

Abstract
Reactive oxygen species attack both base and sugar moieties in DNA with a preference among the bases for reaction at guanine. In the present study, 2′-deoxyguanosine (dG) was oxidized by a copper-mediated Fenton reaction with the reductants ascorbate or N-acetyl-cysteine, yielding oxidation on both the base and the sugar. The primary oxidized lesions observed in these studies include the 2′-deoxyribonucleosides of 8-oxo-7,8-dihydroguanosine (dOG), spiroiminodihydantoin (dSp), guanidinohydantoin (dGh), oxazolone (dZ), and 5-carboxamido-5-formamido-2-iminohydantion (d2Ih), as well as the free base guanine. d2Ih was the major product observed in the nucleoside, single- and double-stranded oligodeoxynucleotide contexts and is proposed to arise from oxidation at C5 of guanine. Product distribution studies provide insight into the role of the reductant in partitioning of dG base oxidation along the C5 and C8 pathways.
Introduction
Cellular damage resulting from oxidative stress has been implicated in many disorders including aging, cancer, and neurological diseases.1,2 Formation of reactive oxygen species (ROS) in vivo results from incomplete reduction of O2 to H2O in mitochondria as well as from inflammation or environmental factors.3,4 These diffusible oxidants can impose damage to cellular macromolecules including DNA.2 H2O2 is a relatively stable ROS produced within mitochondria that is capable of diffusing throughout the cell promoting free radical oxidation reactions via the Fenton reaction.5,6 The Fenton reaction occurs when H2O2 is activated by Fe(II) yielding hydroxyl radical (Equation I), or by Cu(I) yielding a either a free or metal bound hydroxyl radical (Equations II, III). In the presence of DNA, a DNA bound Cu(I)-OOH complex has been proposed.5,7 All of these species are strong one-electron oxidants. Isolated cellular DNA often contains copper,8 and studies suggest that Cu-mediated oxidation of DNA is associated with aging and cancer disease states.9
| (Equation I) |
| (Equation II) |
| (Equation III) |
DNA oxidation by Cu-mediated Fenton chemistry has been shown to induce oxidation of 2-deoxyribose leading to base release and strand scission when Cu is not bound to DNA;6,10 when Cu is bound to DNA, base-centered oxidations have been proposed.6 Binding of both Cu(I) and Cu(II) ions with DNA occurs primarily at N7 of 2′-deoxyguanosine (dG, mass = M)11 in which the coordination of Cu(I) (Ka ~ 109 M−1)12 is ~105 times stronger than Cu(II) (Ka ~104 M−1).13 Furthermore, copper-mediated Fenton oxidation of DNA with bound Cu(I) yields base oxidation at dG sites,6 and nucleosome oxidation studies using Cu(II)/H2O2 indicated formation of more base lesions than single-strand breaks.8 Further studies have suggested that Cu-mediated DNA damage is sequence selective with dG repeats being more prone to base modification than other sequences.14 It has been suggested that hydroxyl radical is not the primary reactive oxidant responsible for base damage.7,15
Guanine is the most electron-rich site in DNA; therefore, oxidation occurs preferentially at dG to yield electrophilic intermediates and a wide spectrum of end products that are typically identified by mass spectrometry (Figure 1).5 These products can be categorized as resulting from initial chemistry at either the C5 or C8 carbons. Alternatively, base release and/or strand scission occur from oxidation of the 2-deoxyribose moiety.5 The oxidation of dG (mass = M) at C8 by HO• (produced by Fenton chemistry or radiolytically),3,16,17 CO3•−,18 chromate,19 and the photoexcited state of riboflavin20 yields 8-oxo-7,8-dihydro-2′-deoxyguanosine (dOG, M+16) as a key product. When dG is oxidized within the DNA context by HO• (produced by Fenton chemistry or radiolytically), the products observed were dOG and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (Fapy-dG, M+18) with dOG observed under oxidizing conditions and Fapy-dG under reducing conditions.21,22 The in vivo concentrations of dOG can be monitored to provide levels of cellular oxidative stress. Basal concentrations of dOG are ~1 in 106 bases,23 but increase under certain disease states.24 dOG lesions in DNA are moderately mutagenic causing G→T transversion mutations in vivo25 when dOG is not removed from the genome by repair processes.26
Fig 1.

Structures of dG oxidation products and their relative mass differences (dR = 2′-deoxyribose).
The oxidation of dOG is much more facile than that of dG as reflected in its lower redox potential (Eo = 0.7 V and 1.3 V vs. NHE, pH 7.0, respectively);27,28 thus, a further two-electron oxidation of dOG occurs readily in the presence of such oxidants as Ir(IV),29,30 chromate,31 CO3•−,18 •NO2,32 or ONOO−33 to yield the hydantoin products, spiroiminodihydantoin (dSp, M+32) and guanidinohydantoin (dGh, M+6). Furthermore, dSp and dGh can be formed directly from dG by four-electron oxidation mediated by singlet oxygen; a mechanistic proposal for this pathway has been reported.34 Yields of dSp and dGh are affected by the reaction pH and the structural context with dSp formation favored at high pH (>6.0) and in nucleoside contexts and dGh favored at low pH (<6.0) and in oligonucleotide contexts.35–37 dSp has been observed in chromate-stressed E. coli,38 and it has been shown that dSp and dGh are highly mutagenic causing G→T and G→C transversion mutations.39
When dG was oxidized by either HO• (formed radiolytically),21 SO4•−, or photoexcited riboflavin, many additional products were observed. Those characterized as resulting from chemistry at C5 of dG were imidazolone (dIz, M-39) and its hydrolysis product oxazolone (dZ, M-21). dZ has been observed in vivo,40 and predominantly causes G→T transversion mutations.41 In the case of dG oxidation by NiCR/KHSO5,42 Mn-TMPyP/KHSO5,43,44 or the epoxidizing agent dimethyldioxirane,45 oxidation at C5 is also observed yielding the product 5-carboxamido-5-formamido-2-iminohydantion (d2Ih, M+34). In work by Karlin and Rokita, a compound with the same mass as d2Ih was observed upon oxidation of an oligodeoxynucleotide (ODN) with a dinuclear copper(II) complex.46 Other coordination compounds of copper formed with phenanthroline and related ligands have been shown to mediate primarily ribose oxidation.10 The large distribution of base and ribose-derived products observed from dG oxidation highlight the complexity in understanding dG oxidation pathways with free radical-generating oxidants.
Fenton oxidation (Fe(II)/H2O2/reductant) of dG in the nucleoside context yields dOG and 5′,8-cyclo-2′-deoxyguanosine (5′,8-cyclo-dG, M-2) in nearly equal amounts.16,47 In the absence of the reductant, 5′,8-cyclo-dG was observed in a 16-fold greater yield.16 In addition, significant amounts (~50%) of sugar chemistry occurred leading to base release that was characterized by the presence of guanine (Gua, M-116); the remaining mass balance was comprised of many sugar oxidation products in low yield.16 These studies suggest that the reductant plays a critical role in determining product distributions from Fenton-mediated oxidation of dG. Radiation-induced oxidation of dG in DNA was shown to yield both 5′,8-cyclo-dG and dOG.48 Copper-mediated Fenton oxidation of dG in DNA was reported to yield both sugar- and base-oxidation products in which the sugar-oxidation products were determined by monitoring strand breaks, and the base-oxidation products dOG and Fapy-dG were quantified using GC-MS.22,49,50 These studies consistently found dOG to be the major oxidation product, with yields at least10-fold greater than that of Fapy-dG. When ODNs or DNA were exposed to Cu(II)/H2O2/ascorbate, intrastrand cross-links between dG and thymidine (T), or dG and 5-methyl-2′-deoxycytidine were observed, pointing to the fact that the DNA context yields new products that were not observed within nucleoside models.51,52 In this study, we have analyzed dG oxidation products within the nucleoside, single- and double-stranded ODN contexts resulting from exposure to Cu(II) and H2O2 in the presence of either ascorbate (Asc) or N-acetyl-cysteine (NAC) as reductant. In these studies a newly identify compound, d2Ih, was observed as the major product of dG oxidation in all contexts.
Results
Product Characterization
The nucleoside oxidation studies were conducted with dG (2.0 mM) in buffer (75 mM NaPi, pH 7.0), followed by stepwise addition of Cu(II) acetate (1.0 mM, final concentration), Asc or NAC (2.0 mM), and then H2O2 (10.0 mM). The reaction was held at 22 °C for 60 min under aerobic conditions, and then terminated by addition of Na2EDTA (10.0 mM). The reaction was analyzed initially by reversed-phase HPLC (RP-HPLC) with a MS detector that allowed the identification of the free base products Gua, 8-oxo-7,8-dihydroguanine (OG) and the nucleoside products dOG and 5′,8-cyclo-dG. The void volume from the previous HPLC run was collected and then reinjected on a Hypercarb HPLC column. This run was monitored by MS to provide initial identification for the free base Gh, and the nucleosides d2Ih, dGh, dSp, and dZ. Under these conditions, dIz elutes past the void volume on the RP-HPLC run and was collected with the void volume. During the process to prepare a sample for the Hypercarb column, dIz hydrolyzed to dZ, therefore only dZ was observed and quantified. Chromatography utilizing the Hypercarb column allowed the separation of diastereomers, and two peaks were observed for both d2Ih and dSp, although the diastereomers of dGh were not separable (see electronic supplementary information). Further support for the products d2Ih, dSp and dZ was obtained through ESI+-MS/MS fragmentation of the free bases derived from in-source fragmentation of the glycosidic bond followed by comparison of the daughter fragments to previously published data (see ESI).30,40,45 Because dGh could not be fragmented, an HRMS was obtained for further characterization. Reactions were also conducted using cysteine as the reductant, and in order to avoid amine adducts to electrophilic dG oxidation intermediates, N-acetyl-cysteine (NAC) was used.53,54
To evaluate the context effect on the oxidation products, the complementary single-stranded ODNs (ODN-1 and ODN-2) and the double-stranded ODN (ODN-12) were allowed to react with the same oxidant systems described above. The ODN and nucleoside oxidation studies were conducted under slightly different conditions; the ODN oxidation utilized 100 μM ODN, 20.0 mM NaPi, pH 7.0, 100 mM NaCl at 37 °C for 8 h reaction with the oxidant system comprising Cu(II) acetate (10.0 μM) reductant (1.0 mM) and H2O2 (1.0 mM). To study the product distributions from dG oxidation in the ODN contexts, the ODNs had to be digested to nucleoside mixtures that could be analyzed by the method outlined for the previous studies. The ODN samples were exhaustively digested with DNaseI, nuclease P1, and snake venom phosphodiesterase, followed by phosphate removal with calf intestinal phosphatase to yield an analyzable nucleoside sample. The digestion method used was shown previously to liberate both diastereomers of dSp and dGh in high yields from ODNs containing synthetic standards of the hydantoins.55 The ODN oxidation studies yielded the same products observed in the nucleoside studies, but in different ratios that will be discussed below. Oxidation of duplex ODN-12 yielded a new product, dGhred. It is assumed that these conditions would have the same efficiency for digesting d2Ih and dGhred from ODN samples allowing a direct comparison of the base oxidation product yields. The sequences of the ODNs used are as follows:
ODN-1: 5′-TCA TGG GTC GTC GGT ATA
ODN-2: 3′-AGT ACC CAG CAG CCA TAT
Product Quantification
Comparison of UV-vis signatures for 5′,8-cyclo-dG, dOG, dSp, dGh, d2Ih, dZ, Gh, Gua, and OG to literature data allowed fast identification and quantification of each compound utilizing an HPLC with a photodiode array (PDA) detector (see ESI).16,30,36,40,45 Quantification of the reactions to determine the yields and product distributions was achieved through integration of reactant and product peak areas while monitoring absorbance at 240 nm, followed by normalization of peak areas using each compound’s unique extinction coefficent ε240. Because no literature data exists for dGhred, the ε240 was assumed to be the same as that of dGh. The individual product yields will be discussed in the following sections.
In the ODN studies, only products derived from oxidation of dG’s base and sugar in which H2O or O2•− trapped the reactive intermediates leading to product formation were quantified, and reactions with another base (i.e., base cross-links) were not analyzed. Because HPLC with a PDA detector was used to quantify products from the ODN-oxidation studies, the levels of 5′,8-cyclo-dG could not be accurately quantified due to its overlap with the large excesses of the other bases in the HPLC chromatogram; therefore, we conclude that this product may exist, but was not quantified. Thus, only relative distributions between the contexts were compared. In the ODN contexts, the mass balance was not quantitative, which appears to result from additional dG oxidation products forming that were not accounted for (e.g., cross-links and 5′,8-cyclo-dG).56,57 With this experimental limitation in mind, the following data should only be interpreted with respect to context effects on dG base oxidation products.
C5 Oxidation Products
In the nucleoside reactions studied with Cu(II)/H2O2/Asc or NAC, d2Ih was the major oxidation product observed when reductant concentrations were near physiological (2 mM), as shown in Table 1. Oxazolone (dZ), also a product of the C5 oxidation pathway, was a minor nucleoside product under both reaction conditions (Table 1). Inspection of the relative product distributions in all nucleoside and ODN contexts yielded d2Ih as the major oxidation product (Figure 2). dZ was a minor product observed in the single-stranded ODN contexts (ODN-1 and ODN-2, Figure 2) and not observed in the double-stranded ODN context (ODN-12, Figure 2). Within the double-stranded ODN context, dGhred was observed in low yields for copper-mediated oxidation in the presence of either Asc or NAC (Figure 2).
Table 1.
Absolute product yields from dG nucleoside oxidations with Cu(II)/H2O2/Asc or NAC at pH 7.0.
| Absolute % Product Yieldsa,b | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Product | Reaction Conditions
|
|||||
| Reductant (−) | Asc 2.0 mM |
Asc 20.0 mM |
NAC (H2O) 2.0 mM |
NAC (D2O) 2.0 mM |
NAC 20.0 mM |
|
| d2Ih | 3.0 | 11.9 | 28.2 | 18.0 | 2.7 | 28.9 |
| dZ | 1.3 | 1.1 | 5.5 | 0.4 | 1.8 | 1.5 |
| Ghred | 0.0 | 0.0 | 0 | 0.0 | 0.0 | 0.0 |
| dOG | 0.0 | 5.4 | 36.2 | 2.6 | 9.6 | 34.2 |
| dSp | 2.0 | 4.7 | 6.3 | 2.9 | 6.3 | 4.7 |
| dGh | 0.2 | 0.7 | 1.1 | 0.8 | 1.4 | 1.6 |
| 5′,8-Cyclo-dG | 1.4 | 1.1 | 1.9 | 0.3 | 0.6 | 2.3 |
| Gua | 1.6 | 8.5 | 5.8 | 6.3 | 5.8 | 8.8 |
| OG | 0.0 | 2.6 | 0.7 | 0.0 | 0.0 | 1.2 |
| Gh | 0.0 | 0.3 | 0 | 0.0 | 0.0 | 0.0 |
|
| ||||||
| Total Conversion | 9.5c | 36.3c | 85.7c | 31.3c | 28.2c | 83.2c |
The average error on each value was ~4% of the value, determined by three independent studies (error = 1 standard deviation). Individual error bars are provided in the ESI.
The reactions were conducted with dG (2.0 mM), Cu(II) acetate (1.0 mM), Asc or NAC (2.0 or 20 mM), and H2O2 (10.0 mM).
After accounting for unreacted dG, the mass balance for each nucleoside reaction was >90%.
Fig 2.

Context effects on relative product distributions of dG when oxidized with (A) Cu(II)/H2O2/Asc or (B) Cu(II)/H2O2/NAC Fenton-like oxidant systems. The calculated mass balance for each context study was: dG nucleoside >90%, ODN-1 and -2 ~70%, and ODN-12 ~60%.
C8 Oxidation Products
The products derived from oxidation at C8 of dG include the initial product dOG, and its further oxidation products dSp and dGh that were all observed with each oxidant system (Table 1) in all contexts studied (Figure 2). Combined yields of dOG, dSp, and dGh provide the amount of initial dG oxidation chemistry that occurs at C8; thus, these combined amounts can be compared to the C5 oxidation product totals to gain insight into the partitioning of products along the C5 and C8 base oxidation pathways. In the nucleoside studies, the combined absolute yields of dOG, dSp, and dGh provided the second largest pathway with Cu(II)/H2O2/Asc and NAC (Table 1, combined yields for Asc (2 mM) = 10.8%, and NAC (2 mM) = 6.3%). In terms of relative yields for which all structural contexts can be compared, the combined yields of dOG, dSp, and dGh were ~30–40% of the total products with Asc as reductant, and ~20–30% with NAC (Figure 2). In other words, more C8 oxidation occurs when Asc is the reductant. The absolute yield of dOG was greatest in the nucleoside studies, and decreased in the ODN studies; whereas, the hydantoin yields increased in the ODN studies. This suggests that dOG was more susceptible to further oxidation in the ODN contexts than in the nucleoside, which likely resulted from the longer reaction time for the ODN studies (8 h vs. 1 h). In addition, the yield of dSp was greater in the nucleoside, and the yield of dGh was greater in the ODN contexts, in accordance with previous studies (Figure 2).
Attempts were made to identify Fapy-dG as a product of nucleoside or ODN oxidation, both at 2.0 and 20 nM concentrations of Asc and NAC. None was detected by reversed-phase LC-MS.
Ribose Oxidation Products
The 2-deoxyribose moiety of dG is also prone to oxidation by Cu-mediated Fenton chemistry.10 In the studies with 2 nM reductant present, two products were identified from sugar oxidation, 5′,8-cyclo-dG (Asc = 1.1%; NAC = 0.3%, Table 1) and Gua (Asc = 8.5%, and NAC = 6.3%, Table 1). In the Fe-mediated Fenton oxidation of dG, both 5′,8-cyclo-dG and Gua were observed along with some minor sugar oxidation products that were not detected in these studies.10,16 In the nucleoside studies using Cu(II)/H2O2/Asc, Gua oxidation leading to OG and Gh free bases was detected in low yields (Asc, OG = 2.6%, and Gh = 0.3%, Table 1). In the ODN oxidation studies, Gua was observed but in lower relative yields than characterized in the nucleoside studies (Figure 2). This effect is most likely a result of the ODN context providing other sugar oxidation sites besides dG, leading to release of the other bases as observed in the HPLC analysis (see ESI).
Partitioning between Pathways
When the reductant concentration was increased 10-fold, the total conversion to products increased dramatically in the nucleoside experiment (Table 1). Interestingly, the increase in the C8 oxidation products was more dramatic than the C5 pathway (four to seven-fold compared to two to three-fold). As expected for the higher reductant concentration, pathways dependent on one-electron chemistry or hydroxyl radical (dOG oxidation to dSp and sugar oxidation to release free bases) were relatively small. To test an earlier hypothesis that singlet oxygen may be involved in the reaction, we also conducted a nucleoside experiment in D2O in which 1O2 has a longer lifetime. Consistent with an earlier study,15 the yield of C8 products (dOG, dSp, dGh) increased by a factor of 3.7 (Table 1). However, the yield of C5 products (d2Ih, dZ) decreased in a compensatory fashion by a factor of 4.1.
Discussion
Mechanisms of Base Oxidation
The C5 oxidation pathway of guanosine leads to two principal products dZ and d2Ih that were observed in all nucleoside reactions. The proposed mechanisms for formation of these products are the topic of many reviews, where the full details of these reactions can be found.5,44,58 Both dZ and d2Ih may originate from the guanine radical intermediate dG• (Scheme 1) that then bifurcates along two proposed coordinates (Scheme 2): (1) Quenching by O2•− leads to 5-HOO-dG, or (2) one-electron oxidation to dG+, followed by addition of H2O at C5 leads to 5-HO-dG.44 The fate of 5-HOO-dG is either reduction to 5-HO-dG or decomposition to dIz and hydrolysis to dZ (Scheme 2).44,59 5-HO-dG is the proposed key intermediate leading to d2Ih via acyl migration giving an intermediate spirocycle that adds H2O at C8, finally yielding d2Ih as a mixture of diastereomers and structural isomers. The ring-opened form of d2Ih shown in Scheme 2 was identified by Ye, et al.45
Scheme 1.

Proposed pathways for the initial one-electron oxidation of dG to dG•.
Scheme 2.

Proposed pathways to form dZ and d2Ih. Compounds characterized in this work are labeled with a dashed box.
An additional pathway can be considered for formation of the key intermediate 5-HO-dG that is particularly relevant to metal-mediated oxidation. Coordination of a high-valent copper species (Eqn. III) to N7 of dG as shown in Scheme 3 could lead to the direct C5 hydroxylation of guanine and commitment to the d2Ih pathway shown in Scheme 2. This mechanism is attractive because it helps explain why the product profiles for copper and nickel-mediated oxidations42 are quite different from those of G radical chemistry initiated by radiation, riboflavin photochemistry, and related processes.3 The former produces d2Ih as a major product via 5-HO-dG while the latter forms dIz/dZ via 5-HOO-dG.
Scheme 3.

Proposed mechanism for oxidation by a high-valent copper species yielding 5-HO-dG.
Oxidation at C8 of guanosine leads to the products dOG and its further oxidation products dSp and dGh. The formation of 5′,8-cyclo-dG is likely initiated by sugar oxidation that will be discussed below. Observation of dOG in the nucleoside reactions is consistent with previous studies in which dG was oxidized in double-stranded DNA under similar reaction conditions to yield dOG.16,22,49,50 A key difference is that most of the prior work was done in the absence of added reductants. In those studies, the role of copper was proposed to be as a vehicle to deliver hydroxyl radical in the vicinity of C8 of dG via Cu binding to N7.3,5,44,58 When HO• adds to C8 of dG, the intermediate radical 8-HO-dG• is formed; this species can yield either dOG under oxidizing conditions or Fapy-dG under reducing conditions (Scheme 4A). This latter point is relevant because all of our studies reported here had reductant present in 2–20 mM concentrations.
Scheme 4.
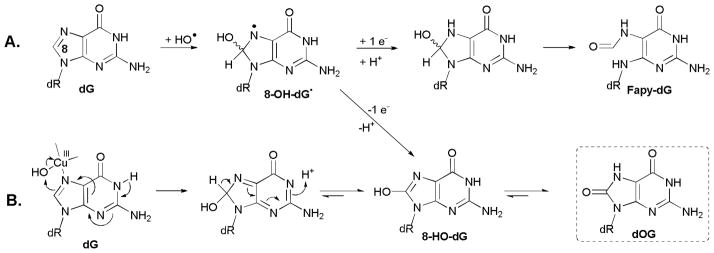
Proposed pathway for HO• addition to C8 of dG leading to dOG and Fapy-dG. Compounds characterized in this work are labeled with a dashed box.
In the present work, dOG was observed from the oxidation of dG by Cu(II)/H2O2/Asc or NAC in yields of 5.4% or 2.6%, respectively (Table 1). We could not detect Fapy-dG as a product of any copper-mediated reaction of dG, either as a nucleoside substrate or in an ODN. Other laboratories report that Fe- and Cu-mediated Fenton reagents produce more dOG (>10-fold) than Fapy-dG.15,22,49 There are two possible explanations for the complete absence of Fapy-dG in our work; either the presence of more oxidant (10.0 mM H2O2) than reductant (2.0 mM Asc or NAC) favors the oxidation pathway of 8-HO-dG• leading to dOG, or Cu facilitates a different mechanism than that proposed in Scheme 4A in which the formation of dOG bypasses the radical intermediate. We present such a possibility in Scheme 4B. In this mechanism, a Cu(III)-OH species mediates 2-electron oxidation of dG directly to a tautomer of dOG without any opportunity for reduction of an intermediate radical. If the mechanism in Scheme 4B predominates, no Fapy-dG would be formed, consistent with our observations. It should be pointed out the role of copper in C8 oxidation as shown in Scheme 4B is completely analogous to that proposed in Scheme 3 for C5 oxidation.
The products dGh and dSp result from subsequent oxidation events on dOG due to its 600 mV lower redox potential.27,28 Oxidation pathways for dOG have been reviewed and are outlined in Scheme 5.3,44,58,60,61 Formation of dSp and dGh is proposed from the common intermediate 5-HO-dOG that then partitions to the hydantoins in a pH and context dependent manner. Rearrangement to dSp is observed at high pH (>6.0) and in the less sterically encumbered nucleoside context,30,35 while low pH (<6.0) and a duplex DNA context favors hydration and decarboxylation to yield dGh (Scheme 5).35,36
Scheme 5.

Proposed pathways for the formation of hydantoin products from dOG. Compounds characterized in this work are labeled with a dashed box.
Analysis of product evolution vs. reaction time shows that dOG is formed before dSp or dGh, supporting previous data that suggest dOG to be the intermediate leading to the hydantoin products (see ESI).3,44,58,60 The reactions investigated in these studies, at 2 mM reductant concentration, consistently gave dSp yields (Asc = 4.7%, and NAC = 2.9%, Table 1) greater than dGh (Asc = 0.7%, and NAC = 0.8%, Table 1) as anticipated because the reactions were conducted under conditions with pH > 6.0 33,36 Also, as expected, the yield of dGh increased in the single- and double-stranded ODN contexts (Figure 2). To further confirm that dSp and dGh result from the intermediate dOG in the Cu(II)/H2O2/Asc reaction, authentic dOG nucleoside was allowed to react with Cu(II)/H2O2/Asc providing the two hydantoins, dSp and dGh (see ESI). Another possible pathway for formation of dSp is through the two-electron oxidation of d2Ih. Purified d2Ih was allowed to react with the Cu-ascorbate oxidant system, and the only product observed was 2Ih free base, suggesting sugar oxidation as the major oxidation product from d2Ih. To further understand if the base of d2Ih could be oxidized, the one-electron oxidant Na2IrCl6 was allowed to react with d2Ih. Reinjection of the oxidation reaction provided no change in d2Ih by HPLC (see ESI). These observations suggest that d2Ih cannot be oxidized to dSp, and that the hydantoins both result from initial oxidation of dG at C8 yielding the intermediate dOG; subsequently, a two-electron oxidation of dOG occurs at C5 yielding the hydantoins, dSp or dGh.
Mechanisms of 2-deoxyribose oxidation
Formation of 5′,8-cyclo-dG is thought to be initiated by oxidation of dG at C5′ yielding a carbon-centered radical 1 that adds to C8 of the heterocyclic ring of dG (Scheme 6); subsequent addition of this radical to C8 of guanine leads to the intermediate radical 2. Radical 2 can then be further oxidized by one electron along with loss of a proton to yield 5′,8-cyclo-dG. Sugar oxidation also causes Gua base release from the nucleoside sugar. Gua base release via the Fenton reaction is proposed to be initiated by the one-electron oxidation of dG at C1′ yielding an intermediate carbon-centered radical 3 that provides peroxyl radical 4 after reaction with dioxygen. Finally, one-electron reduction and proton transfer occur to yield the alcohol 5. Compound 5 decomposes to Gua free base and a sugar lactone (Scheme 6).10,16 OG (M-100) and Gh (M-110) free bases result from the two- and four-electron oxidation of Gua, respectively, following the previously proposed mechanisms (Schemes 4 and 5).5,36,62
Scheme 6.

Proposed pathways for the sugar oxidation products 5′,8-cyclo-dG and Gua. Compounds characterized in this work are labeled with a dashed box.
The free base Gua resulting from sugar oxidation was observed in all Cu-mediated Fenton reactions. This observation parallels previous studies that characterized Gua base release from oxidation of dG nucleoside with Fe-based Fenton reagents.16 Additionally, DNA oxidation with Cu-mediated Fenton reagents led to free base oxidation rather than deglycosylation of dOG or dGh, respectively (see ESI). Only Gh free base was observed and not Sp free base; in previous studies, OG free base oxidation at pH 7.0 yielded Gh, not Sp, consistent with our findings and the fact that uric acid oxidation leads to allantoin in preference to spirodihydantoin.62
Partitioning between Pathways
The overall yields of dG oxidation products (Table 1) were similar in the Cu(II)/H2O2/Asc and Cu(II)/H2O2/NAC reactions (36.3 ± 1.5 and 31.3 ± 1.3%, respectively, 2 mM reductant). However, closer inspection of the product yields grouped by reaction pathway indicates a bigger difference between the two reductants studied than is first apparent. The products can be grouped into three clusters: the C5 pathway (d2Ih and dZ), the C8 pathway (dOG and its two-electron oxidation products dSp and dGh), and the sugar oxidation pathway (Gua, OG and Gh). The ratio of C5:C8:sugar reactivity for Cu(II)/H2O2/Asc was 1.00: 0.84: 0.96, whereas for Cu(II)/H2O2/NAC it was 1.00: 0.34: 0.36, based on the nucleoside values in Table 1. Within error, the Asc-based system gave similar product yields along each reaction channel. In previous studies that quantified product distributions from dG oxidation by Fe(III)-EDTA/H2O2/NADH, similar yields of C8 products (dOG) and sugar oxidation products (5′,8-cyclo-dG, Gua, and OG) were observed,16,47 but C5 oxidation products were not identified because of the HPLC conditions used.16 The Cu(II)/H2O2/Asc and NAC systems both gave equal reactivity at C8 and the ribose of dG, suggesting a common feature to both Fe- and Cu-mediated Fenton oxidation of the nucleoside dG. The present work also permitted the characterization and quantification of C5 oxidation products, and notably, d2Ih yields increased with the stronger reductant, NAC. With Asc, the relative reactivities at C5 and C8 were similar, but NAC provided roughly three-fold more reaction at C5 than C8 or the sugar (Table 1). The reducing agent effect on product distribution was evaluated by Cadet and coworkers when dG was oxidized with HO• (generated by γ irradiation) in the presence of Asc or cysteine to yield the C5 product dZ and the C8 products dOG and Fapy-dG.63 In their studies, the absolute product yields differed from ours, but the trends were the same. With the stronger reductant cysteine, C8 oxidation product yields decreased, and they concluded that cysteine, the stronger reductant, quenched the radicals leading to C8 oxidation products better than Asc.63 This observation was mirrored by our results in which the NAC reactions gave lower yields of C8 oxidation products when compared to the Asc reaction. The base-oxidation pathway dependence on reductant suggests two possibilities: (1) Radical chemistry plays a larger role in dOG formation than in the pathway to d2Ih, and/or (2) the coordination environment for Cu in the Asc reaction is different than the NAC reaction causing a shift in the base oxidation pathway. The latter point is expected based on the propensity for cysteine thiols to coordinate directly to Cu(I) and Cu(II). Of course, the precise structure of the copper complex that carries out the key oxidation step is unknown.
The conclusions above are presented for the copper-mediated Fenton reaction conducted in the presence of physiological concentrations of reductant, either Asc or NAC, of 2 mM. Further mechanistic insight could be gained by increasing the reductant concentration ten-fold. Under these conditions, the total conversion to products increased, indicating that the formation of the reactive oxidant is dependent upon reductant. This step is presumably the reduction of Cu(II) to Cu(I). Consistent with this hypothesis is the observation of very low levels of product formation when the Cu(II) concentration remains the same, but no reductant is added (Table 1, column 1). Curiously, the effect of increasing the reductant concentration is more dramatic for the C8 pathway than for the C5 pathway. This data would argue that the reactive species responsible for oxidation at C5 vs. C8 of dG is not the same. Also pertinent to this point is the fact that the C8 pathway was preferred when the solvent was D2O compared to the C5 pathway dominating in H2O (Table 1). One possibility is that 1O2 contributes to the C8 pathway as suggested by Frelon et al.,15 but not to the formation of C5 products. The formation of singlet oxygen in this system could result from dimerization of a Cu(I) peroxyl species that decomposes to generate 1O2 and 2 Cu(III)-OH complexes, as shown in equations IV–VI.
| (Equation IV) |
| (Equation V) |
| (Equation VI) |
With respect to ribose chemistry, cysteine has been shown to reduce sugar radicals more efficiently than Asc,64,65 consistent with our observation that NAC yields less sugar oxidation chemistry. In the Cu(II)/H2O2/Asc system, yields of base oxidation products (d2Ih + dZ + dOG + dSp + dGh) were approximately two-fold greater than sugar oxidation products. This supports the proposal that Cu coordination to N7 of dG favors base oxidation products, as well as the observation that DNA oxidation with Cu(II)/H2O2 gives more base oxidation than strand breaks.8,11,13
Context Effects on dG Base Oxidation Products
Previous studies have alluded to the observation that reaction context effects the oxidation product distributions; for example, dSp is a major nucleoside product, while dGh is a major DNA product under the same reaction conditions.35 With this thought in mind, the distribution of base oxidation products from dG were compared in the nucleoside, single- and double-stranded ODN contexts in which the products formed were the same, but their relative yields differed. From the data it appears that oxidation of ODNs with Cu(II)/H2O2/reductant, Asc or NAC, is affected most by the double-stranded ODN-12 context (Figure 2). Three differences were observed in these context studies: (1) dZ was not observed in the double-stranded ODN-12 context, but appears in small amounts in the nucleoside and single-stranded ODN-1 and ODN-2 contexts. (2) The ratios for the hydantoin products, dSp and dGh, are context dependent, as expected, favoring more dGh in single- and double-stranded ODN contexts studied. It has been proposed that the ODN context favors dGh because it is less sterically demanding, and the acid/base chemistry of the intermediate 5-HO-dOG in the ODN context favors acid-catalyzed decarboxylation leading to dGh.33,35,61 (3) Importantly, a new product of double-stranded DNA oxidation, dGhred (M+8), was observed by analysis with HPLC-ESI+-MS. This structure was previously proposed by Pratviel and Meunier,44 and in the present studies a mass consistent with dGhred was observed. Because the ODN context affects the reactivity of 5-HO-dOG leading to higher yields of dGh, they proposed that the analogous intermediate 5-HO-dG would be affected in the same way (Scheme 7).44 Our results are consistent with this idea, and the hydrolytic chemistry of 5-HO-dG appears to parallel that of 5-HO-dOG to yield dGhred and dGh, respectively. It is not known if the five-membered ring of dGhred is opened or closed, but the ring-opened form is similar to that of d2Ih, which has been more extensively characterized.45
Scheme 7.

Product comparison for the intermediates 5-HO-dOG and 5-HO-dG.
Lastly, we note that the diastereomers of both dSp and d2Ih were separable on the Hypercarb HPLC column.66,67 Cu-mediated oxidation of dG yielded dSp diastereomer ratios that are approximately 1:1 in all contexts studied. In contrast, it is interesting to note that the Cu-mediated formation of d2Ih, in all reaction contexts, favored the later eluting d2Ih diastereomer by a factor of 2:1. Without further structural analysis, the absolute stereochemistry of each d2Ih diastereomer remains unknown.
Conclusions
DNA isolated from cellular extracts contains high concentrations of associated Cu, and it has been proposed that Cu-mediated oxidation of DNA underlies certain disease states.9 In this study, the major oxidation product of the copper-mediated Fenton reaction observed in all contexts was the recently characterized hydantoin d2Ih. The reductant appears to play a role in defining the overall product distribution between oxidation of guanosine at C5 vs. C8 vs. the ribose group. In these studies, the overall two-electron oxidation of dG at C5 was a major reaction channel leading initially to 5-OH-dG, an isomer of the well-studied dOG. Subsequent hydration of 5-OH-dG led to the characterized product d2Ih. Secondarily, oxidation at C8 of the purine using physiologically-relevant concentrations of reductant yielded dOG and no detectable quantities of Fapy-dG, highlighting a key difference in the mechanisms between radiation vs. metal-mediated oxidation of the guanine base. In radiation-induced damage, a free hydroxyl radical is formed that adds to C8 of dG, yielding an intermediate radical that can partition to dOG under oxidizing conditions or yield Fapy-dG under reducing conditions. Whereas in Cu-mediated oxidation of dG, the intermediate radical leading to dOG is bypassed because Cu is coordinated to N7 of dG for which direct transfer of the hydroxyl group to C8 of dG occurs following Scheme 4B. In the double-stranded ODN context a new product, dGhred, was observed supporting the hypothesis of Pratviel and Meunier for formation of this structure.44 These studies highlight a difference in the product distributions observed between radiation-induced and Cu-mediated damage to guanosine and point to the importance of further studies of d2Ih as a potential lesion leading to mutagenesis.
Experimental Section
Nucleoside Studies
All reagents were obtained from commercially available sources and used without further purification unless otherwise stated. A 200-μL solution of dG (3.0 mM, 0.60 μmol, 0.16 mg) in NaPi buffer (75.0 mM, pH 7.0) was oxidized by addition of copper(II) acetate (1.0 mM, 0.20 μmol, 0.04 mg), Asc or NAC (N-acetyl-cysteine, 2.0 mM, 0.40 μmol, 0.03 mg), followed by addition of H2O2 (10.0 mM, 2.0 μmol, 0.07 mg). The solution was kept at 22 °C for 60 min and then quenched with Na2EDTA (10.0 mM, 2.0 μmol, 0.50 mg). The reaction mixture was first analyzed by RP-HPLC that retained Gua, OG, dOG, and 5′,8-cyclo-dG. The void volume from the RP-HPLC run was collected, dried down, resuspended in the Hypercarb column mobile phase (0.1% acetic acid), and then injected on a Hypercarb HPLC column to analyze Gh, d2Ih, dGh, dSp, and dZ. All HPLC separation parameters are presented in the supporting information. HPLC-ESI+-MS results for each identified compound are as follows: m/z (M+H)+ 152.1 (Gua), 168.1 (OG), 268.1 (dG) 284.1 (dOG), 266.3 (5′,8-cyclo-dG), 158.1 (Gh), 302.1 (d2Ih diastereomers), 274.1 (dGh), 300.1 (dSp diastereomers), and 247.1 (dZ); all found values matched their calculated values. Collected samples provided the following ESI+-MS/MS data for the free bases of the following compounds: d2Ih diastereomers (186, 158, and 141; lit.,45 186, 158, 141) dSp diastereomers (184, 156, 141, 113, 99, and 86; lit.,30 184, 156, 141, 113, 99, and 86) and dZ (247, 203, 131; lit.,40 247, 203, 131). HRMS-ESI+ (m/z) for dSP, C10H13N5O6Na (M)+ calcd 322.0764, found 322.0761; dGh, C9H15N5O5Na (M)+ calcd 296.0971, found 296.0980; dZ, C8H14N4O5Na (M)+ calcd 269.0862, found 269.0870. UV-vis profiles for each compound are shown in the ESI. Integrated peak areas obtained from absorbance at 240 nm on each HPLC run, were used to quantify the reaction yields through normalization of each area by its unique ε240 nm (ddH2O): dG (lit.,16 14,080 dm3 mol−1 cm−1), dOG (lit.,16 14,300 dm3 mol−1 cm−1), 5′,8-cyclo-dG (lit.,16 14,080 dm3 mol−1 cm−1), d2Ih (lit.,42 3,275 dm3 mol−1 cm−1), dGh (lit.,36 2,412 dm3 mol−1 cm−1), dSp (lit.,30 3,275 dm3 mol−1 cm−1), dZ (lit.,40 1,778 dm3 mol−1 cm−1), Gua (lit.,16 14,080 dm3 mol−1 cm−1), OG (lit.,1614,300 dm3 mol−1 cm−1), and Gh (lit.,36 2,412 dm3 mol−1 cm−1).
Oligodeoxynucleotide Studies
The ODNs were synthesized following standard procedures by the DNA/Peptide Core facility at the University of Utah were used after HPLC purification (ESI). Formation of double-stranded ODN-12 was achieved by heating equimolar ratios of ODN-1 and ODN-2 in NaPi buffer (20.0 mM, pH 7.0) with NaCl (100.0 mM), at 90 °C for 5 min, followed by slowly cooling the samples to room temperature over 3 h. Cu-mediated oxidations were conducted in a 100-μL solution of ODN-1, ODN-2, or ODN-12 (100 μM, 10.0 nmol) in NaPi buffer (20.0 mM, pH 7.0) and NaCl (100 mM) at 37 °C. First, the ODN samples were incubated with Cu(II) acetate (10.0 μM, 1.0 nmole) for 5 min. Next, the reaction was initiated by addition of Asc or NAC (1.0 mM, 100.0 nmol) then H2O2 (1.0 mM, 100.0 nmol), and the reaction was allowed to proceed for 8 h. The ODN samples were then digested with a suite of nucleases to liberate the oxidized nucleotides as follows: (1) The ODN samples were lyophilized to dryness and then resuspended in DNase I reaction buffer (20.0 mM Tris (pH 8.4), 2.0 mM MgCl2, 50.0 mM KCl) followed by addition of DNase I (2.0 U), and incubated at 37 °C for 3 h. The antioxidant butylated hydroxytoluene (2.0 mM) along with the deaminase inhibitors pentostatin (100 μM) and tetrahydrouridine (100 μM) were initially added before commencement of the digestion. (2) 10-μL of a NaOAc buffer solution (100.0 mM, pH 5.3) containing zinc acetate (10.0 mM) was added to the digestion solution, followed by nuclease P1 (2.0 U). The reaction was incubated at 45 °C for 9 h, followed by addition of more nuclease P1 (2.0 U) and incubation for another 9 h. (3) 11-μL of tris buffer (100.0 mM, pH 7.8) with MgCl2 (10.0 mM) and snake venom phosphodiesterase (2.0 U) was added to the digestion mixture. The reaction was incubated at 45 °C for 9 h after which snake venom phosphodiesterase (2.0 U) and calf intestinal phosphatase (16.0 U) were added and allowed to react for 9 h to liberate the damaged and undamaged nucleosides from the reacted ODNs. The digestion proteins were removed before HPLC analysis by passing the sample through a 10,000 molecular weight cutoff filter (Millipore), and then analyzed by HPLC following the previous method. The HPLC areas for the canonical DNA bases were normalized by their extinction coeffiencts ε240 nm (ddH2O): 2′-deoxyadenosine (lit.,68 9,500 dm3 mol−1 cm−1), 2′-deoxycytidine (lit.,68 7,400 dm3 mol−1 cm−1), and thymidine (lit.,68 9,900 dm3 mol−1 cm−1). An ODN-12 reaction with Cu(II)/H2O2/Asc was analyzed by HPLC-ESI+-MS running the Hypercarb HPLC column to provide the additional MS data m/z (M+H)+ 276.1 (dGhred).
Supplementary Material
Acknowledgments
We thank Dr. Xin Chen (University of Utah) for contributions to the DNA digestion conditions used in this study and the National Cancer Institute for a grant supporting this work (CA090689). Shared resources were supported by award number P30CA042014 from the National Cancer Institute.
Footnotes
Electronic supplementary information (ESI) available: Experimental details of HPLC separation and mass spectrometric characterization of products, time and pH-dependent studies, and compilations of product yield data. See DOI:.
Dedicated to the memory of Professor Athel Beckwith and his distinguished career in free-radical chemistry.
References
- 1.Cooke MS, Olinski R, Evans ME. Clin Chim Acta. 2006;365:30–49. doi: 10.1016/j.cca.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 2.Adly AAM. Res J Immunol. 2010;3:129–145. [Google Scholar]
- 3.Cadet J, Douki T, Ravanat JL. Free Radical Biol Med. 2010;49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 4.Henkler F, Brinkmann J, Luch A. Cancers. 2010;2:376–396. doi: 10.3390/cancers2020376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burrows CJ, Muller JG. Chem Rev. 1998;98:1109–1152. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 6.Drouin R, Rodriguez H, Gao SW, Gebreyes Z, O’Connor TR, Holmquist GP, Akman SA. Free Radical Biol Med. 1996;21:261–273. doi: 10.1016/0891-5849(96)00037-8. [DOI] [PubMed] [Google Scholar]
- 7.Oikawa S, Kawanishi S. Biochemistry. 1996;35:4584–4590. doi: 10.1021/bi9527000. [DOI] [PubMed] [Google Scholar]
- 8.Liang Q, Dedon PC. Chem Res Toxicol. 2001;14:416–422. doi: 10.1021/tx0002278. [DOI] [PubMed] [Google Scholar]
- 9.Ames BN, Shigenaga MK, Hagen TM. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pogozelski WK, Tullius TD. Chem Rev. 1998;98:1089–1108. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 11.Sorokin VA, Valeev VA, Gladchenko GO, Sysa IV, Blagoi YP, Volchok IV. J Inorg Biochem. 1996;63:79–97. doi: 10.1016/0162-0134(95)00176-x. [DOI] [PubMed] [Google Scholar]
- 12.Prutz WABJ, Land EJ. Int J Radiat Biol. 1990;58:215–234. doi: 10.1080/09553009014551581. [DOI] [PubMed] [Google Scholar]
- 13.Bryan SE, Frieden E. Biochemistry. 1967;6:2728–2734. doi: 10.1021/bi00861a012. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez H, Drouin R, Holmquist GP, O’Connor TR, Boiteux S, Laval J, Doroshow JH, Akman SA. J Biol Chem. 1995;270:17633–17640. doi: 10.1074/jbc.270.29.17633. [DOI] [PubMed] [Google Scholar]
- 15.Frelon S, Douki T, Favier A, Cadet J. Chem Res Toxicol. 2003;16:191–197. doi: 10.1021/tx025650q. [DOI] [PubMed] [Google Scholar]
- 16.Henle ES, Luo Y, Gassmann W, Linn S. J Biol Chem. 1996;271:21177–21186. [PubMed] [Google Scholar]
- 17.Murata-Kamiya N, Kamiya H, Muraoka M, Kaji H, Kasai H. J Radiat Res. 1997;38:121–131. doi: 10.1269/jrr.38.121. [DOI] [PubMed] [Google Scholar]
- 18.Joffe A, Geacintov NE, Shafirovich V. Chem Res Toxicol. 2003;16:1528–1538. doi: 10.1021/tx034142t. [DOI] [PubMed] [Google Scholar]
- 19.Sugden KD, Campo CK, Martin BD. Chem Res Toxicol. 2001;14:1315–1322. doi: 10.1021/tx010088+. [DOI] [PubMed] [Google Scholar]
- 20.Adam W, Saha-Moller CR, Schonberger A. J Am Chem Soc. 1997;119:719–723. [Google Scholar]
- 21.Pouget JPFS, Ravanat JL, Testard I, Odin F, Cadet J. Radiat Res. 2002;157:589–595. doi: 10.1667/0033-7587(2002)157[0589:fomdbi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 22.Dizdaroglu M, Rao G, Halliwell B, Gajewski E. Arch Biochem Biophys. 1991;285:317–324. doi: 10.1016/0003-9861(91)90366-q. [DOI] [PubMed] [Google Scholar]
- 23.Gedik CM, Collins A. FASEB J. 2005;19:82–84. doi: 10.1096/fj.04-1767fje. [DOI] [PubMed] [Google Scholar]
- 24.Mangal D, Vudathala D, Park JH, Lee SH, Penning TM, Blair IA. Chem Res Toxicol. 2009;22:788–797. doi: 10.1021/tx800343c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibutani S, Takeshita M, Grollman AP. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 26.David SS, O’Shea VL, Kundu S. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steenken S, Jovanovic SV. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 28.Steenken S, Jovanovic SV, Bietti M, Bernhard K. J Am Chem Soc. 2000;122:2373–2374. [Google Scholar]
- 29.Luo W, Muller JG, Burrows CJ. Org Lett. 2001;3:2801–2804. doi: 10.1021/ol0161763. [DOI] [PubMed] [Google Scholar]
- 30.Luo W, Muller JG, Rachlin EM, Burrows CJ. Org Lett. 2000;2:613–616. doi: 10.1021/ol9913643. [DOI] [PubMed] [Google Scholar]
- 31.Slade PG, Hailer MK, Martin BD, Sugden KD. Chem Res Toxicol. 2005;18:1140–1149. doi: 10.1021/tx050033y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Misiaszek R, Crean C, Geacintov NE, Shafirovich V. J Am Chem Soc. 2005;127:2191–2200. doi: 10.1021/ja044390r. [DOI] [PubMed] [Google Scholar]
- 33.Niles JC, Wishnok JS, Tannenbaum SR. Chem Res Toxicol. 2004;17:1510–1519. doi: 10.1021/tx0400048. [DOI] [PubMed] [Google Scholar]
- 34.Ye Y, Muller JG, Luo W, Mayne CL, Shallop AJ, Jones RA, Burrows CJ. J Am Chem Soc. 2003;125:13926–13927. doi: 10.1021/ja0378660. [DOI] [PubMed] [Google Scholar]
- 35.Gremaud JN, Martin BD, Sugden KD. Chem Res Toxicol. 2009;23:379–385. doi: 10.1021/tx900362r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo W, Muller JG, Rachlin EM, Burrows CJ. Chem Res Toxicol. 2001;14:927–938. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 37.Duarte V, Muller JG, Burrows CJ. Nucleic Acids Res. 1999;27:496–502. doi: 10.1093/nar/27.2.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hailer MK, Slade PG, Martin BD, Sugden KD. Chem Res Toxicol. 2005;18:1378–1383. doi: 10.1021/tx0501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 40.Matter B, Malejka-Giganti D, Csallany AS, Tretyakova N. Nucleic Acids Res. 2006;34:5449–5460. doi: 10.1093/nar/gkl596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duarte V, Gasparutto D, Jaquinod M, Cadet J. Nucleic Acids Res. 2000;28:1555–1563. doi: 10.1093/nar/28.7.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghude P, Schallenberger MA, Fleming AM, Muller JG, Burrows CJ. Inorg Chim Acta. 2011 doi: 10.1016/j.ica.2010.12.063. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vialas C, Claparols C, Pratviel G, Meunier B. J Am Chem Soc. 2000;122:2157–2167. [Google Scholar]
- 44.Pratviel G, Meunier B. Chem Eur J. 2006;12:6018–6030. doi: 10.1002/chem.200600539. [DOI] [PubMed] [Google Scholar]
- 45.Ye W, Sangaiah R, Degen DE, Gold A, Jayaraj K, Koshlap KM, Boysen G, Williams J, Tomer KB, Mocanu V, Dicheva N, Parker CE, Schaaper RM, Ball LM. J Am Chem Soc. 2009;131:6114–6123. doi: 10.1021/ja8090752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L, Murthy NN, Telser J, Zakharov LN, Yap GPA, Rheingold AL, Karlin KD, Rokita SE. Inorg Chem. 2006;45:7144–7159. doi: 10.1021/ic0605930. [DOI] [PubMed] [Google Scholar]
- 47.Grey CE, Adlercreutz P. J Agric Food Chem. 2006;54:2350–2358. doi: 10.1021/jf052202q. [DOI] [PubMed] [Google Scholar]
- 48.Belmadoui N, Boussicault F, Guerra M, Ravanat JL, Chatgilialoglu C, Cadet J. Org Biomol Chem. 2010;8:3211–3219. doi: 10.1039/c004531d. [DOI] [PubMed] [Google Scholar]
- 49.Aruoma OI, Halliwell B, Gajewski E, Dizdaroglu M. Biochem J. 1991;273:601–604. doi: 10.1042/bj2730601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kennedy LJ, Moore K, Caulfield JL, Tannenbaum SR, Dedon PC. Chem Res Toxicol. 1997;10:386–392. doi: 10.1021/tx960102w. [DOI] [PubMed] [Google Scholar]
- 51.Hong H, Cao H, Wang Y, Wang Y. Chem Res Toxicol. 2006;19:614–621. doi: 10.1021/tx060025x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao H, Wang Y. Nucleic Acids Res. 2007;35:4833–4844. doi: 10.1093/nar/gkm497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu X, Muller JG, Ye Y, Burrows CJ. J Am Chem Soc. 2008;130:703–709. doi: 10.1021/ja077102a. [DOI] [PubMed] [Google Scholar]
- 54.Solivio MJ, Joy TJ, Sallans L, Merino EJ. J Inorg Biochem. 2010;104:1000–1005. [PubMed] [Google Scholar]
- 55.Chen X. PhD Dissertation. University of Utah; Salt Lake City, UT: 2008. [Google Scholar]
- 56.Wang Y. Chem Res Toxicol. 2008;21:276–281. doi: 10.1021/tx700411g. [DOI] [PubMed] [Google Scholar]
- 57.Crean C, Geacintov NE, Shafirovich V. Free Radical Biol Med. 2008;45:1125–1134. doi: 10.1016/j.freeradbiomed.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gimisis T, Cismas C. Eur J Org Chem. 2006:1351–1378. [Google Scholar]
- 59.Raoul S, Berger M, Buchko GW, Joshi PC, Morin B, Weinfeld M, Cadet J. J Chem Soc, Perkin Trans. 1996;2:371–381. [Google Scholar]
- 60.Neeley WL, Essigmann JM. Chem Res Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 61.Munk BH, Burrows CJ, Schlegel HB. J Am Chem Soc. 2008;130:5245–5256. doi: 10.1021/ja7104448. [DOI] [PubMed] [Google Scholar]
- 62.Yu H, Niles JC, Wishnok JS, Tannenbaum SR. Org Lett. 2004;6:3417–3420. doi: 10.1021/ol048547w. [DOI] [PubMed] [Google Scholar]
- 63.Douki T, Spinelli S, Ravanat J-L, Cadet J. J Chem Soc, Perkin Trans. 1999;2:1875–1880. [Google Scholar]
- 64.O’Neill P. Radiat Res. 1993;96:198–210. [PubMed] [Google Scholar]
- 65.O’Neill P, Chapman PW. Int J Radiat Biol. 1985;47:71–80. doi: 10.1080/09553008514550101. [DOI] [PubMed] [Google Scholar]
- 66.Karwowski B, Dupeyrat F, Bardet M, Ravanat JL, Krajewski P, Cadet J. Chem Res Toxicol. 2006;19:1357–1365. doi: 10.1021/tx060088f. [DOI] [PubMed] [Google Scholar]
- 67.Ding S, Jia L, Durandin A, Crean C, Kolbanovskiy A, Shafirovich V, Broyde S, Geacintov NE. Chem Res Toxicol. 2009;22 doi: 10.1021/tx900107q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson NA, McKenzie R, McLean L, Sowers LC, Fletcher HM. J Bacteriol. 2004;186:7697–7703. doi: 10.1128/JB.186.22.7697-7703.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.