

Abstract
Autoimmune Addison's disease (AAD) is caused by selective destruction of the hormone-producing cells of the adrenal cortex. As yet, little is known about the potential role played by environmental factors in this process. Type I and/or type III interferons (IFNs) are signature responses to virus infections, and have also been implicated in the pathogenesis of autoimmune endocrine disorders such as type 1 diabetes and autoimmune thyroiditis. Transient development of AAD and exacerbation of established or subclinical disease, as well as the induction of autoantibodies associated with AAD, have been reported following therapeutic administration of type I IFNs. We therefore hypothesize that exposure to such IFNs could render the adrenal cortex susceptible to autoimmune attack in genetically predisposed individuals. In this study, we investigated possible immunopathological effects of type I and type III IFNs on adrenocortical cells in relation to AAD. Both types I and III IFNs exerted significant cytotoxicity on NCI-H295R adrenocortical carcinoma cells and potentiated IFN-γ-and polyinosine-polycytidylic acid [poly (I : C)]-induced chemokine secretion. Furthermore, we observed increased expression of human leucocyte antigen (HLA) class I molecules and up-regulation of 21-hydroxylase, the primary antigenic target in AAD. We propose that these combined effects could serve to initiate or aggravate an ongoing autoimmune response against the adrenal cortex in AAD.
Keywords: 21-hydroxylase, Addison's disease, CXC chemokine ligand 10, interferon, polyinosine-polycytidylic acid
Introduction
Autoimmune Addison's disease (AAD) is a classic organ-specific autoimmune disease wherein the adrenal cortex is targeted and destroyed by the immune system, resulting in a lack of hormone-producing cells. Consequently, patients suffer from an inability to produce vital steroid hormones such as cortisol and aldosterone, and require continuous replacement therapy. Most patients harbour circulating autoantibodies against steroid cytochrome P450 21-hydroxylase (21OH) [1], a protein expressed exclusively in the adrenal cortex and an enzyme involved in the biosynthesis of steroid hormones [2]. 21OH is also considered to be the main antigen recognized by autoreactive T cells in AAD, supported by reports on T cell proliferation and interferon (IFN)-γ production in response to 21OH protein and 21OH-derived peptides [3,4]. The underlying cause of AAD is believed to be multi-factorial, involving genes, environmental factors and endogenous components [5]. Among these, genetic associations are by far the most studied, with specific human leucocyte antigen (HLA) class II alleles representing the highest risk factors known to date [1]. Environmental factors and the role played by adrenocortical cells themselves have received less attention.
An increasing number of autoimmune diseases have been linked with virus infections in recent years, including both systemic diseases such as systemic lupus erythematosus (SLE) [6] and organ-specific conditions such as type 1 diabetes (T1D) [7] and autoimmune thyroid disease (AITD) [8]. Virus infections are often accompanied by the induction of a host type I and/or type III IFN response – most notably IFN-α, IFN-β (both type I) and IFN-λ (type III), referred to collectively as types I/III IFNs from here onwards. These are potent anti-viral cytokines that play key roles in combating viral infections [9,10]. Induction of IFNs is typically triggered by the recognition of viral nucleic acids by intracellular host receptors present in the cytosol or endosomes of many cells [11]. In particular, the presence of dsRNA structures, which are formed upon infection with both DNA and RNA viruses [12], can be sensed by Toll-like receptor 3 (TLR-3) and activate the expression of types I/III IFNs [11,13]. IFN-α and IFN-β engage the two subunits of the IFN-α/-β receptor (IFN-AR), IFN-AR1 and IFN-AR2, to form an active dimer and activate the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway [14]. This, in turn, initiates a variety of events associated with an ‘anti-viral state’, including RNA degradation, inhibition of protein synthesis, major histocompatibility complex (MHC) class I up-regulation [10], chemokine secretion [15] and apoptosis [16,17]. Type III IFNs signal through a different heterodimeric receptor complex (referred to as IFN-λR) composed of IFN-λR1 [interleukin (IL)-28Rα] and IFN-λR2 (IL-10Rβ) chains [10,18], yet activate much of the same signalling pathways as type I IFNs [10,18,19].
The IFNs have also been implicated directly in autoimmune diseases through elevated serum levels [7,20], IFN signatures in peripheral blood [21] and disease phenotypes in transgenic mouse models [22,23]. Numerous case reports involving therapeutic use of type I IFNs have substantiated this link. In particular, treatment of chronic hepatitis C virus (HCV) infections with type I IFNs have been reported to precipitate autoimmunity towards endocrine organs such as the pancreas and thyroid gland, as evident by the induction of specific autoantibodies and development of clinically overt disease (T1D and AITD) [24–28].
Suggestive of a potential role in triggering adrenal autoimmunity, IFN-α treatment of chronic HCV infection has been reported to induce the production of 21OH autoantibodies [26]. One report described transient occurrence of 21OH autoantibodies and clinical adrenocortical insufficiency in a woman carrying the HLA class II high-risk genotype for AAD over a 2-year period of IFN-α treatment [29]. Furthermore, exacerbation of subclinical and established Addison's disease with an increased need for glucocorticoid replacement has been noted in individuals on IFN-α therapy [30–32]. The adrenal cortex is also permissive to certain herpesviruses [33], which could trigger a local IFN response in the case of an infection. To delineate potential immunopathological effects of types I and III IFNs on the adrenal cortex in relation to AAD, we performed in-vitro cell culture studies with NCI-H295R adrenocortical carcinoma cells.
Materials and methods
Cell culture
Human adrenocortical carcinoma NCI-H295R cells (referred to as H295R) were cultured in Dulbecco's modified Eagle's medium (DMEM)/F12 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 2·5% Nu-Serum™ IV Serum Replacement (BD Biosciences, San Jose, CA, USA), 1% insulin, transferrin, selenium (ITS+ Premix; BD Biosciences) and 100 U/ml penicillin/100 μg/ml streptomycin (Lonza, Basel, Switzerland) at 37°C with 5% CO2. In stimulation experiments, H295R cells were seeded in supplemented medium at 3 × 105 cells/well in 24-well plates or 5–10 × 105 cells/well in six-well plates, as indicated. The cells were left untreated or stimulated with recombinant cytokines and/or polyinosine-polycytidylic acid [poly (I : C)] for 24 h before being used in downstream applications (detailed below). Final concentrations were 100 μg/ml for poly (I : C) (Sigma-Aldrich, St Louis, MO, USA) and 1 μg/ml for IFN-γ (Biolegend, San Diego, CA, USA), while IFN-α (IFN-α2b; PBL Interferon Source, Piscataway, NJ, USA), IFN-β (IFN-β1a; PBL Interferon Source) and IFN-λ (IL-29/IFNλ1; R&D Systems, Minneapolis, MN, USA) were used at varying concentrations as indicated. Cell culture supernatants were harvested from the 24-well set-up and stored at −80°C until further use.
Immunofluorescence
For staining of IFN-AR1 and IFN-λR1 chains, H295R cells were treated as described previously [34], but without the permeabilization step. Primary antibodies were mouse anti-human IFN-AR1 (R&D Systems; clone no. 85221) and mouse anti-human IFN-λR1 (R&D Systems; clone no. 601106), both applied at a 1:100 dilution. Positive structures were visualized with Alexa 488-conjugated donkey anti-mouse immunoglobulin (Ig)G (Molecular Probes, Invitrogen) secondary antibodies, applied at a 1:1000 dilution. Microscope slides were examined under a Nikon TE2000 wide-field fluorescence microscope equipped with a ×60 objective, and the images were acquired with a Nikon DS-U2/L2 camera controlled by NIS-Elements AR version 3·10 software. The imaging was performed at the Molecular Imaging Center (Fuge, Norwegian Research Council), University of Bergen.
Immunohistochemistry
Slides mounted with 5-μm sections of paraformaldehyde (PFA)-fixed and paraffin-embedded adrenal tissue (Abcam, Cambridge, UK) were deparaffinized in Neo-Clear (Merck, Darmstadt, Germany) and rehydrated in a graded ethanol series and Milli-Q water. The antigen retrieval was performed with ethylenediamine tetraacetic acid (EDTA) buffer pH 8 (Abcam) at 120°C for 20 min in an autoclave. Endogenous peroxidase activity was blocked for 10 min with 0·03% H2O2 (Sigma-Aldrich) in Tris-buffered saline (TBS), pH 7·6. From this point onwards, we used instructions and solutions from the mouse-and rabbit-specific HRP Plus (ABC) detection IHC kit (Abcam) with regard to incubation times and washes. The primary antibodies were diluted in 1% bovine serum albumin (BSA)/TBS (w/v) and incubated overnight at 4°C. All other incubation steps were performed at room temperature (RT). Both IFN-AR1 and IFN-λR1 antibodies (see Immunofluorescence section) were diluted 1:100. Between the different incubations the slides were washed in TBS with 0·025% Triton X-100 (Sigma-Aldrich). The slides were developed with 3-amino-9-ethylcarbazole (AEC; BD Biosciences) for 5 min and counterstained with haematoxylin (Merck) for 1 min. Finally, the slides were washed under running tapwater for 5 min before mounting with IMMU-MOUNT aqueous mounting medium (Thermo Scientific, Runcorn, UK). Slides were examined under an Olympus BX51 bright-field microscope and images acquired with an Olympus DP71 camera controlled by Cell P (version 2·6) software.
Cytotoxicity assay and chemokine production
Cytotoxic effects of IFNs and poly (I : C) on H295R cells were evaluated by a lactate dehydrogenase (LDH) release assay (Clontech, Mountain View, CA, USA) in accordance with the manufacturer's instructions. Relative cytotoxicity was normalized against cells treated with 0·1% Triton X-100 as a measure of maximum cell death.
Chemokine secretion from H295R cells following 24 h IFN and/or poly (I : C) stimulation was measured in culture supernatants using enzyme-linked immunosorbent assay (ELISA) DuoSet kits specific for CXCL9, CXCL10 and CXCL11 (R&D Systems). All assays were performed in accordance with the manufacturer's description, with samples run in duplicate.
Flow cytometry
For the assessment of HLA class I expression, cells stimulated with IFN-α, IFN-β or IFN-λ were detached from 24-well plates by 5 min incubation with TrypLE Select (Life Technologies, Paisley, UK), resuspended in supplemented medium and centrifuged for 5 min at 300 g. Cells were then resuspended in 100 μl of 1% BSA/phosphate-buffered saline (PBS) (w/v) and stained with 2 μg mouse anti-human HLA-A,-B,-C antibodies (Biolegend; clone W6/32) for 30 min on ice. Following two washes with 1% BSA/PBS, cells were again resuspended in 100 μl of 1% BSA/PBS and incubated with Alexa 488-conjugated donkey anti-mouse IgG at a 1:100 dilution for 30 min on ice. Finally, the cells were washed twice with 1% BSA/PBS, resuspended in 200 μl of the same buffer, and analysed immediately on an Accuri C6 flow cytometer.
To detect IFN-AR1 and IFN-λR1 expression, cells grown in six-well plates were fixed with 1·3% PFA/PBS (w/v, pH 7·2), scraped and centrifuged for 5 min at 300 g. Cells were then resuspended in 100 μl 1% BSA/PBS and treated as described above for the detection of HLA class I expression, except for primary antibodies, which were mouse anti-human IFN-AR1 and mouse anti-human IFN-λR1 (see Immunofluorescence section), both diluted at 1:10.
Real-time polymerase chain reaction (RT–PCR)
H295R cells were stimulated in 24-well plates as described above. After 24 h, total RNA was extracted from the cells using the RNeasy Mini Kit (Qiagen) in accordance with the manufacturer's protocol. Sufficient RNA quality was confirmed by running samples on an Agilent 2100 Bioanalyzer. cDNA was synthesized from 500 ng of total RNA using a high-capacity RNA-to-cDNA kit (Applied Biosystems, Warrington, UK). Quantitative RT–PCR was performed on cDNA samples using TaqMan Gene Expression Master Mix (Applied Biosystems) on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems). Primer/probe sets for CYP21A2 (Hs00416901_g1), i.e. the gene encoding 21OH and β-actin (catalogue no. 4333762T) were obtained from Life Technologies. The amplification efficiency of the target (CYP21A2) and reference gene (β-actin) was validated by running standard curves for both primer/probe sets. Relative mRNA expression was calculated by the comparative CT method (Applied Biosystems).
T cell migration and enzyme-linked immunospot (ELISPOT) assay
Peripheral blood mononuclear cells (PBMCs) were isolated from a patient recently diagnosed with AAD by Ficoll-Paque PLUS (GE Healthcare, Little Chalfont, UK) density centrifugation. The patient had signed informed consent approved by the Health Region West ethics committee (149/96-47·96). In order to raise a short-term polyclonal T cell line against 21OH, 7 × 106 PBMCs were resuspended in 200 μl of RPMI-1640 (Lonza) supplemented with 10% AB serum (Sigma-Aldrich), 2 mM L-glutamine, 10 mM HEPES buffer, 1 mM sodium pyruvate, 1% non-essential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin and 5 × 10−5 M 2-mercaptoethanol (Sigma-Aldrich), and stimulated with recombinant 21OH (2·5 μg/ml) purified from baculovirus-infected insect cells, as described previously [35] for 1 h at 37°C with 5% CO2. After pulsing with 21OH, the cells were diluted to 3 × 106 PBMC per ml in cell culture medium enriched with 25 ng/ml IL-7 (Biolegend) and transferred to two wells in a 24-well culture plate. At day 3, cells were expanded with enriched RPMI-1640 (as described above) with 1000 U/ml of IL-2 (Peprotech, London, UK). From this point onwards, IL-2 containing medium was renewed and cells were split as necessary.
The expression of CXCR3 on CD4+ and CD8+ T cells among 21OH-stimulated PBMCs was evaluated by flow cytometry using anti-human CD4 allophycocyanin (APC) (Biolegend; clone OKT4), CD8 APC and CXCR3 phycoerythrin (PE) (Biolegend; clone G025H7) antibodies (staining performed as described above).
On day 20, the ability of CD4+ and CD8+ T cells to migrate towards CXCL10-containing supernatants from H295R cultures was evaluated in a transwell assay. Five μm polycarbonate membrane transwell inserts (Corning, Inc., Corning, MY, USA) in 24-well plates were pre-wetted with 50 μl AIM-V (Life Technologies/Gibco, Carlsbad, CA, USA) for 1 h at 37°C. Supernatants from H295R cultures stimulated with IFN-γ and poly (I : C) to produce large amounts of CXCL10, and supernatants from unstimulated (blank) cultures were heated to 37°C and transferred to the bottom wells. To evaluate the relative contribution of CXCL10 to T cell migration, supernatants were also incubated with 5 μg/ml neutralizing anti-human CXCL10 antibodies (R&D Systems; catalogue no. AF-266-NA) for 1 h prior to the assay. Five × 105 PBMCs from 21OH stimulation were added to each transwell insert and allowed to migrate towards supernatants for 1 h at 37°C. Cells that migrated to the bottom wells were harvested by extensive washing with PBS and stained with anti-human CD4 fluorescein isothiocyanate (FITC) and CD8 APC (Biolegend; clone OKT4 and SK1, respectively) antibodies in 1% BSA/PBS buffer for 30 min on ice. Following washing, the cells were resuspended in a total of 200 μl of the same buffer and analysed immediately on Accuri C6, running the samples for a fixed time-period. Each condition was performed in duplicate.
On the same day, the T cell response against selected 21OH-derived peptides was tested in an IFN-γ enzyme-linked immunospot (ELISPOT) assay (ELISpotPRO; Mabtech, Nacka Strand, Sweden). The 21OH-stimulated PBMCs were initially rested overnight in enriched RPMI-1640 without IL-2 before 3 × 105 cells were restimulated with 21OH-derived peptides (1 μg/ml) alone or in the presence of 10 μg/ml of blocking anti-CD4 (eBioscience, San Diego, CA, USA; clone RPA-T4) or anti-CD8 antibodies (eBioscience; clone OKT8). The cells were then transferred to ELISPOT wells coated with anti-IFN-γ antibodies and analysed in triplicate (105 cells per well). After 16 h of incubation, the ELISPOT assay was carried out as recommended by the manufacturer. Spots were developed for 15 min and counted manually using a dissecting microscope.
Statistics
All quantitative data except from T cell experiments are expressed as means ± standard deviation (s.d.) of three independent experiments. Statistical significance between different treatments and controls was determined by one-way analysis of variance (anova) with Dunnett's post-hoc test. Synergism between two variables was tested for by performing a two-way anova. For both statistical operations, P < 0·05 was considered significant. All tests were performed with GraphPad Prism version 5·02.
Results
Expression of type I and type III IFN receptors in the adrenal cortex
We initially confirmed the expression of IFN-AR1 in H295R adrenocortical carcinoma cells by flow cytometry and extended earlier findings [16] with the demonstration of IFN-λR1 (Fig. 1). The expression of IFN-AR1 was considerably lower than that of IFN-λR1, and this was also evident by immunofluorescence; both IFN-λR1 and IFN-AR1 stained in a dot-like pattern throughout the cells, with the former producing the strongest signal (data not shown).
Figure 1.
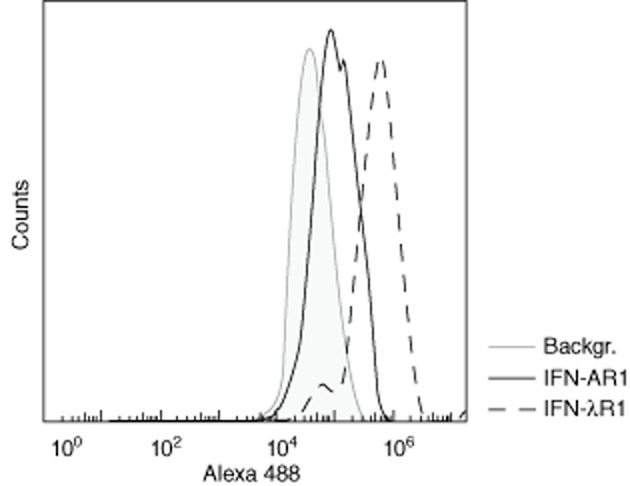
Interferon alpha/beta receptor chain 1 (IFN-AR1) and IFN lambda receptor 1 (IFN-λR1) expression in H295R cells. H295R cells were fixed and stained with mouse anti-human IFN-AR1 (solid line) or mouse anti-human IFN-λR1 (dashed line) followed by Alexa 488-conjugated donkey anti-mouse immunoglobulin (Ig)G and analysed on an Accuri C6 flow cytometer. The figure shows a representative histogram from one of two experiments. Shaded background: cells stained with Alexa 488 anti-mouse IgG only.
To investigate whether these receptors are also expressed in normal adrenal cortex, we performed immunohistochemistry on commercially available human adrenal tissue sections. As shown in Fig. 2, IFN-AR1 (Fig. 2a) and IFN-λR1 (Fig. 2b) were clearly expressed in the adrenal cortex. Both chains stained positively throughout the majority of the sections, covering both the zona glomerulosa and zona fasciculata. Cells from the zona reticularis may also be among the positively stained cells near the right edges of the sections, but the identity of these cells was difficult to determine.
Figure 2.
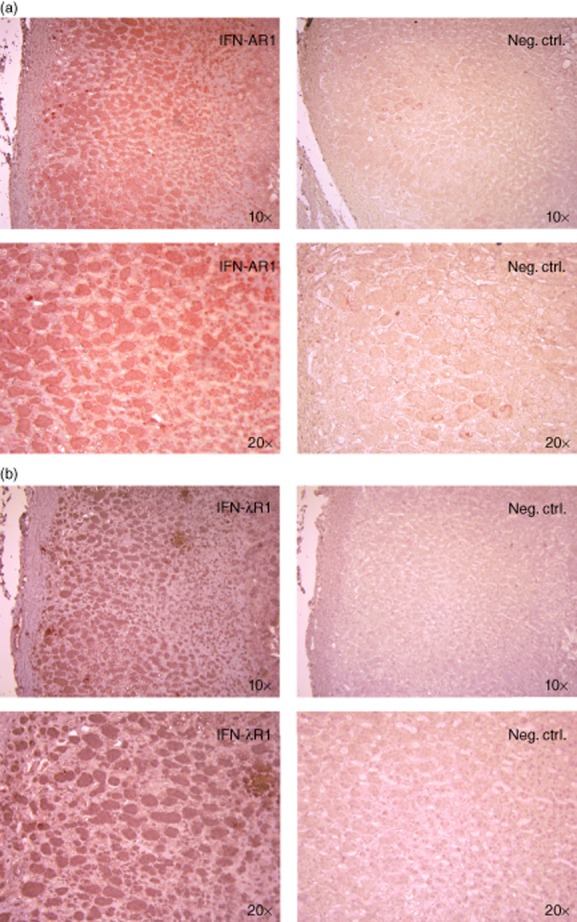
Expression of interferon alpha/beta receptor chain 1 (IFN-AR1) and IFN lambda receptor 1 (IFN-λR1) in human adrenal cortex. Human adrenal tissue sections were stained with (a) anti-human IFN-AR1 or (b) anti-human IFN-λR1 antibodies followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. Positive structures were visualized with 3-amino-9-ethylcarbazole substrate (red) and cell nuclei counterstained with haematoxylin (blue). Neg. ctrl = staining performed with secondary antibody only. Images were obtained with ×10 and ×20 objectives as indicated.
Types I and III IFNs are cytotoxic for H295R adrenocortical cells
To investigate the ability of types I and III IFNs to exert direct cytotoxicity on adrenocortical cells, we stimulated H295R cells with IFN-α, IFN-β and IFN-λ for 24 h and evaluated the membrane integrity by means of LDH release. Both types of IFN induced significant LDH release in a dose-dependent manner (data not shown), reaching a plateau at about 20% when stimulated with 100 U/ml IFN-α, 10 U/ml IFN-β or 100 ng/ml IFN-λ (Fig. 3). Combining these IFNs did not result in any significant increase in LDH release, indicating maximum stimulation of overlapping signalling pathways. We therefore used these IFN concentrations for further experiments.
Figure 3.
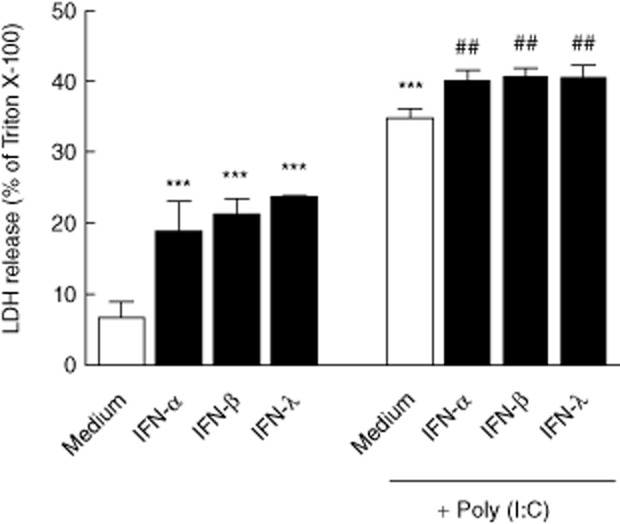
Types I/III interferons exhibit cytotoxic effects on H295R cells. H295R cells were stimulated with interferons (IFNs) and/or polyinosine-polycytidylic acid [poly (I : C)] for 24 h and lactate dehydrogenase (LDH) release, as a measure of direct cytotoxicity, was estimated in culture supernatants. Results are expressed as mean percentage of LDH release normalized to cells treated with 0·1% Triton X-100 (maximum lysis) ± standard deviation (s.d.) of three independent experiments. Asterisks indicate statistical significance versus unstimulated (medium) cells (***P < 0·001), while hash signs indicate statistical significance versus poly (I : C)-stimulated cells (##P < 0·01), as determined by one-way analysis of variance (anova) followed by Dunnett's post-hoc test.
We have shown recently that H295R cells express functional TLR-3 and respond to poly (I : C), an analogue of viral dsRNA, with minimal or no induction of type I IFNs [34]. However, in the case of a viral infection, the adrenal cortex may be exposed to both viral nucleic acids and type I and/or type III IFNs derived from other cells (e.g. infiltrating immune cells or tissue macrophages residing in the adrenal cortex). Stimulation of H295R cells with 100 μg/ml poly (I : C) alone resulted in significantly more LDH release than from cells stimulated with IFNs alone or in combination (P < 0·001, Fig. 3 and data not shown). When poly (I : C) was added in combination with either of the three IFNs, a small but significant increase in LDH release was observed (P < 0·01).
Increased HLA class I expression on H295R cells following IFN stimulation
Hyperexpression of HLA class I, a well-documented effect of IFN stimulation, is evident in autoimmune disorders such as T1D [36] and AITD [37]. To assess such a potential effect of IFNs on adrenocortical cells, we evaluated HLA class I surface expression on H295R cells by flow cytometry after 24 h of stimulation. IFN-α, IFN-β and IFN-λ all enhanced HLA class I expression, resulting in two-to threefold increase in median fluorescence intensity (MFI) compared to unstimulated cells (P < 0·05 for IFN-α and IFN-λ, Fig. 4). The effect of poly (I : C) was less pronounced, causing only a marginal increase in MFI (not significant). Conversely, IFN-γ treatment gave a greater than fourfold increase in MFI (P < 0·001 versus unstimulated cells), and thus the highest expression levels observed.
Figure 4.
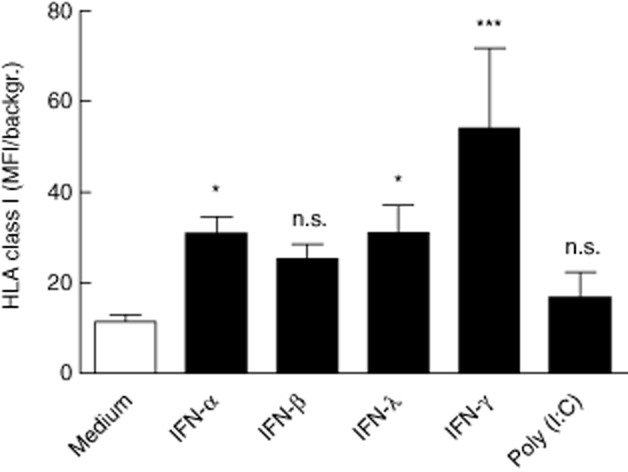
Increased expression of human leucocyte antigen (HLA) class I on H295R cells following interferon stimulation. After 24 h of incubation with the indicated stimuli, H295R cells were stained with mouse anti-human HLA-A,-B,-C antibodies followed by Alexa 488-conjugated donkey anti-mouse immunoglobulin (Ig)G, and analysed on an Accuri C6 flow cytometer. Results are expressed as mean fold increase in mean fluorescence intensity (MFI) compared to background [cells stained with Alexa 488 anti-mouse immunoglobulin (Ig)G only] ± standard deviation (s.d.) of three independent experiments. Asterisks indicate statistical significance versus unstimulated (medium) cells (*P < 0·05; ***P < 0·001) as determined by one-way analysis of variance (anova) followed by Dunnett's post-hoc test.
Stimulation of 21-hydroxylase mRNA expression by IFNs and TLR-3 activation
Increased expression of autoantigens may serve as a contributing factor to the induction of autoimmune responses [37,38]. Therefore, we determined the effect of IFNs [and poly (I : C)] on the expression of 21-hydroxylase (21OH) mRNA in H295R cells by quantitative PCR. Stimulation with types I/III IFNs increased 21OH expression two-to threefold compared to unstimulated cells (P < 0·01 for IFN-α and P < 0·001 for IFN-β and IFN-λ, Fig. 5). In contrast to HLA class I, IFN-γ had little effect on 21OH expression, while poly (I : C) stimulation resulted in a threefold up-regulation of 21OH (P < 0·001). Co-stimulation of H295R cells with either IFN-γ or poly (I : C), along with types I/III IFNs, did not enhance 21OH expression compared to single-stimulated cells (data not shown).
Figure 5.
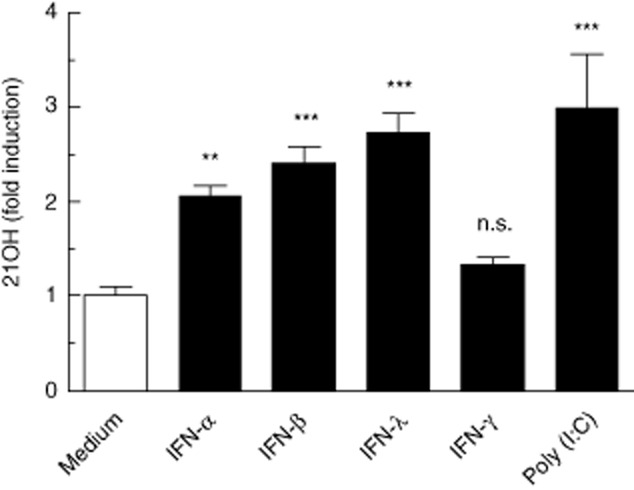
Interferons (IFNs) and polyinosine-polycytidylic acid [poly (I : C)] stimulates 21OH mRNA expression in H295R cells. Total RNA was isolated from H295R cells following 24 h stimulation with IFNs or poly (I : C). 21OH expression was measured by reverse transcription–quantitative polymerase chain reaction (RT–qPCR) and normalized to β-actin. Results are expressed as mean fold induction over unstimulated cells (medium) ± standard deviation (s.d.) of three independent experiments. Asterisks indicate statistical significance versus unstimulated (medium) cells (**P < 0·01; ***P < 0·001) as determined by one-way analysis of variance (anova) followed by Dunnett's post-hoc test.
Types I and III IFNs synergize with poly (I : C) and IFN-γ to induce chemokine secretion
Patients with AAD have elevated serum levels of the CXC chemokine ligand 10 (CXCL10) [34,39], which has also been implicated in other endocrine autoimmune conditions [40]. As CXCL10 is secreted by adrenocortical cells upon stimulation with proinflammatory cytokines IFN-γ and tumour necrosis factor (TNF)-α [39], and following TLR-3 stimulation [34], we investigated whether types I/III IFNs could have similar effects, as has been shown for thyrocytes in the context of AITD [15]. IFN-α, IFN-β and IFN-λ alone were not able to induce significant amounts of CXCL10 from H295R cells following 24 h of stimulation (data not shown). However, when combined with either poly (I : C) or IFN-γ (1 μg/ml), CXCL10 secretion was increased several-fold compared to poly (I : C) and IFN-γ treatment alone (Fig. 6). Thus, there appeared to be a synergistic effect of types I/III IFNs and both poly (I : C) and IFN-γ, although not statistically significant for all combinations as calculated by two-way anova (Fig. 6).
Figure 6.
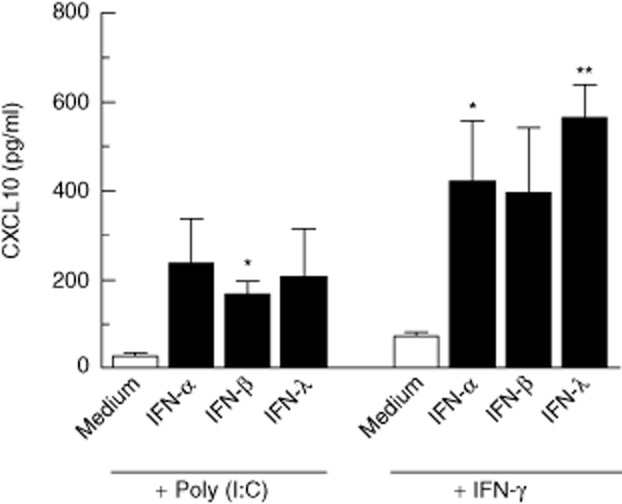
CXC chemokine ligand 10 (CXCL10) production by H295R cells upon co-stimulation with interferons and polyinosine-polycytidylic acid [poly (I : C)]. H295R cells were incubated with the indicated combinations of stimuli for 24 h and CXCL10 production was measured in culture supernatants by enzyme-linked immunosorbent assay (ELISA). Results are expressed as mean CXCL10 concentration ± standard deviation (s.d.) of three independent experiments. Asterisks indicate synergistic effects of two specific stimuli (* P < 0·05; ** P < 0·01) as determined by two-way analysis of variance (anova).
H295R cells did not produce significant amounts of CXCL11 following stimulation with the above-mentioned ligands (data not shown). For CXCL9, only IFN-γ had a slight stimulating effect (P < 0·05), yet the levels were still very low (9·1 ± 1·2 pg/ml versus 5·7 ± 1·8 pg/ml for unstimulated cells), and no significant increases were observed in co-stimulation experiments (data not shown).
CXCL10 produced by H295R cells induces migration of 21OH-stimulated T cells
To evaluate whether CXCL10 produced by H295R cells is chemotactic to CXCR3-expressing T cells, we stimulated PBMCs from a patient with confirmed AAD with recombinant 21OH and measured the migration of CD4+ and CD8+ T cells towards H295R supernatants containing CXCL10. On the day of the assay, the relative expression of CXCR3 among CD4+ and CD8+ T cells from 21OH stimulation cultures were 63·4 and 84·2%, respectively (Fig. 7a). Using a transwell culture system, we observed increased migration of both CD4+ (Fig. 7b) and CD8+ (Fig. 7c) T cells towards supernatants from poly (I : C)-and IFN-γ-stimulated H295R cells (CXCL10 concentration: 1568·6 pg/ml by ELISA) versus supernatants from unstimulated cells (CXCL10 undetectable by ELISA). A similar trend was seen in an experiment performed with supernatants from co-stimulation with types I/III IFNs and IFN-γ (data not shown). The specific migration towards these supernatants was not as pronounced, yet this could be a reflection of lower CXCL10 concentration as measured by ELISA (below 200 pg/ml). The increase in CD4+ and CD8+ migration in the present experiment was neutralized by preincubating supernatants with monoclonal anti-CXCL10 antibodies (Fig. 7b,c).
Figure 7.
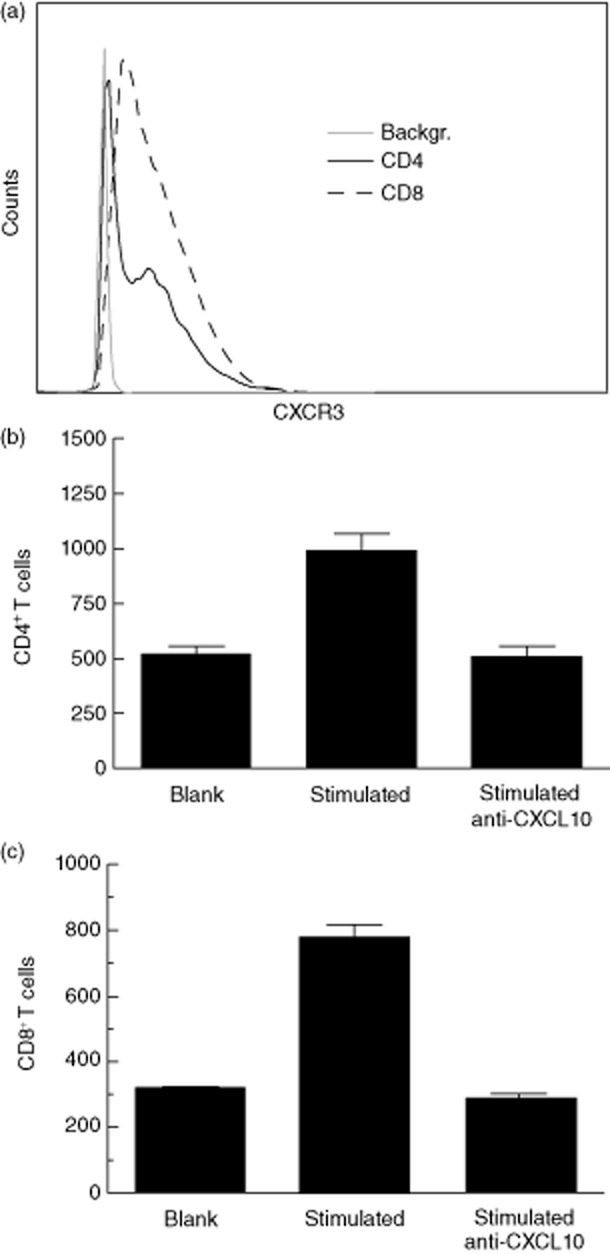
Increased T cell migration towards CXC chemokine ligand 10 (CXCL10)-containing supernatants from H295R cells. Peripheral blood mononuclear cells (PBMCs) from a patient with autoimmune Addison's disease (AAD) were stimulated with recombinant 21OH and expanded with interleukin (IL)-2. (a) The expression of CXCR3 among CD4+ (solid line) and CD8+ T cells (dashed line) was measured by flow cytometry. The ability of CD4+ (b) and CD8+ (c) T cells to migrate towards supernatants from H295R cells was tested in a 1 h transwell assay; 5 × 105 PBMCs were loaded in each transwell insert, and supernatants from H295R cells stimulated to produce CXCL10 (stimulated) or supernatants from unstimulated cells (blank) were added to the bottom wells. Migrated cells were harvested, stained with anti-CD4 and anti-CD8 antibodies and analysed by flow cytometry. The relative contribution of CXCL10 to T cell migration was evaluated by preincubating ‘stimulated’ supernatants with anti-CXCL10 for 1 h prior to the assay (right bars). Results are expressed as number of migrated cells ± standard deviation (s.d.) of two parallels.
The specificity of the T cell line was confirmed by ELISPOT with selected 21OH-derived peptides (data not shown). The highest frequency of peptide-specific T cells was detected against a peptide consisting of amino acid residues 209–226 of 21OH [416 ± 52 spot-forming units (SFU) per 1 million cells]. This response was partially blocked in the presence of both anti-CD4 (117 ± 34 SFU per 1 million cells) and anti-CD8 (193 ± 60 SFU per 1 million cells) antibodies, suggesting the presence of both CD4+ and CD8+ reactive T cells.
Discussion
While most autoimmune diseases are founded on a genetic basis, there is probably still the need for a precipitating event (e.g. a microbial infection, vaccination or chemical exposure) to initiate an autoimmune reaction in susceptible individuals. Viral infections induce a strong inflammatory response with activation of pattern recognition receptors and induction of types I/III IFNs, and a growing number of links between autoimmune disease and both viruses and types I/III IFNs suggests that these factors may promote an environment that is suited for autoimmune responses.
The adrenal cortex expresses several molecules of immunological importance, notably many of the TLRs [41,42], and a range of cytokines and chemokines under various conditions [34,43]. We hypothesize that exposure to types I/III IFNs through IFN therapy or as part of an infection could render the adrenal cortex vulnerable to autoimmune insult in genetically susceptible individuals. To evaluate such potential mechanisms, we studied the effect of types I/III IFNs on relevant parameters in adrenocortical cells. With the lack of primary adrenocortical cells and tissue from patients with AAD, which is unavailable for practical and ethical reasons, we chose to use the well-differentiated and-characterized H295R adrenocortical carcinoma cell line for our studies.
The presence of both IFN-AR1 and IFN-λR1 was demonstrated in H295R cells by flow cytometry and immunofluorescence analyses. Upon the identification of IFN-λR in 2003, IFN-λR1 expression was detected at RNA level in the adrenal gland [18]. In this study, we showed by immunohistochemistry that IFN-λR1 protein is expressed throughout the normal human adrenal cortex, including the two outer cell layers and possibly also the zona reticularis. We also showed that IFN-AR1 is expressed in a pattern similar to IFN-λR1 in adrenal sections, and is more comparable to IFN-λR1 in terms of staining intensity than shown in H295R cells. Induction of cell death is a well-known effect of type I IFNs, and a trait shared with type III IFNs [19]. Accordingly, types I/III IFNs exerted significant cytotoxicity on H295R cells as measured by LDH release. Our data are in concordance with those of Koetsveld et al. showing an approximately 10-fold higher potency of IFN-β than IFN-α [16]. The fact that similar maximum levels of LDH release was reached by types I and III IFN treatment, and not increased by co-stimulation, probably reflects converging signalling pathways activated by the different IFNs.
Some case reports have described the development of adrenocortical insufficiency in connection with herpesvirus infections [44–47], and among these, one reported extensive necrosis in the adrenal cortex caused by herpes simplex virus [44]. Having previously demonstrated the expression of TLR-3 in H295R cells, which was also reported recently in the adrenal gland of a patient with co-existing Cushing syndrome and AAD [42], we tested the ability of poly (I : C), a TLR-3 stimulant able to mimic viral infections [48], to exert cytotoxicity on H295R cells. The effect of poly (I : C) was significantly greater than types I/III IFNs, which could be due to nuclear factor kappa B (NF-κB)-mediated apoptosis, as has been shown for pancreatic beta cells [49]. A slight but significant increase in LDH release was noted upon co-stimulation with poly (I : C) and types I/III IFNs compared to poly (I : C) alone. This effect was less than their individual effects combined, possibly reflecting the activation of overlapping signalling pathways. In relation to IFN-α therapy, a direct cytotoxic effect of IFN-α on the adrenal cortex, resulting in loss of hormone-producing cells, may contribute to the increased need for glucocorticoid replacement observed in some patients with subclinical or established AAD [30–32]. In individuals with a normal adrenocortical function this would probably only have a minor influence on the hormone-producing capacity of the organ, as clinical adrenocortical insufficiency does not occur before 90% of the hormone-producing cells are destroyed [50], and progenitor cells from the adrenal capsule or subcapsular region might replace the lost cells [51].
Upon virus infections, types I/III IFN-mediated up-regulation of HLA class I expression facilitates the presentation of virus-derived peptides and killing of infected cells by cytotoxic T lymphocytes (CTLs). While this represents an important mechanism to eliminate viral infections, such mechanisms may promote the development of autoimmunity in susceptible individuals. In line with this notion, high HLA class I expression was found to correspond with the onset of disease in a mouse model of virus-induced T1D [52]. Hyperexpression of HLA class I by insulin-containing cells of the pancreas is also a hallmark of inflamed human islets in patients with T1D [53,54]. Furthermore, the co-ordinate up-regulation of HLA class I and autoantigens by IFNs, as was demonstrated in a transgenic mouse model for thyroid autoimmunity and in human thyrocytes [23,55], could vastly increase the likelihood of autoimmune responses to take place.
In H295R cells, types I/III IFNs increased both HLA class I (at protein level) and 21OH expression (mRNA). Meanwhile, IFN-γ and poly (I : C) appeared to only affect one or the other, with IFN-γ inducing high levels of HLA class I and poly (I : C) promoting the up-regulation of 21OH. These data may suggest that exposure to types I/III IFNs could evoke autoimmune reactions in individuals predisposed to AAD through increased presentation of 21OH peptides on adrenocortical HLA class I molecules. Viral dsRNA structures formed under infection of the adrenal cortex could contribute to this increased presentation by stimulating 21OH expression, and IFN-γ produced by infiltrating autoreactive T cells could help to sustain high levels of HLA class I. In support of this hypothesis, adenovirus infection in H295R cells was shown previously to increase the expression of several key steroidogenic enzymes, including 21OH [56]. Upon cessation of types I/III IFN stimulation (in the case of IFN therapy withdrawal or virus infection clearance), T cell-derived IFN-γ could still stimulate HLA class I expression, while increasing levels of adrenocorticotrophic hormone (ACTH), as a result of autoimmune adrenocortical failure, would up-regulate 21OH [57].
Along with their potential direct effects on adrenocortical cells, types I/III IFNs could further act to shape an immune response against the adrenal cortex through mechanisms similar to those that promote viral elimination. Specifically, the presence of type I IFNs (and possibly type III IFNs) could facilitate cross-presentation of 21OH peptides by dendritic cells (DCs) following uptake of apoptotic cells (e.g. as a result of IFN-mediated cytotoxicity). In a normal situation, DCs sampling apoptotic material would probably reside in an immature state with a lack of co-stimulatory molecules on their surface and promote tolerance to adrenal cortex self-antigens (e.g. by inducing T cell anergy or deletion) [58]. As suggested by Banchereau and Pascural for SLE, it could be speculated that type I IFN stimulation might lead to improper activation of these otherwise tolerogenic DCs, cross-priming of naive autoreactive T cells and the development of effector CTLs that could inflict damage on the target organ [58]. In individuals with a genetic susceptibility to AAD, such a mechanism could be envisioned as the initial event that breaks tolerance to self-antigens of the adrenal cortex (i.e. 21OH or other targets), triggering an autoimmune attack.
CXCL10 is a ligand for the chemokine receptor CXCR3, which is highly expressed on activated T cells, and is associated with the recruitment of T cells to sites of inflammation [59]. In both T1D and AITD, patients have elevated serum levels of CXCL10 [60,61] and there is strong evidence to suggest that cells within the affected organs are responsible, at least in part, for the recruitment of CXCR3-expressing T cells through the production of CXCL10 [62,63]. Interestingly, CXCL10 expression was increased markedly in thyrocytes from transgenic mice overexpressing IFN-α in the thyroid gland [23], and was also secreted by human primary thyrocytes upon in-vitro stimulation with type I IFNs [15]. We have previously suggested a potential amplification loop for CXCL10 production by adrenocortical cells in AAD involving viral TLR-3 stimulation [poly (I : C)] and proinflammatory cytokines (IFN-γ and TNF-α) derived from infiltrating autoreactive T cells [34]. Investigating whether types I/III IFNs could play a part in such a circuit, we did not observe significant CXCL10 production upon stimulation of H295R cells with these IFNs alone. However, in combination with poly (I : C) or IFN-γ, types I/III IFNs exhibited a stimulating effect on CXCL10 production, although not statistically significant for all combinations. With regard to other CXCR3-binding chemokines, stimulation of H295R cells with types I/III IFNs, poly (I : C) or IFN-γ (alone or in combination) had little effect on the production of chemokines CXCL9 and CXCL11. Although it remains to be seen whether this selective induction of CXCL10 also applies to primary adrenocortical cells, this could reflect a specific mechanism involving types I/III IFNs that may be operative in AAD.
Using a polyclonal T cell line raised against 21OH, we assessed the functionality of CXCL10 produced by H295R cells in a transwell migration assay. We observed increased migration of both CD4+ and CD8+ T cells towards supernatants from H295R cells stimulated to produce CXCL10 versus supernatants from unstimulated cells. The relative increase in migration towards CXCL10-containing supernatants versus blank supernatants was slightly bigger for CD8+ T cells (∼2·4-fold) than CD4+ T cells (∼1·9-fold), possibly reflecting the higher percentage of CXCR3-positive cells within the CD8+ population (Fig. 7a). By preincubating supernatants with anti-CXCL10, the increase in both CD4+ and CD8+ T cell migration was neutralized, suggesting that the CXCL10 produced by H295R cells is indeed chemotactic for T cells.
In the case of a viral infection in the adrenal cortex, types I/III IFNs may thus act in concert with viral dsRNA structures to induce (in the initial phase) and/or increase the infiltration of immune cells by means of CXCL10 secretion. This could lead to the recruitment of potentially harmful lymphocytes (in individuals predisposed to AAD) that produces proinflammatory cytokines such as IFN-γ and TNF-α, whose action on adrenocortical cells would serve to amplify the CXCL10 production, thus establishing a vicious cycle that could grossly expand the mononuclear infiltrate.
In summary, we have demonstrated the expression of types I and III IFN receptors in the human adrenal cortex, and shown specific effects of types I/III IFNs, both alone and in combination with TLR-3 or IFN-γ stimulation, on adrenocortical cells that may have implications for the pathogenesis of AAD. Accordingly, we provide potential mechanisms by which types I/III IFNs triggered by a viral infection or IFN therapy could serve to initiate or fuel an ongoing autoimmune attack on the adrenal cortex in genetically susceptible individuals. Taken together, exposure of the adrenal cortex to types I/III IFNs could lead to the co-ordinate up-regulation 21OH (autoantigen) and HLA class I expression, increased recruitment of immune cells through enhanced CXCL10 production and apoptosis/necrosis of adrenocortical cells. Increased autoantigen expression combined with a cytotoxic effect on adrenocortical cells would increase the probability of 21OH peptides being cross-presented to CD8+ T cells by dendritic cells (DCs) which, in such a proinflammatory environment, could lead to the activation and expansion of 21OH-specific autoreactive T cells. Such cells could be attracted to the adrenal cortex by CXCL10 (and other chemokines), and effectively kill adrenocortical cells expressing high levels of HLA class I.
For future studies it will be important to establish whether the present findings are evident in the adrenal cortex of patients with AAD. In the lack of primary cells from patients, this could be answered by performing post-mortem analysis of diseased tissue. Such analyses may reveal the possible hyperexpression of HLA class I and 21OH, as well as the production of CXCL10 by the adrenal cortex and the presence of CXCR3-expressing T cells. The effect of types I/III IFNs on cellular responses against 21OH can be studied using isolated T cells and antigen-presenting cells from patients with AAD.
Acknowledgments
The human adrenocortical carcinoma cell line NCI-H295R was a kind gift from Professor Marit Bakke (Institute of Biomedicine, University of Bergen, Norway). This work was supported by funding from the Western Norway Regional Health Authority (grant no. 911701) and the Research Council of Norway (grant no. 213704).
Disclosure
The authors declare no conflicts of interest.
Author contributions
A. H. and E. B. designed the study and A. H., K. E., L. B. and E. B. performed the experiments. All authors contributed to the analysis of the data and writing of the paper.
References
- 1.Erichsen MM, Lovas K, Skinningsrud B, et al. Clinical, immunological, and genetic features of autoimmune primary adrenal insufficiency: observations from a Norwegian registry. J Clin Endocrinol Metab. 2009;94:4882–4890. doi: 10.1210/jc.2009-1368. [DOI] [PubMed] [Google Scholar]
- 2.Winqvist O, Karlsson FA, Kampe O. 21-Hydroxylase, a major autoantigen in idiopathic Addison's disease. Lancet. 1992;339:1559–1562. doi: 10.1016/0140-6736(92)91829-w. [DOI] [PubMed] [Google Scholar]
- 3.Bratland E, Skinningsrud B, Undlien DE, Mozes E, Husebye ES. T cell responses to steroid cytochrome P450 21-hydroxylase in patients with autoimmune primary adrenal insufficiency. J Clin Endocrinol Metab. 2009;94:5117–5124. doi: 10.1210/jc.2009-1115. [DOI] [PubMed] [Google Scholar]
- 4.Rottembourg D, Deal C, Lambert M, et al. 21-Hydroxylase epitopes are targeted by CD8 T cells in autoimmune Addison's disease. J Autoimmun. 2010;35:309–315. doi: 10.1016/j.jaut.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Bratland E, Husebye ES. Cellular immunity and immunopathology in autoimmune Addison's disease. Mol Cell Endocrinol. 2011;336:180–189. doi: 10.1016/j.mce.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 6.James JA, Kaufman KM, Farris AD, Taylor-Albert E, Lehman TJ, Harley JB. An increased prevalence of Epstein–Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J Clin Invest. 1997;100:3019–3026. doi: 10.1172/JCI119856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chehadeh W, Weill J, Vantyghem MC, et al. Increased level of interferon-alpha in blood of patients with insulin-dependent diabetes mellitus: relationship with coxsackievirus B infection. J Infect Dis. 2000;181:1929–1939. doi: 10.1086/315516. [DOI] [PubMed] [Google Scholar]
- 8.Antonelli A, Ferri C, Pampana A, et al. Thyroid disorders in chronic hepatitis C. Am J Med. 2004;117:10–13. doi: 10.1016/j.amjmed.2004.01.023. [DOI] [PubMed] [Google Scholar]
- 9.Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology. 2006;44:896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- 10.Kotenko SV, Gallagher G, Baurin VV, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 11.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 12.Jacobs BL, Langland JO. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology. 1996;219:339–349. doi: 10.1006/viro.1996.0259. [DOI] [PubMed] [Google Scholar]
- 13.Coccia EM, Severa M, Giacomini E, et al. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol. 2004;34:796–805. doi: 10.1002/eji.200324610. [DOI] [PubMed] [Google Scholar]
- 14.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 15.Antonelli A, Ferrari SM, Fallahi P, et al. Interferon-alpha,-beta and-gamma induce CXCL9 and CXCL10 secretion by human thyrocytes: modulation by peroxisome proliferator-activated receptor-gamma agonists. Cytokine. 2010;50:260–267. doi: 10.1016/j.cyto.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 16.van Koetsveld PM, Vitale G, de Herder WW, et al. Potent inhibitory effects of type I interferons on human adrenocortical carcinoma cell growth. J Clin Endocrinol Metab. 2006;91:4537–4543. doi: 10.1210/jc.2006-0620. [DOI] [PubMed] [Google Scholar]
- 17.Chawla-Sarkar M, Leaman DW, Borden EC. Preferential induction of apoptosis by interferon (IFN)-beta compared with IFN-alpha2: correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin Cancer Res. 2001;7:1821–1831. [PubMed] [Google Scholar]
- 18.Sheppard P, Kindsvogel W, Xu W, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 19.Li W, Lewis-Antes A, Huang J, Balan M, Kotenko SV. Regulation of apoptosis by type III interferons. Cell Prolif. 2008;41:960–979. doi: 10.1111/j.1365-2184.2008.00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Q, Yang Q, Lourenco E, Sun H, Zhang Y. Interferon-lambda1 induces peripheral blood mononuclear cell-derived chemokines secretion in patients with systemic lupus erythematosus: its correlation with disease activity. Arthritis Res Ther. 2011;13:R88. doi: 10.1186/ar3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Baarsen LG, van der Pouw Kraan TC, Kragt JJ, et al. A subtype of multiple sclerosis defined by an activated immune defense program. Genes Immun. 2006;7:522–531. doi: 10.1038/sj.gene.6364324. [DOI] [PubMed] [Google Scholar]
- 22.Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA. 2008;105:12439–12444. doi: 10.1073/pnas.0806439105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akeno N, Smith EP, Stefan M, et al. IFN-alpha mediates the development of autoimmunity both by direct tissue toxicity and through immune cell recruitment mechanisms. J Immunol. 2011;186:4693–4706. doi: 10.4049/jimmunol.1002631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Betterle C, Fabris P, Zanchetta R, et al. Autoimmunity against pancreatic islets and other tissues before and after interferon-alpha therapy in patients with hepatitis C virus chronic infection. Diabetes Care. 2000;23:1177–1181. doi: 10.2337/diacare.23.8.1177. [DOI] [PubMed] [Google Scholar]
- 25.Fabris P, Betterle C, Floreani A, et al. Development of type 1 diabetes mellitus during interferon alfa therapy for chronic HCV hepatitis. Lancet. 1992;340:548. doi: 10.1016/0140-6736(92)91744-s. [DOI] [PubMed] [Google Scholar]
- 26.Wesche B, Jaeckel E, Trautwein C, et al. Induction of autoantibodies to the adrenal cortex and pancreatic islet cells by interferon alpha therapy for chronic hepatitis C. Gut. 2001;48:378–383. doi: 10.1136/gut.48.3.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayashi M, Kataoka Y, Tachikawa K, Koguchi H, Tanaka H. Dual onset of type 1 diabetes mellitus and Graves' disease during treatment with pegylated interferon alpha-2b and ribavirin for chronic hepatitis C. Diabetes Res Clin Pract. 2009;86:e19–21. doi: 10.1016/j.diabres.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Yamazaki M, Sato A, Takeda T, Komatsu M. Distinct clinical courses in type 1 diabetes mellitus induced by peg-interferon-alpha treatment for chronic hepatitis C. Intern Med. 2010;49:403–407. doi: 10.2169/internalmedicine.49.2656. [DOI] [PubMed] [Google Scholar]
- 29.Krysiak R, Boldys A, Okopien B. Autoimmune polyglandular syndrome type 2 induced by treatment with interferon alpha. Am J Med Sci. 2011;341:504–507. doi: 10.1097/MAJ.0b013e31820ff7af. [DOI] [PubMed] [Google Scholar]
- 30.Oshimoto K, Shimizu H, Sato N, Mori M. [A case of Addison's disease which became worse during interferon therapy: insulin secretion under hyposmolarity] Nihon Naibunpi Gakkai Zasshi. 1994;70:511–516. doi: 10.1507/endocrine1927.70.5_511. [DOI] [PubMed] [Google Scholar]
- 31.Tran HA, Song S, Lojewski RJ, Reeves GE. Exacerbation of hepatitis C induced subclinical hypoadrenalism by interferon-alpha2beta: a case report. Cases J. 2008;1:157. doi: 10.1186/1757-1626-1-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knost JA, Sherwin S, Abrams P, Oldham RK. Increased steroid dependence after recombinant leucocyte interferon therapy. Lancet. 1981;2:1287–1288. doi: 10.1016/s0140-6736(81)91522-1. [DOI] [PubMed] [Google Scholar]
- 33.Paolo WF, Jr, Nosanchuk JD. Adrenal infections. Int J Infect Dis. 2006;10:343–353. doi: 10.1016/j.ijid.2005.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bratland E, Hellesen A, Husebye ES. Induction of CXCL10 chemokine in adrenocortical cells by stimulation through toll-like receptor 3. Mol Cell Endocrinol. 2013;365:75–83. doi: 10.1016/j.mce.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Bratland E, Bredholt G, Mellgren G, Knappskog PM, Mozes E, Husebye ES. The purification and application of biologically active recombinant steroid cytochrome P450 21-hydroxylase: the major autoantigen in autoimmune Addison's disease. J Autoimmun. 2009;33:58–67. doi: 10.1016/j.jaut.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 36.Itoh N, Hanafusa T, Miyazaki A, et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993;92:2313–2322. doi: 10.1172/JCI116835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang H, Li X, Lin L, et al. Upregulation of thyroid transcription factor-1 and human leukocyte antigen class I in Hashimoto's disease providing a clinical evidence for possible triggering autoimmune reaction. Eur J Endocrinol. 2011;164:795–800. doi: 10.1530/EJE-10-0960. [DOI] [PubMed] [Google Scholar]
- 38.Arvan P, Pietropaolo M, Ostrov D, Rhodes CJ. Islet autoantigens: structure, function, localization, and regulation. Cold Spring Harbour Perspect Med. 2012;2:a007658. doi: 10.1101/cshperspect.a007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rotondi M, Falorni A, De Bellis A, et al. Elevated serum interferon-gamma-inducible chemokine-10/CXC chemokine ligand-10 in autoimmune primary adrenal insufficiency and in vitro expression in human adrenal cells primary cultures after stimulation with proinflammatory cytokines. J Clin Endocrinol Metab. 2005;90:2357–2363. doi: 10.1210/jc.2004-1062. [DOI] [PubMed] [Google Scholar]
- 40.Rotondi M, Chiovato L, Romagnani S, Serio M, Romagnani P. Role of chemokines in endocrine autoimmune diseases. Endocr Rev. 2007;28:492–520. doi: 10.1210/er.2006-0044. [DOI] [PubMed] [Google Scholar]
- 41.Kanczkowski W, Zacharowski K, Wirth MP, Ehrhart-Bornstein M, Bornstein SR. Differential expression and action of Toll-like receptors in human adrenocortical cells. Mol Cell Endocrinol. 2009;300:57–65. doi: 10.1016/j.mce.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 42.Colucci R, Jimenez RE, Farrar W, Malgor R, Kohn L, Schwartz FL. Coexistence of Cushing syndrome from functional adrenal adenoma and Addison disease from immune-mediated adrenalitis. J Am Osteopath Assoc. 2012;112:374–379. [PubMed] [Google Scholar]
- 43.Bornstein SR, Chrousos GP. Clinical review 104: adrenocorticotropin (ACTH)-and non-ACTH-mediated regulation of the adrenal cortex: neural and immune inputs. J Clin Endocrinol Metab. 1999;84:1729–1736. doi: 10.1210/jcem.84.5.5631. [DOI] [PubMed] [Google Scholar]
- 44.Brain RT, Pugh RC, Dudgeon JA. Adrenal necrosis in generalized herpes simplex. Arch Dis Child. 1957;32:120–126. doi: 10.1136/adc.32.162.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hertel NT, Jacobsen BB, Pedersen FK, Heilmann C. Adrenocortical insufficiency associated with Epstein–Barr virus infection in a patient with the Wiskott–Aldrich syndrome. Eur J Pediatr. 1987;146:603–604. doi: 10.1007/BF02467365. [DOI] [PubMed] [Google Scholar]
- 46.Schmitt K, Deutsch J, Tulzer G, Meindi R, Aberle S. Autoimmune hepatitis and adrenal insufficiency in an infant with human herpesvirus-6 infection. Lancet. 1996;348:966. doi: 10.1016/s0140-6736(05)65385-8. [DOI] [PubMed] [Google Scholar]
- 47.Guinovart RM, Carrascosa JM, Bielsa I, Rodriguez C, Ferrandiz C. A black tongue in a young woman. Clin Exp Dermatol. 2011;36:429–430. doi: 10.1111/j.1365-2230.2010.03946.x. [DOI] [PubMed] [Google Scholar]
- 48.Caskey M, Lefebvre F, Filali-Mouhim A, et al. Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J Exp Med. 2011;208:2357–2366. doi: 10.1084/jem.20111171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu D, Cardozo AK, Darville MI, Eizirik DL. Double-stranded RNA cooperates with interferon-gamma and IL-1 beta to induce both chemokine expression and nuclear factor-kappa B-dependent apoptosis in pancreatic beta-cells: potential mechanisms for viral-induced insulitis and beta-cell death in type 1 diabetes mellitus. Endocrinology. 2002;143:1225–1234. doi: 10.1210/endo.143.4.8737. [DOI] [PubMed] [Google Scholar]
- 50.Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison's disease. BMJ. 1978;1:1591–1592. doi: 10.1136/bmj.1.6127.1591-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wood MA, Hammer GD. Adrenocortical stem and progenitor cells: unifying model of two proposed origins. Mol Cell Endocrinol. 2011;336:206–212. doi: 10.1016/j.mce.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohashi PS, Oehen S, Aichele P, et al. Induction of diabetes is influenced by the infectious virus and local expression of MHC class I and tumor necrosis factor-alpha. J Immunol. 1993;150:5185–5194. [PubMed] [Google Scholar]
- 53.Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG, Gamble DR. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med. 1985;313:353–360. doi: 10.1056/NEJM198508083130604. [DOI] [PubMed] [Google Scholar]
- 54.Foulis AK, Farquharson MA, Meager A. Immunoreactive alpha-interferon in insulin-secreting beta cells in type 1 diabetes mellitus. Lancet. 1987;2:1423–1427. doi: 10.1016/s0140-6736(87)91128-7. [DOI] [PubMed] [Google Scholar]
- 55.Stefan M, Jacobson EM, Huber AK, et al. Novel variant of thyroglobulin promoter triggers thyroid autoimmunity through an epigenetic interferon alpha-modulated mechanism. J Biol Chem. 2011;286:31168–31179. doi: 10.1074/jbc.M111.247510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matkovic U, Pacenti M, Trevisan M, Palu G, Barzon L. Investigation on human adrenocortical cell response to adenovirus and adenoviral vector infection. J Cell Physiol. 2009;220:45–57. doi: 10.1002/jcp.21727. [DOI] [PubMed] [Google Scholar]
- 57.Xing Y, Parker CR, Edwards M, Rainey WE. ACTH is a potent regulator of gene expression in human adrenal cells. J Mol Endocrinol. 2010;45:59–68. doi: 10.1677/JME-10-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 59.Qin S, Rottman JB, Myers P, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101:746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Antonelli A, Fallahi P, Rotondi M, et al. Increased serum CXCL10 in Graves' disease or autoimmune thyroiditis is not associated with hyper-or hypothyroidism per se, but is specifically sustained by the autoimmune, inflammatory process. Eur J Endocrinol. 2006;154:651–658. doi: 10.1530/eje.1.02137. [DOI] [PubMed] [Google Scholar]
- 61.Shimada A, Morimoto J, Kodama K, et al. Elevated serum IP-10 levels observed in type 1 diabetes. Diabetes Care. 2001;24:510–515. doi: 10.2337/diacare.24.3.510. [DOI] [PubMed] [Google Scholar]
- 62.Uno S, Imagawa A, Saisho K, et al. Expression of chemokines, CXC chemokine ligand 10 (CXCL10) and CXCR3 in the inflamed islets of patients with recent-onset autoimmune type 1 diabetes. Endocr J. 2010;57:991–996. doi: 10.1507/endocrj.k10e-076. [DOI] [PubMed] [Google Scholar]
- 63.Garcia-Lopez MA, Sancho D, Sanchez-Madrid F, Marazuela M. Thyrocytes from autoimmune thyroid disorders produce the chemokines IP-10 and Mig and attract CXCR3+ lymphocytes. J Clin Endocrinol Metab. 2001;86:5008–5016. doi: 10.1210/jcem.86.10.7953. [DOI] [PubMed] [Google Scholar]