

Abstract
Migration of circulating leukocytes from the vasculature into the surrounding tissue is an important component of the inflammatory response. Among the cell surface molecules identified as contributing to leukocyte extravasation is VCAM-1, expressed on activated vascular endothelium, which participates in all stages of leukocyte–endothelial interaction by binding to leukocyte surface expressed integrin VLA-4. However, not all VLA-4-mediated events can be linked to VCAM-1. A novel interaction between VLA-4 and endothelial Lutheran (Lu) blood group antigens and basal cell adhesion molecule (BCAM) proteins has been recently shown, suggesting that Lu/BCAM may have a role in leukocyte recruitments in inflamed tissues. Here, we assessed the participation of Lu/BCAM in the immunopathogenesis of crescentic glomerulonephritis. High expression of Lu/BCAM in glomeruli of mice with rapidly progressive glomerulonephritis suggests a potential role for the local expression of Lu/BCAM in nephritogenic recruitment of leukocytes. Genetic deficiency of Lu/BCAM attenuated glomerular accumulation of T cells and macrophages, crescent formation, and proteinuria, correlating with reduced fibrin and platelet deposition in glomeruli. Furthermore, we found a pro-adhesive interaction between human monocyte α4β1 integrin and Lu/BCAM proteins. Thus, Lu/BCAM may have a critical role in facilitating the accumulation of monocytes and macrophages, thereby exacerbating renal injury.
Keywords: adhesion molecule, crescentic glomerulonephritis, endothelium, macrophage
Necrotizing crescentic rapidly progressive glomerulonephritis (RPGN) is a class of acquired renal disease that remains one of few human autoimmune diseases that represent an acute threat to survival.1 This has stimulated investigation into the immunobiology of the condition in the hope of understanding the pathogenesis not only of anti-glomerular basement membrane disease, but also of other forms of glomerulonephritis (GN) in which the aggravating antigen(s) is as yet unknown. Focal necrotizing crescentic GN is the renal lesion typically associated with the clinical syndrome of RPGN and is a medical emergency that requires side effect–prone immunosuppressive therapies. Untreated RPGN progresses rapidly to renal insufficiency. This process is almost always associated with severe interstitial and periglomerular inflammation. The inflammatory infiltrate gives way to a progressive fibrotic process involving the crescents and the periglomerular and peritubular interstitium, accompanied by tubular atrophy and progressive renal failure. Although the pathogenesis of crescentic RPGN is incompletely understood and likely involves several convergent pathways, there is general agreement that circulating mononuclear phagocytes have a central role. Administration of nephrotoxic serum to rodents or rabbits results in a severe proliferative and necrotizing GN that is characterized by glomerular crescent formation and accumulation of leukocytes.2, 3, 4 These infiltrating cells may then release inflammatory mediators that influence the behavior of glomerular, tubular, and interstitial cells. This interaction between infiltrating and resident cells leads to cellular proliferation, matrix expansion, and may ultimately lead to glomerular sclerosis and interstitial fibrosis. Monocytes and macrophages have a critical role as shown by ablation of macrophages in murine crescentic GN that reduced renal injury and improved renal function.5 Part of the deleterious action of glomerular macrophages could be directly linked to the augmented glomerular procoagulant activity as a result of their expression of surface membrane procoagulant activity and by their potential to indirectly augment glomerular procoagulant activity6 by the production of cytokines capable of enhancing endothelial cell procoagulant activity.7 In addition, infiltrating glomerular macrophages are the major source of IL-18 and tumor necrosis factor (TNF).9 TNF-alpha was shown to promote VCAM-1 and ICAM-1 glomerular expression and the recruitment of PMNs and lymphocytes that were markedly reduced in TNF-deficient mice in experimental RPGN induced by anti-glomerular basement membrane (GBM) antibody.10 Thus, strategies that reduce monocyte infiltrates could be a promising avenue for complementary therapy of RPGN. For example, neutralization of the chemokine monocyte chemoattractant protein-1 (MCP-1) resulted in a marked decrease in both glomerular crescent formation and deposition of type I collagen.11 Another strategy to prevent leukocyte infiltration in the kidney could be to target endothelial molecules involved in cell adhesion. Among the candidates, Lutheran (Lu) blood group antigens and basal cell adhesion molecule (BCAM) antigen in endothelial cells are carried by Lu/BCAM (CD239) glycoproteins of the immunoglobulin superfamily. Lu/BCAM glycoproteins are receptors of laminin alpha-5 chains, a major component of the extracellular matrix.12 Notably, the second ligand of Lu/BCAM is integrin α4β1. The α4β1 integrin or very late antigen (VLA-4) or CD49d/CD29 is expressed mainly on monocytes, lymphocytes, eosinophils, and immature circulating sickle red cells.13 A novel interaction of α4β1 integrin in sickle red cells with endothelial Lu/BCAM proteins has been recently shown to mediate sickle cell adhesion to the endothelium.13 The expression of Lu/BCAM in the mouse kidney has been localized in the glomerulus, distal tubule, collecting duct, and in blood vessels. Within glomeruli, Lu/BCAM is expressed at cell contact with GBM, in particular, at glomerular endothelium.14 Thus, we hypothesized that an interaction between integrin α4β1 and Lu/BCAM could be considered to promote endothelial inflammation through α4β1-mediated adhesion of leukocytes in experimental RPGN. We tested the effect of the genetically determined Lu/BCAM deficiency on crescentic RPGN induced by the infusion of nephrotoxic serum (NTS) in mice.15
RESULTS
Attenuated RPGN and no renal failure in Lu−/− mice
A total of 12 Lu−/− and 14 wild-type (WT) male littermates of mixed 129/Ola-C57BL/6J genetic background had similar renal histology and functional parameters (albuminuria to creatinine ratio, serum creatinine, and blood urea nitrogen) at baseline (Figure 1). Injection of anti-glomerular basement membrane (anti-GBM) nephrotoxic serum (NTS) induced nephrotic syndrome in WT animals. Nephrotic syndrome is caused by hypoproteinemia due to massive urinary loss of large proteins, particularly albumin, leading to hypoalbuminemia and ascites. Lu/BCAM deficiency significantly prevented both the incidence and severity of ascites (not shown) as well as the renal dysfunction reflected by albuminuria (Figure 1a) and blood urea nitrogen and serum creatinine concentrations (Figure 1b and c), which were normal in Lu−/− mice.
Figure 1.

Deletion of Lu gene prevents severe renal failure. Albuminuria (a), blood urea nitrogen (BUN) concentrations (b), and serum creatinine concentration (c) in NTS-challenged Lu+/+ and Lu−/− animals on day 4 and 21 after NTS, and in unchallenged controls (day 0). Values are mean±s.e.m. (n=10–14 per group). *P<0.05 versus Lu−/− at same time point. NTS, nephrotoxic serum.
As an index of early renal microvascular damage, we measured renal blood flow velocity in the renal artery before and on day 4 of NTS-induced RPGN. Whereas renal blood flow velocity remained normal in NTS-challenged Lu−/− animals, Lu+/+ animals displayed a significantly more profound diminution of mean renal blood flow on day 4 than that measured in Lu−/− counterparts (Figure 2). More severe alteration of renal blood flow in Lu+/+ animals was concomitant to equal, and later significantly higher, systolic blood pressure levels compared with those measured in Lu−/− mice (Figure 2). This suggests that Lu/BCAM deficiency limited the early rise in renal vascular resistance.
Figure 2.

Deletion of Lu gene prevents early fall in renal perfusion. (a) Lu/BCAM deficiency significantly prevents the fall in renal blood flow velocity (RBFV) in NTS-challenged nephritic mice on day 4. Only wild-type Lu+/+ mice displayed relative reduction of renal blood flow velocity (P=0.036 for comparison of RBFV in Lu +/+ on day 0 versus Lu+/+ on day 4, paired sample Wilcoxon signed-rank test), whereas Lu−/− mice did not (P=0.147). On day 4, mean RBFV are different between groups (Mann–Whitney's test; P=0.018) despite a similar degree of systolic pressure (b). *P<0.05 vs. Lu+/+ NTS-treated mice on day 4. #P<0.05 vs. day 0 (paired sample). NTS, nephrotoxic serum.
We histologically examined WT mice injected with NTS, and found severe GN by day 21 (Figures 3a–f), whereas Lu−/− littermates had significantly less renal damage (Figure 3g–l). Overall, Lu−/− mice displayed significantly fewer (3.5-fold less) crescentic glomeruli (Figure 3m). Lu−/− mice also exhibited fewer fibrocellular crescents (Figure 3n), no increases in glomerular diameter, and virtually no rupture of Bowman's capsule (Supplementary Figure S1A and B online).
Figure 3.

Deletion of Lu gene prevents fatal glomerular destruction. Photomicrographs illustrating Masson trichrome staining of kidney sections on different days after NTS in Lu+/+ (a–f) and Lu−/− (g–l) mice. In unchallenged animals (controls, CTR) and on day 4 no significant damage was observed in both groups, but on day 21 Lu−/− NTS-treated mice displayed fewer and less severe glomerular and tubulointerstitial lesions than their Lu+/+ counterparts that displayed 3.5 times as many crescents in glomeruli and more tubules with proteinaceous casts (a–c and g–i; bar=80 μm); (d–f and j–l; bar=20 μm). (m and n) Glomerular damage assessed by histopathological evaluation of total crescent formation (m) and fibrocellular crescent formation (n) on day 21 after NTS infusion in NTS-injected Lu+/+ and Lu−/− mice. **P<0.01 and ***P<0.001 versus Lu+/+ NTS-treated mice. (o and p) Ultrastructural analysis of podocytes and the glomerular basement membrane by transmission electron microscopy (TEM) in NTS-treated Lu+/+ glomerulus (o) with representative effacement of the foot processes (thick arrows) and focal thickening of the GBM (thin arrow). Such features were limited in Lu−/− glomeruli (p). Bars=0.05 μm. (q) Representative photomicrographs illustrating the progressive loss of WT-1 expression in Lu+/+ glomeruli (upper panels) and, to a milder extent, in Lu−/− glomeruli upon NTS challenge. (r) Average number of WT-1-positive cells per glomerular section on day 21. *P<0.05 versus Lu−/− on day 21, ##P<0.01 vs. respective group on day 0. Data are means±sem. NTS, nephrotoxic serum.
Reduced ultrastructural alterations in Lu−/− glomeruli
Because glomerular expression of Lu/BCAM is constitutive and differential loss of glomerular permselectivity with heavy albuminuria preceded the development of the crescents already on day 7, we evaluated the morphological features of podocytes in Lu+/+ and Lu−/− mice on day 4 after an injection of NTS. Notably, podocyte ultrastructure was normal and identical in Lu+/+ and Lu−/− mice under control conditions with focal thickening of the external lamina of the GBM in Lu−/− condition as previously described.14 In response to NTS, WT Lu+/+ mice displayed mild-to-severe effacement of the foot processes of podocytes (Figure 3o). These ultrastructural alterations were markedly attenuated in Lu−/− animals (Figure 3p). Consistently Lu/BCAM deficiency was associated with fewer loss of differentiated podocytes than in Lu+/+ mice after NTS challenge, as assessed by WT-1 immunostaining on day 21 (Figure 3q and r).
Role of Lu/BCAM in the immuno-inflammatory response associated with RPGN
Although T cells and macrophages are central players both in our mouse model of NTS-induced GN and in human crescentic RPGN,4, 11, 16 antibody deposition may also have a pathophysiological role during the early stages of the disease, promoting activation of complement.16, 17 Therefore, we assessed the humoral response of Lu−/− and Lu+/+ mice to sheep IgG. Sheep IgG deposition in glomerular basement membranes in the kidneys of both groups after NTS injection displayed similar intensity and pattern (Figure 4a). Glomerular deposition of mouse IgG was also similar in both NTS-injected groups (Figure 4b). Serial dilutions of sera from pre-immunized animals showed similar titers of mouse anti-sheep antibodies (Figure 4c). Thus, we found no evidence that Lu/BCAM deficiency altered the humoral immune response in this model.
Figure 4.

Lu/BCAM deficiency does not affect the humoral immune response to sheep IgG. (a) Immunostaining for sheep IgG in renal cortex from Lu−/− and Lu+/+ mice after immunization and NTS injection on day 4 and 21, and from untreated normal mice (day 0). Bar=20 μm. (b) Immunostaining for mouse IgG in renal cortex from Lu−/− and Lu+/+ mice after immunization and NTS injection on day 4 and 21, and from untreated normal mice (day 0). Bar=20 μm. (c) Titers of mouse IgG to sheep IgG measured in plasma from Lu−/− and Lu+/+ mice immunized with sheep NTS and from non-immunized normal mice. AU: arbitrary unit of optical density (OD) resulting from peroxidase-associated activity. Data are means±s.e.m. (n=6 per group). NTS, nephrotoxic serum.
Lu/BCAM glomerular expression remains localized in endothelial cells during the course of RPGN
We evaluated whether change in Lu/BCAM expression would occur in vivo during RPGN. Sequential assessment of immunoreactive Lu/BCAM by immunofluorescence on day 0, 4, and 21 of experimental RPGN demonstrated prominent endocapillary expression (Figure 5a). This pattern is consistent with Lu/BCAM expression in endothelium and was also observed by immunohistochemistry (Figure 5b). No significant change in Lu/BCAM expression was found on day 4 in glomeruli (Figure 5a) and cortex (Figure 5e and f). Lu/BCAM glomerular expression did become faint on day 21 when extensive glomerular capillary damage was found (Figure 5a).
Figure 5.

Immunostaining for Lu/BCAM in normal and RPGN mouse kidneys, showing strong expression in the glomerular endothelium. (a) Representative photomicrographs after immunofluorescence staining for Lu/BCAM and nidogen in healthy control condition and on day 4 and 21 of NTS-induced RPGN. Bar=20 μm. Note that prominent Lu/BCAM expression is endocapillary. Some fainter mesangial expression is also observed. (b) Immunochemistry confirmed prominent endothelial pattern of Lu/BCAM expression that is consistent in healthy and diseased glomeruli. Representative photomicrograph after immunofluorescence staining (c) and immunohistochemistry (d) for Lu/BCAM in Lu−/− kidney cortex. (e and f) Western blot analysis of Lu/BCAM and tubulin ratio in renal cortex from wild-type Lu+/+ mice treated or not by NTS (n=5 per group) and Lu−/− control cortex (n=1). Data are means±s.e.m. NTS, nephrotoxic serum; RPGN, rapidly progressive glomerulonephritis.
Lu/BCAM expression favors glomerular fibrin and platelet deposition
At sites of glomerular damage, the primary hemostatic reaction involves platelet and fibrin deposition. At these sites, circulating leukocytes marginate and become activated. Furthermore, glomerular fibrin deposition is important in the pathogenesis of renal failure and crescent formation in GN.18 In addition, adhered platelets can support leukocyte localization.19, 20 Remarkably, although Lu+/+ nephritic kidney displayed prominent fibrin immunoreactivity in 80% of glomeruli after 3 weeks (Figure 6), Lu/BCAM-deficient mice exhibited fewer fibrin and platelet deposition in glomeruli than their WT counterparts 21 days after NTS challenge (Figures 6 and 7). In contrast, glomeruli from Lu−/− mice exhibited better preservation of podocin expression than that observed in Lu+/+ mice (Figures 6c and 7c).
Figure 6.

Deletion of Lu gene prevents glomerular deposition of fibrin and loss of podocin expression. (a) Immunohistochemical detection of fibrin formation in kidney cryosections from Lu+/+ and Lu−/− mice without (day 0) or with NTS-induced RPGN (on day 4 and day 21). Bar=50 μm. (b) Comparative evolution of immunoreactive fibrin deposition in glomeruli from NTS-challenged Lu+/+ and Lu−/− animals. Semiquantiative scoring showed significantly increased fibrin deposition score in nephritic cortex from Lu+/+ mice as compared with Lu−/− mice on day 21. *P<0.05; **P<0.01. (c) Representative immunofluorescence photomicrographs after double staining for podocin (red) and fibrin (green) in kidney cryosections from Lu+/+ and Lu−/− mice without (day 0) or with NTS-induced RPGN (on day 4 and day 21). Note that podocin expression was markedly more blunted in Lu+/+ than in Lu−/− on day 21. Bar=80 μm. NTS, nephrotoxic serum; RPGN, rapidly progressive glomerulonephritis.
Figure 7.

Deletion of Lu gene prevents glomerular deposition of platelets and loss of podocin expression. (a) Comparative evolution of thrombocyte deposition in glomeruli from NTS-challenged Lu+/+ and Lu−/− animals. Bar=50 μm. (b) Image analysis showed significantly increased fibrin deposition score in nephritic cortex from Lu+/+ mice as compared with Lu−/− mice on day 21. **P<0.01 vs. Lu−/− condition on day 21. (c) Double immunofluorescence staining for podocin (red) and platelets (green) in kidney cryosections from Lu+/+ and Lu−/− mice without (day 0) or with NTS-induced RPGN (on day 4 and day 21). Note that podocin expression was markedly more blunted in Lu+/+ than in Lu−/− on day 21. Bar=80 μm. NTS, nephrotoxic serum; RPGN, rapidly progressive glomerulonephritis.
Lu/BCAM in the immune-inflammation associated with RPGN. Lu/BCAM deficiency prevents macrophage and T-lymphocyte infiltrates in the kidney
Macrophages and T cells are central players both in our mouse model of NTS-induced GN and in human crescentic RPGN.4, 5, 21, 22 Therefore, to determine whether the differences in immune cell infiltration may explain the augmented glomerular and interstitial damage of the WT group to NTS, the kidneys were stained for the immune cell markers CD3 for T lymphocytes (Figure 8a and b) and F4/80 for resident macrophages (Figure 8c and d). CD3-positive lymphocytes were present around the glomeruli and throughout the renal interstitium in the NTS-infused WT group, but not in the Lu−/− group (P<0.05; Figure 8b). Lu/BCAM-deficient mice displayed fewer F4/80+ macrophages in the kidney cortex and around glomeruli than their WT counterparts on day 21 after NTS injection (P<0.01; Figure 8d). A similar picture was observed when using other macrophage markers such as CD11b, CD68, and Mac-3 (Figure 9 and Figure 10), although at least clearly two distinct populations of myeloid cells were recruited in Lu+/+ nephritic kidneys but not in Lu−/− kidneys. F4/80- and CD11b-positive cells surrounded parietal basal membrane of diseased glomeruli and infiltrated the kidney interstitium. By contrast, staining for CD68 and Mac-3 recognized a population of not only interstitial but also intraglomerular cells (Figures 9 and 10). Mac-3-positive cells were prominently a part of crescentic structures (Figure 9c, detail). Again, Lu/BCAM-deficient animals displayed significantly fewer infiltrates of Mac-3-expressing cells in glomeruli (Figure 10c). Although not fully co-localized, Mac-3 expression and fibrin deposition in glomeruli were highly associated, irrespective of genotype (Supplementary Figure S2). Thus, Lu/BCAM deficiency on renal endothelial cells leads to marked diminution of the accumulation of macrophages and T lymphocytes in the kidney despite severe stimulus for experimental RPGN. In blood, Lu+/+ animals displayed lymphopenia and granulopenia after 2 weeks of experimental RPGN, whereas no such alterations were observed in Lu−/− animals (Table 1).
Figure 8.

Lu/BCAM deficiency prevents T lymphocyte- and F4/80-positive macrophage infiltrates. (a) Immunostaining for CD3+ lymphocytes and (b) quantification by image analysis of CD3-positive infiltrates in renal cortex from NTS-injected mice (on day 0, 4, and 21). (c) Immunostaining for F4/80+ macrophages and (d) quantification by image analysis of F4/80-positive infiltrates in renal cortex from NTS-injected mice. Data are means±s.e.m. (n=12 per group). *P<0.05 and **P<0.01 versus baseline on day 0 and versus Lu −/− on day 21. Bar=80 μm. NTS, nephrotoxic serum.
Figure 9.

Lu/BCAM deficiency prevents CD11b, CD68, and MAC-3-positive macrophage infiltrates. (a) Representative immunostaining for CD11b-positive macrophages, (b) for CD68-positive macrophages and (c) for MAC-3-positive macrophages in renal cortex from NTS-injected Lu+/+ and Lu−/− mice. CD11b-positive cells are primarily periglomerular and in the interstitium with a pattern close to F4/80-positive cells. In contrast, CD68-positive cells are also intraglomerular in Lu+/+ nephritic kidneys only. (a and b): magnification x200, Bar=80 μm. (c) MAC-3-positives cells are also intraglomerular and within cellular crescents in nephritic renal cortex from Lu+/+ animals. Magnification x400, Bar=50 μm. Right panels: higher magnification for detailed view of representative glomeruli from NTS-treated mice. NTS, nephrotoxic serum.
Figure 10.

Quantification of Lu/BCAM effect on macrophage infiltrates in nephrotic kidneys. Quantification by image analysis of CD11b (a), CD68 (b), and MAC-3 (c) positive macrophages infiltrates in renal cortex from NTS-injected Lu+/+ and Lu−/− mice. *P<0.05, **P<0.01 versus Lu−/−. NTS, nephrotoxic serum.
Table 1. Hematological parameters indicate that Lu+/+ animals displayed lymphopenia and granulopenia after 2 weeks of experimental RPGN; such alterations were not observed in Lu−/− animals (P<0.05 vs. Lu+/+ at baseline).
|
Baseline |
Day 14 post NTS |
|||
|---|---|---|---|---|
| Hematological parameters | Lu+/+ (n=11) | Lu−/− (n=13) | Lu+/+ (n=10) | Lu−/− (n=10) |
| Erythrocytes (x106/ml) | 8.85±0.34 | 8.07±0.27 | 5.91±0.77 | 7.22±0.15 |
| White blood cells (x103/ml) | 8.35±0.66 | 7.77±0.34 | 5.04±0.76a | 6.07±0.45 |
| Monocyte (x103/ml) | 0.62±0.05 | 0.76±0.11 | 0.59±0.15 | 0.74±0.12 |
| Granulocytes (x103/ml) | 0.46±0.06 | 0.49±0.05 | 0.25±0.07a | 0.32±0.05 |
| Lymphocytes (x103/ml) | 7.14±0.43 | 7.42±0.52 | 3.83±0.58a | 5.52±0.35 |
| Platelets (x103/ml) | 962.3±65.6 | 955.3±77.0 | 745.80±82.99 | 914.00±49.41 |
Abbreviations: Lu, Lutheran; NTS, nephrotoxic serum.
P<0.05 vs. Lu+/+ at baseline.
Glomerular protection conferred by Lu/BCAM deficiency is confirmed in congenic C57Bl6/J mice
To exclude genetic differences beside the Lu gene as an explanation for the observed differences between Lu−/− and WT Lu+/+ littermates of mixed 129/Ola-C57BL/6J background, we applied the same passive anti-GBM nephritis protocol to induce RPGN in Lu/−/−and Lu+/+ of congenic C57Bl6/J background. Again, Lu/BCAM deficiency blunted part of the increase in albumin urinary excretion and prevented renal failure as assessed by blood urea nitrogen level (Supplementary Figure S3 online). Accordingly, Lu−/− animals displayed fewer crescentic glomeruli and lower fibrin and platelet deposition and glomerular infiltrates with Mac-3-expressing cells (Supplementary Figure S4 online).
Lu/BCAM deficiency prevents increase in circulating classical monocyte Ly6Chi subset and kidney entry of both the classical and nonclassical monocyte subsets
Mouse monocytes comprise at least two phenotypically distinct subsets: Ly6Chi 7/4hi CCR2+ CX3CR1lo and Ly6Clo 7/4lo CCR2− CX3CR1hi monocytes. ‘Inflammatory' Ly6Chi monocytes rapidly enter sites of inflammation, whereas ‘Resident' Ly6Clo monocytes enter lymphoid and non-lymphoid organs under homeostatic conditions and patrol across the vascular endothelium.23 The specific role of each monocyte subset in anti-GBM disease remains unclear. We performed multicolor FACS analysis of classical (Ly6Chi Ly6G− CD11b+ CD115+) and nonclassical (Ly6Clow Ly6G− CD11b+ CD115+) subsets of monocytes in blood and kidneys. In kidneys, Lu−/− displayed fewer monocyte infiltrates of both subsets (−36.5±7.2% and −29.6±5.8% for Ly6Chi Ly6G− CD11b+ CD115+ and Ly6Clow Ly6G− CD11b+ CD115+, respectively, P<0.05 vs. Lu+/+ group), with no significant change in subsets ratio. Lu+/+ mice displayed higher Ly6Chi CD11b+ to Ly6Clow CD11b+ populations ratio in blood on day 8 as compared with Lu−/− animals (P<0.05) (Supplementary Figure S5 online).
Adhesion of peripheral human leukocytes to Lu/BCAM-Fc under flow conditions
To determine whether Lu/BCAM glycoprotein could interact with integrin α4β1, we performed flow adhesion assays of human PBMCs using Lu/BCAM-Fc protein coated on plastic microchannels. PBMCs, primarily monocytes, adhered to coated Lu/BCAM-Fc in the presence of Ca2+ and Mg2+ even in high shear stress conditions (4 dynes/cm2) (Figure 11a). Preincubation of freshly isolated PBMCs with recombinant Lu-Fc protein significantly reduced leukocyte adhesion as compared with cells incubated with control Fc fragment. As a positive control, leukocytes adhered to VCAM-1-Fc (Vascular Cell Adhesion Molecule-1), a known ligand for integrin α4β1. Under flow conditions, human monocytes expressing integrin α4β1 adhered specifically to immobilized Lu/BCAM-Fc proteins. Cell adhesion to Lu/BCAM-Fc was inhibited by blocking monoclonal antibodies anti-β1 integrin (Figure 11b) through a large range of physiological shear stress (analysis of variance, ANOVA: P<0.001 vs. adhesion after incubation with isotype-matched control antibody).
Figure 11.
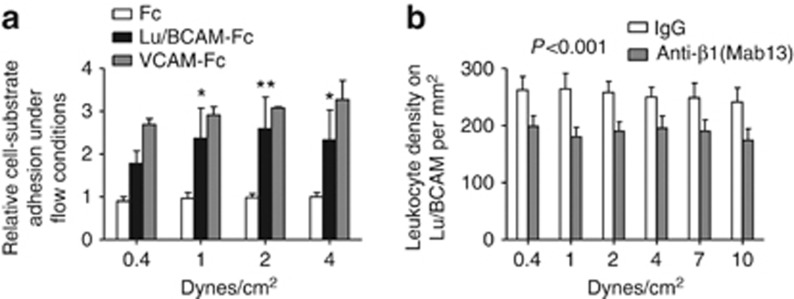
Lu/BCAM interaction with β1 integrin mediates monocyte adhesion in shear stress conditions. (a) Human monocytes adhered to coated Lu/BCAM-Fc in the presence of Ca2+ and Mg2+ even in high shear stress conditions. As a positive control, leukocytes adhere to VCAM-1-Fc protein, a known ligand for integrin α4β1. White bars: adhesion to immobilized Fc proteins; black bars: adhesion to immobilized Lu/BCAM-Fc proteins; gray bars: adhesion to immobilized VCAM-1-Fc proteins. (b) Part of adhesion to Lu/BCAM-Fc but not to Fc alone was blunted when leukocytes were preincubated with an anti-β1 antibody, suggesting specific interaction (analysis of variance, ANOVA: P<0.001 versus adhesion after incubation with isotype-matched control antibody).
DISCUSSION
Necrotizing crescentic RPGN is an inflammatory process directed by cognate immune responses, which results in severe glomerular injury and renal failure.1 In the past, the participation of antibody and complement in human RPGN was emphasized, leading to the view that humoral immune effectors, principally immune complexes or anti-glomerular basement membrane (GBM) antibody, were the predominant pathogenic mediators in this severe form of GN. In fact, B cell–deficient mice that have normal cell-mediated immunity but cannot produce Ig develop crescentic anti-GBM GN to the same extent as their normal littermates, confirming that crescent formation can occur completely independent of humoral immune responses.24 Thus, the involvement of T cells and macrophages has been recognized, suggesting an additional contribution of cell-mediated immunity.1, 11, 21, 25, 26, 27, 28, 29, 30, 31 Although numerous studies have examined the role of soluble factors in mediating recruitment of lymphocytes and macrophages in the kidney, the mechanisms whereby myeloid cells interact with the renal endothelium in the progression of RPGN are still not fully understood. Considering the constitutive expression of Lu/BCAM on the surface of endothelial cells in the kidney, we postulated that they could be involved in the binding of integrins on myeloid cell, contributing to their reinforced adhesiveness to vascular endothelium. Our hypothesis was supported by the presence of α4β1 consensus-binding motifs in the extracellular domain of Lu/BCAM.32 Furthermore, endothelial Lu/BCAM was recently shown to be a receptor for the α4β1 integrin in red blood cells (RBCs), thereby promoting adhesion of sickle RBCs to the endothelium.13 Regulation of α4β1 activation in vivo may promote leukocyte adhesion.33 Likewise, a pathological role for the integrin receptor Lu/BCAM is being realized in SS RBC aggregation to monocytes in sickle cell disease.34 Erythroid Lu/BCAM proteins are implicated in these aggregates through their interaction with α4β1 integrin on peripheral blood mononuclear cells. In the context of RPGN, monocyte adhesion to the α4β1 ligands thrombospondin, VCAM-1, and fibronectin or other factors in the vasculature may markedly impact glomerular damage. Because mice erythrocytes do not express Lu/BCAM, we hypothesize that Lu/BCAM promotes glomerular inflammation and demolition through its endothelial expression. Therefore, to directly examine the actions of Lu/BCAM, a VLA-4/α4β1 integrin receptor on the endothelium in the pathogenesis of RPGN, we used Lu−/− and Lu+/+ littermate mice and then infused these mice with anti-GBM NTS. We observed that genetic deficiency in Lu/BCAM prevents renal injury (albuminuria, glomerular fibrinoid necrosis, and crescent formation) along with reduced kidney infiltrates of T lymphocytes and macrophages. In many kidney diseases, the density of mononuclear cell accumulation correlates with the degree of renal dysfunction and is predictive of disease progression.31, 35, 36 Previous examination of the expression of VCMA-1 in experimental RPGN has been performed during the very early stages of nephritis. Infusion of antibody to the glomerular basement membrane in rats induced glomerular upregulation of ICAM-1, ELAM-1, and VCAM-1 in a TNF-alpha-dependent manner.37 Anti-VLA-4 mAb modestly inhibited PMN migration and had no significant effect on proteinuria. We did not observe any change in neutrophil infiltrates in Lu/BCAM-deficient nephritic animals. However, our study may have missed transient action on neutrophils as studies of similar models of acute GN have revealed that neutrophil recruitment to the glomerulus follows a rapid, yet restricted, time course. Neutrophils accumulate in glomeruli within hours of induction of inflammation, but are not present 4–8 h later.38 More efficient than VLA-4 antagonism, inhibition of Mac-1 was found to alleviate the GBM antibody–associated increase in glomerular injury, demonstrating an essential role for the elevation in neutrophil dwell time in mediating glomerular injury.39, 40 Thus, this latter step is supposed to be independent of Lu/BCAM–VLA-4 interaction. Interestingly, a subsequent work demonstrated that mAbs to VCAM-1 had no significant effect on glomerular damage in a rat model of NTS-induced RPGN, whereas mAbs to VLA-4 significantly attenuated renal injury.41 This study concluded that α4β1 integrin/VLA-4 is an important mediator of glomerular injury, operating after transendothelial leukocyte migration, and that kidney endothelial VCAM-1 has little part in early leukocyte influx into glomeruli. In fact, the marked protection conferred by Lu/BCAM deficiency suggests that this endothelial glycoprotein could be an alternative ligand promoting inflammation of the kidney vasculature.
To examine the type of macrophage recruited within glomeruli, we performed immunohistochemistry for several markers of macrophage, including CD68, Mac-3, CD11b, and F4/80. Macrosialin/CD68 may not be a specific marker for macrophages but rather an antigen indicative of phagocytosis.42 It is expressed on tissue macrophages, Langerhans cells, and at low levels on dendritic cells. Nevertheless, in crescentic RPGN, this marker was previously found to be expressed by virtually all CD45+monocytes/macrophages,43 whereas F4/80-expressing cells represented a minority of CD45+monocytes. Although immunostaining techniques failed to provide evidence of monocyte intraglomerular influx on day 4 when recruitment of the innate immunity is intense, significant alleviation of platelet and fibrin deposition at a later time point in glomeruli from NTS-challenged Lu/BCAM-deficient mice suggests that the Lu/BCAM molecule has a major pathophysiological role in the process of microvascular injury leading to RPGN. Accordingly, early concomitant increase in renal vascular resistance index measured in WT animals was alleviated in Lu/BCAM-deficient mice. Macrophages have been shown to be responsible for glomerular fibrin deposition in anti-GBM antibody–induced GN.6, 7 Likewise, significant fibrin deposition was observed to be associated with glomerular macrophage accumulation in our passive model of RPGN. Glomerular tissue factor gene and protein expression were strikingly increased in human crescentic RPGN, in particular within the crescents and in the mesangial area in the proximity of monocytes.44 Furthermore, fibrin deposition also translates thrombin activity that has been shown to aggravate RPGN per se.45
Interestingly, Lu−/− animals displayed significantly fewer intraglomerular CD68+ and Mac-3+ cells and much fewer periglomerular and interstitial CD11b+ and F4/80+ cells than did the Lu+/+ animals upon NTS challenge. This suggests that Lu/BCAM may contribute to the recruitment of distinct macrophage populations with a potential impact on activation of the coagulation cascade.
By using cell sorting and FACS analysis, we then examined whether Lu/BCAM expression would have influenced egress of CD11b+Ly6Chi monocytes from blood into inflamed kidneys after NTS infusion. However, we found that the recruitment of nonclassical monocytes approached the rate of classical monocyte entry into the nephritic kidney with no obvious effect of Lu genotype. It is uncertain whether both subsets participate in glomerular disease progression from the onset or whether the contribution of nonclassical monocytes increases later. Surprisingly, a significantly higher proportion of Ly6Chi monocytes from unchanged total monocyte count was found in wild-type Lu+/+ nephritic mice as compared with NTS-challenged Lu−/− mice, suggesting that mobilization of this pro-inflammatory monocyte subset was altered in Lu/BCAM-deficient animals. This could be an indirect effect due to the blunted kidney damage with less release of endogenous danger signal by injured kidney cells. Overall, we could not find a sorting role for Lu/BCAM on specific monocyte and macrophage populations in the kidney. Further studies will be required to determine whether Lu/BCAM expression may promote preferential recruitment of specific monocyte subsets at different time points. Deciphering whether Lu/BCAM had an impact on the later F4/80 high population that originates from the resident population derived from the yolk sac and is not recruited from the circulation as reported recently by Geissmann and coll.46 will also require further studies. As F4/80+ macrophages develop and stay in close association with epithelial structures and are associated with capillaries throughout the microcirculation,47 they may be secondarily responsive to Lu/BCAM-dependent capillary and epithelial injury. Similarly, alleviation of endothelial and tissue injury in Lu/BCAM-deficient kidneys may have had an impact on the VLA-4 independent recruitment of T lymphocytes in the interstitium. Overall, Lu/BCAM deficiency was sufficient to prevent severe glomerular damage and renal failure in mice. Furthermore, we describe a novel pathophysiological interaction between leukocyte integrin α4β1 and endothelial Lu/BCAM proteins. Integrin α4β1 is known to have high affinity for VCAM-1, which is only expressed on the surface of activated endothelial cells. As Lu/BCAM proteins are expressed on both resting and activated cells, we hypothesize that they could contribute to abnormal adhesion of myeloid cells to resting endothelium either by tethering myeloid cells or by reinforcing their adhesion along with other described interactions to the glomerular endothelium. These results indicate for the first time that Lu/BCAM exhibits a broad pro-nephritic and pro-inflammatory action in the course of experimental RPGN and may represent a future therapeutic target in inflammatory diseases.
MATERIALS AND METHODS
Experimental animal models
Homozygous Lu−/− and their littermates WT mice Lu+/+ of hybrid 129/Ola-C57BL/6J genetic background were generated by heterozygous mating at the Institut Clinique de la Souris (ICS, Strasbourg, France) as previously described.14 We used male mice between 12 and 16 weeks of age.
After eight generations of backcrossing, by using marker-assisted technologies on a C57Bl6/J background (N8), 10 homozygous Lu−/− and 10 congenic littermate Lu+/+ mice were used in another set of experiments.
Passive anti-GBM nephritis protocol in mice has been popularized and induced as described previously.11, 48 Anti-GBM nephrotoxic serum was injected through the retro-orbital venous sinus at 6μl/g body weight for 3 days continuously as previously described.15 We collected urine and blood on day 4, 7, 10, 14, and 21. On day 14 (C57Bl6/J mice) or on day 21 (129/Ola-C57BL/6J mice), animals were euthanized. Experiments were conducted according to the French veterinary guidelines and those formulated by the European Community for experimental animal use (L358-86/609EEC), and were approved by the Institut National de la Santé et de la Recherche Médicale (INSERM).
Physiological parameter measurements
Cardiac and renal hemodynamic studies were assessed by echocardiography (Vivid 7, GE Medical Systems ultrasound, Velizy-Villacoublay, France) equipped with a 12-MHz linear transducer (12 l). The measurement procedure was described previously.49
Biochemical analysis
Urinary concentrations of albumin were normalized by creatinine concentrations in individual samples. Albumin excretion was expressed as grams per mol of creatinine. Serum creatinine and urea concentrations were expressed as μmol/l and mmol/l, respectively. Analyses were performed by the IBiSA core facility at Institut Claude Bernard, Paris. Assessment of creatinine levels was performed by using a mouse creatinine ELISA kit (Cusabio Biotech, Wuhan, P.R. China).
Blood cell count analysis
The blood samples were collected through retro-orbital venous sinus with 75 μl sodium heparinized hematocrit capillaries (Hirschmann Laborgerate, Eberstadt, Germany) into 1 ml ETDA Minicollect tubes at room temperature. The proportion of white blood cells, RBCs, and platelets was assessed with the help of an electronic hematology counter (type MS9-5; Melet Schloesing Laboratories, Osny, France).
Renal histology and morphological evaluation
The kidneys were fixed in formalin 10% v/v (Labonord, Templemars, France) for at least 24 h, dehydrated, and sections of 3 μm were realized and stained with Masson's trichrome. Glomerular crescents were defined as glomeruli containing two or more layers of cells in Bowman's space and scored as severe glomerular involvement when present. The crescent glomerulus ratio was calculated by counting injury and normal glomeruli (60–70 glomeruli for each kidney sample). The proportion of pathological glomeruli was evaluated by examination of at least 60 glomeruli per section, by an examiner masked to the experimental conditions (PLT). The Histotlab software (Microvision, Evry, France) was used for measurement of average glomerular radius and 50 to 60 glomeruli were measured for each kidney sample (n=12 per group).
Transmission electron microscopy
The preparation of the kidneys for transmission electron microscopy was performed according to procedures as previously described.14
Western blotting
After 6 days of culture, the cell outgrowths from decapsulated glomeruli were carefully separated and placed in 200 μl PhosphoSafeExtraction Reagent (Merck Biochemistry, Fontenay-sous-bois, France) on ice, and lyzed with a sonicator. Total protein concentration was measured with Novagen BCA Protein Assay Kit (Merck Biochemistry). Protein amounting to 20 μg was subjected to electrophoresis in Criterion XT precast gel (4–12% Bis-tris, Bio-Rad). The proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Marnes-la-coquette, France). After washing three times with phosphate-buffered saline Tween-20 (PBST), the membrane was blocked for 1 hour at room temperature with 5% milk in PBST and then incubated with rabbit polyclonal anti-murine (m) Lu antibody 455 (1:15,000, kindly provided by Yves Colin-Aronovicz, Inserm, INTS), rabbit polyclonal to NPHS2 antibody (podocin) (1:2000, Abcam, Paris, France), rat monoclonal anti-tubulin antibody (1:5000, Abcam) in PBST overnight at 4 °C. After washing three times with PBST, the membrane was incubated with ELC horseradish peroxidase–linked whole antibody (1:5000, Amersham Biosciences, Glattbrugg, Switzerland) for 60 min at room temperature. The detection of specific signals was performed using the Immun-Star WesternC Chemiluminescent Kit (Bio-Rad) with an LAS-4000 imaging system (Fuji, Stamford, CT) used to reveal the bands; densitometric analysis was used for quantification.
Immunofluorescence detection
For indirect immunofluorescence staining, kidney sections were embedded in OCT (Sakura, AJ Alphen aan den Rijn, The Netherlands) compound frozen in isopentane cooled in liquid nitrogen. The frozen sections (5 μm) were fixed with cold acetone for 10 min and then washed three times in PBS, blocked in 5% BSA in PBS for 1 h at room temperature, and then incubated with primary antibodies diluted in 1% BSA-PBS overnight at 4 °C: rabbit polyclonal anti-murine (m) Lu antibody 455 (1:1500, kindly provided by Yves Colin-Aronovicz), rabbit polyclonal to NPHS2 (podocin) antibody (1:2000, Abcam). After washing the unbound antibodies three times in PBS, Cy3-conjugated secondary antibodies (Chemicon International, Millipore SAS, Molsheim, France) were used at a 1:1000 dilution for 1 h at room temperature. After three washes, the cells were treated with the second primary antibody FITC-conjugated polyclonal goat antiserum to mouse fibrinogen (Nordic Immunological Laboratories, Susteren, The Netherlands, 1:10) or anti-thrombocyte-FITC Ab (Cedarlane, TEBU-BIO, Le Perray en Yvelines, France, 1:200) diluted in 1% BSA-PBS and incubated for 30 min at room temperature for double immunofluorescence staining. The slides were mounted with Fluoprep mounting medium, and analyzed using the Zeiss 2 fluorescent microscope, AxioCam HRC camera, and the Axiovision 4.3 software (Carl Zeiss S.A.S., Marly le Roi, France).
To detect Lu expression in podocyte, cells were cultured on μ-slides (Ibidi). After a 15 min fixation with 2% paraformaldehyde, cells were washed twice with PBS and incubated for 5 min with 0.2% triton (Sigma–Aldrich Chimie S.a.r.l., Saint-Quentin Fallavier, France) in PBS. The same protocol for frozen kidney sections was used for cultured cells.
To assess podocyte differentiation and leukocyte infiltration in the kidneys, 4 μm paraffin-embedded kidney sections were cut, dehydrated, and treated with target retrieval solution (Dako, Dako France S.A.S., Les Ulis, France) for 20 min at 98 °C in a water bath to unmask specific antigens. After cooling, the sections were incubated with peroxidase blocking reagent (Dako) for 20 min and then in 10% goat serum-1% BSA in PBS for 1 h. The sections were incubated overnight at 4 °C with anti-Wilms tumor-1 (WT-1) (1:500, Santa Cruz Biotechnology, Heidelberg, Germany), anti-CD3 (1:100, Dako), anti-F4/80 (1:500, Serotec, Oxford, UK), MAC-3 (1:50, BD Pharmingen, Le Pont de Claix, France), CD11b (1:100, Abcam), CD68 (1:100, Serotec), or fibrinogen (1:500, Dako) antibody. Histofine reagents (Nichirei Biosciences, Tokyo, Japan) were used and signal was detected in the presence of AEC substrate chromogen (Dako). Sections were counterstained with hematoxylin (Vector Laboratories, Eurobio, Courtaboeuf, France). The number of WT-1-positive cells was counted in 30 glomeruli for each kidney sample. The leukocyte infiltration quantification was accessed using Image J software (NIH). Positively stained Mac-3 cells per 50 glomeruli of each mouse were counted and expressed per glomerular cross-section. Glomerular fibrin deposition was assessed by studying at least 50 glomeruli per mouse with the help of polyclonal rabbit antihuman fibrin(ogen) Ab (1:500, Dako) according to a previously published protocol.50 The degree of glomerular fibrin deposition was scored semiquantitatively (on a scale of 0–3+) as follows: 0, no fibrin deposition; 1, fibrin occupying up to one-third of the glomerular cross-sectional area; 2, fibrin occupying one-third to two-thirds of the glomerulus; and 3, greater than two-thirds of the glomerular cross-section covered by fibrin.
For sheep IgG and mouse IgG staining, 5 μm kidney cryosections were fixed in cold acetone for 10 min, then washed in PBS and blocked with 5% BSA-PBS for 1h at room temperature. Kidney sections were then stained with Alexa Fluor 568 anti-sheep IgG, Alexa Fluor 594 anti-mouse IgG (Invitrogen, Life Technologies SAS, Saint Aubin, France, 1:500). After washing, the slides were mounted with Fluoprep (Biomérieux, Craponne, France) and analyzed using the Zeiss 2 fluorescent microscope, AxioCam HRCcamera and the Axiovision 4.3 software.
For double immunofluorescent staining, frozen kidney sections (5 μm) were fixed with cold acetone for 10 min and then washed three times in PBS, blocked in 5% BSA in PBS for 1 h at room temperature, and then incubated overnight at 4 °C with single or mixed primary antibodies diluted in 1% BSA-PBS: rabbit polyclonal anti-murine (m) Lu antibody 455 (1:1500, kindly provided by Yves Colin-Aronovicz, Inserm, INTS) and rat anti-nidogen antibody (1:500, Chemicon International) or rabbit polyclonal to NPHS2 antibody (podocin) (1:2000, Abcam). After washing three times, unbound antibodies—secondary FITC- and Cy3-conjugated antibodies (Chemicon International)—were added at a 1:500 and 1:1000 dilution for 1 h at room temperature.
Direct immunofluorescence microscopy was performed with goat IgG against mouse fibrinogen, conjugated with FITC (Nordic Immunology, 1:10) and FITC-conjugated anti-mouse thrombocyte rat IgM (Cedarlane, 1:200) diluted in 1% BSA-PBS.
To examine the localization of fibrin and macrophages in glomeruli, double fluorescent labeling was performed on 4 μm kidney paraffin-embedded sections. Macrophage were detected with anti-Mac-3 Ab (1:50, BD Pharmingen) and fibrin was detected with polyclonal rabbit antihuman fibrin(ogen) Ab (1:500, Dako). The secondary antibodies used were donkey anti-rabbit conjugated with FITC and donkey anti-rat conjugated with Cy3 (both 1:500, Jackson ImmunoResearch Europe, Suffolk, UK). The slides were mounted with Fluoprep (Biomérieux), and analyzed using the Zeiss 2 fluorescent microscope, AxioCam HRCcamera and the Axiovision 4.3 software.
Measurement of the murine anti-sheep IgG immune response
Serum mouse anti-sheep IgG levels were measured by enzyme-linked immunosorbent assay15 by using plates (Nunc Maxisorb; Fisher Scientific, Illkirch, France) coated overnight at 4 °C with 100 μg/ml of sheep IgG (Sigma). After blocking with 3% bovine serum albumin, diluted serum samples were incubated for 1 h at 37 °C. For each experiment a range of serum dilutions was tested with a standard curve of a known positive sample. After washing, peroxidase-coupled anti-mouse IgG (Fc-specific) (Rockland Immunochemicals, Gilbertsville, PA) and peroxidase substrate were added.
Flow cytometry analysis
Kidney cortex were weighed, minced, and digested in 450 U/ml Collagenase I, 125 U/ml Collagenase XI, 60 U/ml DNAseI, and 60 U/ml hyaluronidase (Sigma–Aldrich) for 1 h at 37 °C. Cells were also isolated from venous blood. All cell suspensions were layered on Histopaque 1083 (Sigma–Aldrich) for gradient density centrifugation. The mononuclear cell fraction was counted and stained using anti-mouse CD11b-PercPCy5.5, Ly6G-PE, CD115-PE (BD Biosciences, Le Pont de Claix), Ly6C-FITC or Ly6C-APC, or the corresponding isotypes, and analyzed on a LSRII Flow Cytometer (BD Biosciences) with the FacsDiva software (BD Biosciences). CD11bhiLy6G− NK1.1− CD115+ cells were considered to be monocytes, and subset discrimination was made upon Ly6C expression.
Adhesion assay under flow conditions
All adhesion assays were performed with 5 ml freshly drawn EDTA-anticoagulated venous blood from three healthy donors after informed consent was obtained in accordance with the Declaration of Helsinki. The study has been approved by the Scientific Committee of the Institut National de la Transfusion Sanguine. Lu-Fc was also purchased with human VCAM-1-Fc from R&D Systems Europe (Lille, France).
Leukocyte adhesion to microslides coated with fusion protein was measured under flow conditions with a capillary flow chamber. Fc, Lu/BCAM-Fc, VCAM-Fc were coated into capillaries (microslides: Sigma–Aldrich; internal channel dimensions: length 50 mm, width 5 mm, height 0.2 mm) at 4 °C overnight and microslides were mounted as described.51 The leukocytes were separated from the peripheral blood of healthy donors using Histopaque 1119 (Sigma–Aldrich). After hypotonic RBC lysis, 5 × 106 cells/ml were incubated with 2 μg/ml murine monoclonal antihuman IgG (Immunotech, Marseille, France, clone 8a4) for 30 min at 37 °C in order to block Fc receptors. Washed cells were suspended in Hanks buffer (10 mM HEPES, 1 mM MgCl2, 1 mM CaCl2) with 5%BSA and then incubated with1 mM MnCl2 for 15 min at 37 °C. Stimulated cells were injected for 10 min at shear stress of 0.2 dyne/cm2, and 5-minute washouts were carried out with Hanks buffer at 0.4, 1, 2, and 4 dyne/cm2. After each washing step, adherent cells were quantified in five representative areas along the centerline of microslide with the help of the AxioObserver Z1 microscope and AxioVision 4 analysis software (Carl Zeiss). Adhesion was totally abolished when 1 mM EDTA was added, indicating that the interaction is dependent on divalent cations as expected with integrins. For inhibition assays, cells were preincubated for 20 min at room temperature with 10 μg/ml anti-CD29/integrin β1–blocking antibody (clone Mab13) or isotype-matched control antibody (BD Pharmingen). Each adhesion experiment was performed three times.
Statistical analysis
Data are expressed as means+SEM. The two-tailed Mann–Whitney test, the Wilcoxon signed-rank test and the Student t-test were used as appropriate. For experiments with more than two subgroups, the nonparametric Kruskal–Wallis analysis of variance (ANOVA) followed by Dunn's multiple comparison test were used. Values of P<0.05 were considered significant. All analyses were performed using Prism version 5.04 for Windows, (GraphPad Software, La Jolla, CA).
Acknowledgments
This work was supported by Institut National de Santé et de la Recherche Médicale (INSERM), by Institut National de la Transfusion Sanguine (INTS), research grant ANR-MRAR SCADHESION 2007, (Caroline Le Van Kim, Yves Colin, and Pierre-Louis Tharaux) from l'Agence Nationale de la Recherche (ANR) and Région Ile-de-France (SESAME program). JH was supported by a fellowship from the Ministère de l'Enseignement Supérieur et de la Recherche. We are grateful to Elizabeth Huc and the ERI970 team for excellent assistance in animal care and handling. We acknowledge the outstanding administrative support from Veronique Oberweiss, Annette De Rueda, Martine Autran, and Philippe Coudol.
All the authors declared no competing interests.
Footnotes
SUPPLEMENTARY MATERIAL
Figure S1. (A). Average glomerular diameter before (day 0) and after (day 4 and 21) the onset of RPGN.
Figure S2. Glomerular expressions of macrophage and fibrin in mice with anti-GBM GN.
Figure S3. Lu/BCAM deficiency prevents functional features of RPGN in C57Bl6/J mice.
Figure S4. Lu/BCAM deficiency attenuates glomerular histopathological damage in C57Bl6/J mice.
Figure S5. (A) Representative FACS dot plots of CD45+ Ly6G−CD11b+ LyYC+ cells in blood from Lu+/+ and Lu−/− mice on day 8 after NTS administration.
Supplementary material is linked to the online version of the paper at http://www.nature.com/ki
Supplementary Material
References
- Lionaki S, Jennette JC, Falk RJ. Anti-neutrophil cytoplasmic (ANCA) and anti-glomerular basement membrane (GBM) autoantibodies in necrotizing and crescentic glomerulonephritis. Semin Immunopathol. 2007;29:459–474. doi: 10.1007/s00281-007-0093-0. [DOI] [PubMed] [Google Scholar]
- Morita T, Suzuki Y, Churg J. Structure and development of the glomerular crescent. Am J Pathol. 1973;72:349–368. [PMC free article] [PubMed] [Google Scholar]
- Holdsworth SR, Allen DE, Thomson NM, et al. Histochemistry of glomerular cells in animal models of crescentic glomerulonephritis. Pathology. 1980;12:339–346. doi: 10.3109/00313028009077095. [DOI] [PubMed] [Google Scholar]
- Hancock WW, Atkins RC. Cellular composition of crescents in human rapidly progressive glomerulonephritis identified using monoclonal antibodies. Am J Nephrol. 1984;4:177–181. doi: 10.1159/000166800. [DOI] [PubMed] [Google Scholar]
- Duffield JS, Tipping PG, Kipari T, et al. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol. 2005;167:1207–1219. doi: 10.1016/S0002-9440(10)61209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth SR, Tipping PG. Macrophage-induced glomerular fibrin deposition in experimental glomerulonephritis in the rabbit. J Clin Invest. 1985;76:1367–1374. doi: 10.1172/JCI112112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping PG, Lowe MG, Holdsworth SR. Glomerular macrophages express augmented procoagulant activity in experimental fibrin-related glomerulonephritis in rabbits. J Clin Invest. 1988;82:1253–1259. doi: 10.1172/JCI113723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping PG, Lowe MG, Holdsworth SR. Glomerular interleukin 1 production is dependent on macrophage infiltration in anti-GBM glomerulonephritis. Kidney Int. 1991;39:103–110. doi: 10.1038/ki.1991.13. [DOI] [PubMed] [Google Scholar]
- Tipping PG, Leong TW, Holdsworth SR. Tumor necrosis factor production by glomerular macrophages in anti-glomerular basement membrane glomerulonephritis in rabbits. Lab Invest. 1991;65:272–279. [PubMed] [Google Scholar]
- Le Hir M, Haas C, Marino M, et al. Prevention of crescentic glomerulonephritis induced by anti-glomerular membrane antibody in tumor necrosis factor-deficient mice. Lab Invest. 1998;78:1625–1631. [PubMed] [Google Scholar]
- Lloyd CM, Minto AW, Dorf ME, et al. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J Exp Med. 1997;185:1371–1380. doi: 10.1084/jem.185.7.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Nemer W, Gane P, Colin Y, et al. The Lutheran blood group glycoproteins, the erythroid receptors for laminin, are adhesion molecules. J Biol Chem. 1998;273:16686–16693. doi: 10.1074/jbc.273.27.16686. [DOI] [PubMed] [Google Scholar]
- El Nemer W, Wautier MP, Rahuel C, et al. Endothelial Lu/BCAM glycoproteins are novel ligands for red blood cell alpha4beta1 integrin: role in adhesion of sickle red blood cells to endothelial cells. Blood. 2007;109:3544–3551. doi: 10.1182/blood-2006-07-035139. [DOI] [PubMed] [Google Scholar]
- Rahuel C, Filipe A, Ritie L, et al. Genetic inactivation of the laminin alpha5 chain receptor Lu/BCAM leads to kidney and intestinal abnormalities in the mouse. Am J Physiol Renal Physiol. 2008;294:F393–F406. doi: 10.1152/ajprenal.00315.2007. [DOI] [PubMed] [Google Scholar]
- Bollee G, Flamant M, Schordan S, et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat Med. 2011;17:1242–1250. doi: 10.1038/nm.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping PG, Holdsworth SR. T cells in glomerulonephritis. Springer Semin Immunopathol. 2003;24:377–393. doi: 10.1007/s00281-003-0121-7. [DOI] [PubMed] [Google Scholar]
- Lin F, Salant DJ, Meyerson H, et al. Respective roles of decay-accelerating factor and CD59 in circumventing glomerular injury in acute nephrotoxic serum nephritis. J Immunol. 2004;172:2636–2642. doi: 10.4049/jimmunol.172.4.2636. [DOI] [PubMed] [Google Scholar]
- Wu X, Helfrich MH, Horton MA, et al. Fibrinogen mediates platelet-polymorphonuclear leukocyte cooperation during immune-complex glomerulonephritis in rats. J Clin Invest. 1994;94:928–936. doi: 10.1172/JCI117459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuligowski MP, Kitching AR, Hickey MJ. Leukocyte recruitment to the inflamed glomerulus: a critical role for platelet-derived P-selectin in the absence of rolling. J Immunol. 2006;176:6991–6999. doi: 10.4049/jimmunol.176.11.6991. [DOI] [PubMed] [Google Scholar]
- Evangelista V, Pamuklar Z, Piccoli A, et al. Src family kinases mediate neutrophil adhesion to adherent platelets. Blood. 2006;109:2461–2469. doi: 10.1182/blood-2006-06-029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XR, Kitching AR, Tipping PG, et al. Interleukin-10 inhibits macrophage-induced glomerular injury. J Am Soc Nephrol. 2000;11:262–269. doi: 10.1681/ASN.V112262. [DOI] [PubMed] [Google Scholar]
- Huang XR, Tipping PG, Apostolopoulos J, et al. Mechanisms of T cell-induced glomerular injury in anti-glomerular basement membrane (GBM) glomerulonephritis in rats. Clin Exp Immunol. 1997;109:134–142. doi: 10.1046/j.1365-2249.1997.4091307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Li S, Holdsworth SR, Tipping PG. Antibody independent crescentic glomerulonephritis in mu chain deficient mice. Kidney Int. 1997;51:672–678. doi: 10.1038/ki.1997.97. [DOI] [PubMed] [Google Scholar]
- Bhan AK, Schneeberger EE, Collins AB, et al. Evidence for a pathogenic role of a cell-mediated immune mechanism in experimental glomerulonephritis. J Exp Med. 1978;148:246–260. doi: 10.1084/jem.148.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham PS, Csizmadia V, Soler D, et al. Lack of chemokine receptor CCR1 enhances Th1 responses and glomerular injury during nephrotoxic nephritis. J Clin Invest. 1999;104:1549–1557. doi: 10.1172/JCI7707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitching AR, Kuligowski MP, Hickey MJ. In vivo imaging of leukocyte recruitment to glomeruli in mice using intravital microscopy. Methods Mol Biol. 2009;466:109–117. doi: 10.1007/978-1-59745-352-3_8. [DOI] [PubMed] [Google Scholar]
- Tipping PG, Kitching AR. Glomerulonephritis, Th1 and Th2: what's new. Clin Exp Immunol. 2005;142:207–215. doi: 10.1111/j.1365-2249.2005.02842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitching AR, Turner AL, Wilson GR, et al. IL-12p40 and IL-18 in crescentic glomerulonephritis: IL-12p40 is the key Th1-defining cytokine chain, whereas IL-18 promotes local inflammation and leukocyte recruitment. J Am Soc Nephrol. 2005;16:2023–2033. doi: 10.1681/ASN.2004121075. [DOI] [PubMed] [Google Scholar]
- Kitching AR, Tipping PG, Kurimoto M, et al. IL-18 has IL-12-independent effects in delayed-type hypersensitivity: studies in cell-mediated crescentic glomerulonephritis. J Immunol. 2000;165:4649–4657. doi: 10.4049/jimmunol.165.8.4649. [DOI] [PubMed] [Google Scholar]
- Nikolic-Paterson DJ, Atkins RC. The role of macrophages in glomerulonephritis. Nephrol Dial Transplant. 2001;16 (Suppl 5:3–7. doi: 10.1093/ndt/16.suppl_5.3. [DOI] [PubMed] [Google Scholar]
- Holness CL, Simmons DL. Structural motifs for recognition and adhesion in members of the immunoglobulin superfamily. J Cell Sci. 1994;107 (Pt 8:2065–2070. doi: 10.1242/jcs.107.8.2065. [DOI] [PubMed] [Google Scholar]
- Rose DM, Han J, Ginsberg MH. Alpha4 integrins and the immune response. Immunol Rev. 2002;186:118–124. doi: 10.1034/j.1600-065x.2002.18611.x. [DOI] [PubMed] [Google Scholar]
- Chaar V, Picot J, Renaud O, et al. Aggregation of mononuclear and red blood cells through an {alpha}4{beta}1-Lu/basal cell adhesion molecule interaction in sickle cell disease. Haematologica. 2010;95:1841–1848. doi: 10.3324/haematol.2010.026294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikezumi Y, Hurst LA, Masaki T, et al. Adoptive transfer studies demonstrate that macrophages can induce proteinuria and mesangial cell proliferation. Kidney Int. 2003;63:83–95. doi: 10.1046/j.1523-1755.2003.00717.x. [DOI] [PubMed] [Google Scholar]
- Atkins RC. Macrophages in renal injury. Am J Kidney Dis. 1998;31:xlv–xlvii. doi: 10.1016/s0272-6386(14)70003-4. [DOI] [PubMed] [Google Scholar]
- Mulligan MS, Johnson KJ, Todd RF, 3rd, et al. Requirements for leukocyte adhesion molecules in nephrotoxic nephritis. J Clin Invest. 1993;91:577–587. doi: 10.1172/JCI116237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping PG, Huang XR, Berndt MC, et al. A role for P selectin in complement-independent neutrophil-mediated glomerular injury. Kidney Int. 1994;46:79–88. doi: 10.1038/ki.1994.246. [DOI] [PubMed] [Google Scholar]
- Tang T, Rosenkranz A, Assmann KJ, et al. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcgamma receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J Exp Med. 1997;186:1853–1863. doi: 10.1084/jem.186.11.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi S, Li A, Westhorpe CL, et al. Multiphoton imaging reveals a new leukocyte recruitment paradigm in the glomerulus. Nat Med. 2013;19:107–112. doi: 10.1038/nm.3024. [DOI] [PubMed] [Google Scholar]
- Allen AR, McHale J, Smith J, et al. Endothelial expression of VCAM-1 in experimental crescentic nephritis and effect of antibodies to very late antigen-4 or VCAM-1 on glomerular injury. J Immunol. 1999;162:5519–5527. [PubMed] [Google Scholar]
- Kunisch E, Fuhrmann R, Roth A, et al. Macrophage specificity of three anti-CD68 monoclonal antibodies (KP1, EBM11, and PGM1) widely used for immunohistochemistry and flow cytometry. Ann Rheum Dis. 2004;63:774–784. doi: 10.1136/ard.2003.013029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki T, Chow F, Nikolic-Paterson DJ, et al. Heterogeneity of antigen expression explains controversy over glomerular macrophage accumulation in mouse glomerulonephritis. Nephrol Dial Transplant. 2003;18:178–181. doi: 10.1093/ndt/18.1.178. [DOI] [PubMed] [Google Scholar]
- Grandaliano G, Gesualdo L, Ranieri E, et al. Tissue factor, plasminogen activator inhibitor-1, and thrombin receptor expression in human crescentic glomerulonephritis. Am J Kidney Dis. 2000;35:726–738. doi: 10.1016/s0272-6386(00)70022-9. [DOI] [PubMed] [Google Scholar]
- Cunningham MA, Rondeau E, Chen X, et al. Protease-activated receptor 1 mediates thrombin-dependent, cell-mediated renal inflammation in crescentic glomerulonephritis. J Exp Med. 2000;191:455–462. doi: 10.1084/jem.191.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- Hume DA, Perry VH, Gordon S. The mononuclear phagocyte system of the mouse defined by immunohistochemical localisation of antigen F4/80: macrophages associated with epithelia. Anat Rec. 1984;210:503–512. doi: 10.1002/ar.1092100311. [DOI] [PubMed] [Google Scholar]
- Salant DJ, Cybulsky AV. Experimental glomerulonephritis. Methods Enzymol. 1988;162:421–461. doi: 10.1016/0076-6879(88)62096-9. [DOI] [PubMed] [Google Scholar]
- Sabaa N, de Franceschi L, Bonnin P, et al. Endothelin receptor antagonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle-cell disease. J Clin Invest. 2008;118:1924–1933. doi: 10.1172/JCI33308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping PG, Erlich JH, Apostolopoulos J, et al. Glomerular tissue factor expression in crescentic glomerulonephritis. Correlations between antigen, activity, and mRNA. Am J Pathol. 1995;147:1736–1748. [PMC free article] [PubMed] [Google Scholar]
- Wautier MP, El Nemer W, Gane P, et al. Increased adhesion to endothelial cells of erythrocytes from patients with polycythemia vera is mediated by laminin alpha5 chain and Lu/BCAM. Blood. 2007;110:894–901. doi: 10.1182/blood-2006-10-048298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.