doi: 10.1006/brcg.1995.1256. [DOI] [PubMed] [Google Scholar]
- 24.Woltjer RL, Sonnen JA, Sokal I, Rung LG, Yang W, Kjerulf JD, Klingert D, Johnson C, Rhew I, Tsuang D, Crane PK, Larson EB, Montine TJ. Quantitation and mapping of cerebral detergent-insoluble proteins in the elderly. Brain Pathol. 2008;19:365–374. doi: 10.1111/j.1750-3639.2008.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marlatt MW, Lucassen PJ, Perry G, Smith MA, Zhu X. Alzheimer’s disease: cerebrovascular dysfunction, oxidative stress, and advanced clinical therapies. J Alzheimers Dis. 2008;15:199–210. doi: 10.3233/jad-2008-15206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milatovic D, VanRollins M, Li K, Montine KS, Montine TJ. Suppression of murine cerebral F2-isoprostanes and F4-neuroprostanes from excitotoxicity and innate immune response in vivo by alpha- or gamma-tocopherol. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;827:88–93. doi: 10.1016/j.jchromb.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 27.Biessels GJ, Kappelle LJ. Increased risk of Alzheimer’s disease in Type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology? Biochem Soc Trans. 2005;33:1041–1044. doi: 10.1042/BST0331041. [DOI] [PubMed] [Google Scholar]
- 28.MacKnight C, Rockwood K, Awalt E, McDowell I. Diabetes mellitus and the risk of dementia, Alzheimer’s disease and vascular cognitive impairment in the Canadian Study of Health and Aging. Dement Geriatr Cogn Disord. 2002;14:77–83. doi: 10.1159/000064928. [DOI] [PubMed] [Google Scholar]
- 29.Heitner J, Dickson D. Diabetics do not have increased Alzheimer-type pathology compared with age-matched control subjects: a retrospective postmortem immunocytochemical and histofluorescent study. Neurology. 1997;49:1306–1311. doi: 10.1212/wnl.49.5.1306. [DOI] [PubMed] [Google Scholar]
- 30.Beeri MS, Silverman JM, Davis KL, Marin D, Grossman HZ, Schmeidler J, Purohit DP, Perl DP, Davidson M, Mohs RC, Haroutunian V. Type 2 diabetes is negatively associated with Alzheimer’s disease neuropathology. J Gerontol A Biol Sci Med Sci. 2005;60:471–475. doi: 10.1093/gerona/60.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 32.Arvanitakis Z, Schneider JA, Wilson RS, Li Y, Arnold SE, Wang Z, Bennett DA. Diabetes is related to cerebral infarction but not to ADpathology in older persons. Neurology. 2006;67:1960–1965. doi: 10.1212/01.wnl.0000247053.45483.4e. [DOI] [PubMed] [Google Scholar]
- 33.Alafuzoff I, Aho L, Helisalmi S, Mannermaa A, Soininen H. beta-Amyloid deposition in brains of subjects with diabetes. Neuropathol Appl Neurobiol. 2009;35:60–68. doi: 10.1111/j.1365-2990.2008.00948.x. [DOI] [PubMed] [Google Scholar]
- 34.Sonnen JA, Larson EB, Brickell K, Crane PK, Woltjer R, Montine TJ, Craft S. Different patterns of cerebral injury in dementia with or without diabetes. Arch Neurol. 2009;66:315–322. doi: 10.1001/archneurol.2008.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yaffe K, Barrett-Connor E, Lin F, Grady D. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch Neurol. 2002;59:378–384. doi: 10.1001/archneur.59.3.378. [DOI] [PubMed] [Google Scholar]
- 36.Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356:1627–1631. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 37.Rockwood K, Kirkland S, Hogan DB, MacKnight C, Merry H, Verreault R, Wolfson C, McDowell I. Use of lipidlowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol. 2002;59:223–227. doi: 10.1001/archneur.59.2.223. [DOI] [PubMed] [Google Scholar]
- 38.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 39.Arvanitakis Z, Schneider JA, Wilson RS, Bienias JL, Kelly JF, Evans DA, Bennett DA. Statins, incident Alzheimer disease, change in cognitive function, and neuropathology. Neurology. 2008;70:1795–1802. doi: 10.1212/01.wnl.0000288181.00826.63. [DOI] [PubMed] [Google Scholar]
- 40.Gray SL, Anderson ML, Crane PK, Breitner JC, McCormick W, Bowen JD, Teri L, Larson E. Antioxidant vitamin supplement use and risk of dementia or Alzheimer’s disease in older adults. J Am Geriatr Soc. 2008;56:291–295. doi: 10.1111/j.1532-5415.2007.01531.x. [DOI] [PubMed] [Google Scholar]
- 41.Sonnen JA, Larson EB, Gray SL, Wilson A, Kohama SG, Crane PK, Breitner JC, Montine TJ. Free radical damage to cerebral cortex in Alzheimer’s disease, microvascular brain injury, and smoking. Ann Neurol. 2009;65:226–229. doi: 10.1002/ana.21508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haneuse S, Schildcrout J, Crane P, Sonnen J, Breitner J, Larson E. Adjustment for selection bias in observational studies with application to the analysis of autopsy data. Neuroepidemiology. 2009;32:229–239. doi: 10.1159/000197389. [DOI] [PMC free article] [PubMed] [Google Scholar]