

Abstract
Steroidogenic enzyme autoantibodies (SEAbs) are frequently present and are markers of autoimmune premature ovarian failure (POF) in females with autoimmune Addison's disease (AAD). The prevalence and significance of SEAbs in males with AAD have not yet been defined. We studied the prevalence of SEAbs in a large cohort of males with AAD and assessed the relationship between SEAbs positivity and testicular function. A total of 154 males with AAD (mean age 34 years) were studied. SEAbs included autoantibodies to steroid-producing cells (StCA), detected by immunofluorescence, and steroid 17α-hydroxylase (17α-OHAbs) and side chain cleavage enzyme (SCCAbs) measured by immunoprecipitation assays. Gonadal function was evaluated by measuring follicle-stimulating hormone (FSH), luteinizing hormone (LH), total testosterone (TT), sex hormone-binding globulin (SHGB), anti-müllerian hormone (AMH) and inhibin-B (I-B). Twenty-six males, 10 SEAbs(+) and 16 SEAbs(–), were followed-up for a mean period of 7·6 years to assess the behaviour of SEAbs and testicular function. SEAbs were found in 24·7% of males with AAD, with the highest frequency in patients with autoimmune polyendocrine syndrome type 1 (APS-1). The levels of reproductive hormones in 30 SEAbs(+) males were in the normal range according to age and were not significantly different compared to 55 SEAbs(–) males (P > 0·05). During follow-up, both SEAbs(+) and SEAbs(–) patients maintained normal testicular function. SEAbs were found with high frequency in males with AAD; however, they were not associated with testicular failure. This study suggests that the diagnostic value of SEAbs in males with AAD differs compared to females, and this may be related to the immunoprivileged status of the testis.
Keywords: Addison's disease, autoantibodies to steroidogenic enzymes, autoimmune polyendocrine syndromes, gonadal function, testis autoimmunity
Introduction
Addison's disease (AD) is a rare condition, with a prevalence of 9·4–14·4 cases per 100 000 population [1]. Autoimmunity is the most frequent cause of AD in developed countries and is responsible for approximately 80% of the cases [2]. Autoimmune AD (AAD) is often associated with other autoimmune diseases, and different combinations of AAD with other autoimmune diseases present as autoimmune polyglandular syndrome (APS) type 1, type 2 or type 4 [1,2]. AAD is more prevalent in females than in males, with a female : male ratio of 2:1. Women with AAD are often positive for steroidogenic enzymes autoantibodies (SEAbs), which include autoantibodies to steroid-producing cells (StCA), to steroid 17α-hydroxylase (17α-OHAbs) and to side chain cleavage enzyme (SCCAbs). In females with AAD, SEAbs are markers of premature ovarian failure (POF) [3–7]. In these patients, POF is characterized by hypergonadotrophic hypogonadism, normal or atrophic ovaries and the presence of lymphocytic infiltration of the ovarian tissue characteristic of lymphocytic oophoritis [8]. Furthermore, in women with AAD, SEAbs are also predictive markers for POF [7,9,10].
In contrast, males with AAD SEAbs have been studied in only a limited number of patients. Furthermore, the studies were limited to StCA only, which were found with varying prevalence from 3·7 to 55% [11–13]. More recently, 17α-OHAbs and/or SCCAbs were also studied in AAD males and found to be positive from 5·4–10% and 2·7–35% of the patients, respectively [1,14]. However, to date there are no reports on the relationship between the presence of SEAbs and testicular function in males with AAD [11–15].
This study aimed to assess the prevalence of SEAbs (StCA, 17α-OHAbs and SCCAbs) in a large cohort of Italian males with AAD referred to our clinic from 1967 to 2012. In addition, groups of SEAbs(+) and SEAbs(–) patients were evaluated for gonadal function, and some SEAbs(+) and SEAbs(–) patients were also followed-up to assess SEAbs positivity and gonadal function over time. Furthermore, the overall value of SEAbs for the detection and prediction of hypergonadotrophic hypogonadism in males with AAD was evaluated.
Materials and methods
Patients
A total of 154 males with AAD referred to our clinic from 1967 to 2012, with a mean age of 34 years (range 2–84), were included in the study. All patients were Caucasian, mainly from North-East Italy and were representative of male Italian patients with AAD. AAD was defined by the presence of at least one of the serological markers: autoantibodies to adrenal cortex (ACA) (detected by indirect immunofluorescence using normal human adrenal tissue) and/or steroid 21-hydroxylase (21-OHAbs) (detected by radioimmunoassay).
The patients were further classified as APS type 1 (APS-1) when AAD was associated with candidiasis and/or chronic hypoparathyroidism, as APS type 2 (APS-2) when AAD was associated with thyroid autoimmune diseases (TAD) and/or type 1 diabetes mellitus (type 1 DM), as APS type 4 (APS-4) when AAD was associated with any other autoimmune disease excluding the previous associations, and as isolated AAD in the absence of other autoimmune diseases [2]. In all, 28 males had APS-1, 80 had APS-2, 11 had APS-4 and 35 had isolated AAD.
The study was approved by the Ethical Committee of the Azienda Ospedaliera – University of Padova and all the patients gave written informed consent to the study. The study was performed according to the principles of the Helsinki declaration.
Autoantibodies to SEAbs
Serum samples from each of the 154 males with AAD were tested for StCA, 17α-OHAbs and SCCAbs at entry to the study after AAD was diagnosed, and in 26 patients the immunological tests were repeated during follow-up. StCA were determined using the indirect immunofluorescent complement fixation (IICF) technique on unfixed cryostatic sections of normal human testis, ovary and adrenal tissue, as described previously [6,7]. 17α-OHAbs and SCCAbs were measured by immunoprecipitation assay (IPA) using recombinant autoantigens labelled with [35S]-methionine, as described previously (≤ 1·0 units/ml were considered negative in both assays) [4–6,22]. All sera were stored in small aliquots at −80°C until testing without freeze/thaw.
Gonadal function
Gonadal function was evaluated in 85 males with AAD [30 SEAbs(+) and 55 SEAbs(–)] at the start of the study by measurements of serum follicle-stimulating hormone (FSH), luteinizing lormone (LH), total testosterone (TT), sex hormone binding globulin (SHBG), anti-müllerian hormone (AMH) and inhibin-B (I-B).
Twenty-six patients with normal gonadal function at the start of the study (10 SEAbs(+) and 16 SEAbs(–)) were followed-up for a mean period of 7·6 years (range 1–19 years) by re-evaluating autoantibody positivity and gonadal function.
The immunometric assay for FSH and LH was a sandwich-type assay using monoclonal labelled antibodies directed against two different epitopes of FSH or LH (ECLIA, Cobas E601 analyzer; Roche Diagnostics, Milan, Italy). The measuring range was 0·1–200 U/l. Normal values for FSH were 1·5–12·4 U/l with intra-and interseries coefficient of variation (CV) of, respectively, 2·6 and 3·7%. Normal values for LH were 1·7–8·6 U/l with intra-and interseries CV, respectively, of 0·9 and 2·0%.
TT was measured by a competitive assay using high-affinity monoclonal antibody specifically directed against testosterone (ECLIA, Cobas E601 analyzer; Roche Diagnostics). The measuring range was 0·087–52 nmol/l. Normal values were 8·64–29 nmol/l (for males aged 20–49 years) and 6·68 to 25·7 nmol/l (for males aged more than 50 years) with intra-and interseries CV, respectively, of 2·4 and 6·1%.
SHBG was measured with a solid-phase, two-site chemiluminescent immunoassay (Immulite 2000 system; Siemens Healthcare Diagnostics, Milan, Italy). Results were expressed in nmol/l with an analytical sensitivity of 0·02 nmol/l. Normal values were 18–114 nmol/l with intra-and interseries CV, respectively, of 2·5 and 5·8%.
AMH was measured with an enzymatically amplified two-site immunoassay [enzyme-linked immunosorbent assay (ELISA) Gen II; Beckman Coulter Clinical Diagnostics, Milan, Italy]. Limit of detection was 0·08 ng/ml, with a normal range of 0·0–12·6 ng/ml. Intra-and interseries CV was 5·4 and 10%, respectively.
Inhibin-B was measured with an enzymatically amplified three-step ‘sandwich’ assay (ELISA Gen II; Beckman Coulter Clinical Diagnostics). Limit of detection was 2·6 pg/ml, with a normal range of 25–325 pg/ml. Intra-and inter-series CV was 3·8 and 5·6%, respectively.
All determinations for gonadal hormones were carried out in the same Clinical Laboratory at the Azienda Ospedaliera of Padova by M. P. and D. F.
Statistical analysis
The concentrations of FSH, LH, TT, SHBG, AMH and I-B were expressed as mean ± standard deviation (s.d.) for the age-related patient groups. The mean values were analysed using Student's t-test (α = 0·05).
Results
Prevalence of SEAbs in patients with AAD
The results of measurement of different SEAbs in patients with AAD, subdivided into APS-1 and non-APS-1 (including APS-2, APS-4 and isolated AAD), are summarized in Table 1 and Fig. 1.
Table 1.
Prevalence of steroid-enzyme autoantibodies (SEAbs) in male patients with autoimmune polyendocrine syndrome type 1 (APS-1) APS-2, APS-4 and isolated autoimmune Addison's disease (AAD).
| Autoantibodies | APS-1 | APS-2 | APS-4 | Isolated AAD |
|---|---|---|---|---|
| StCA | 60·7% (17/28) | 12·5% (10/80) | 9% (1/11) | 3% (1/35) |
| 17αOHAbs | 46·4% (13/28) | 8·75% (7/80) | 9% (1/11) | 0% (0/35) |
| SCCAbs | 46·4% (13/28) | 2·6% (4/80) | 0% (0/11) | 3% (1/35) |
StCA = steroid-producing cells autoantibodies; 17α-OHAbs = steroid 17α-hydroxylase autoantibodies; SCCAbs = side-chain-cleavage enzyme autoantibodies.
Figure 1.
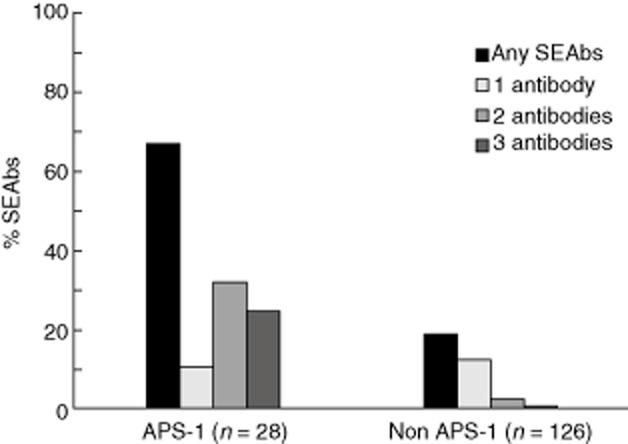
Prevalence of steroid-enzyme autoantibodies (SEAbs) in male patients with autoimmune polyendocrine syndrome type 1 (APS-1) and non APS-1 [including APS-2, APS-4 and isolated autoimmune Addison's disease (AAD)].
The individual results of different SEAbs measurements for each SEAbs(+) patient are summarized in Fig. 2. The mean concentrations of 17α-OHAbs in APS-1 patients were 30·9 units/ml compared to mean concentrations of 4·9 units/ml in non-APS-1 patients, while the mean concentrations of SCCAbs in APS-1 patients were 19·4 units/ml compared to 1·7 units/ml in non-APS-1 patients. SEAbs concentrations were higher in patients with APS-1 than in non-APS-1 patients (P < 0·05).
Figure 2.
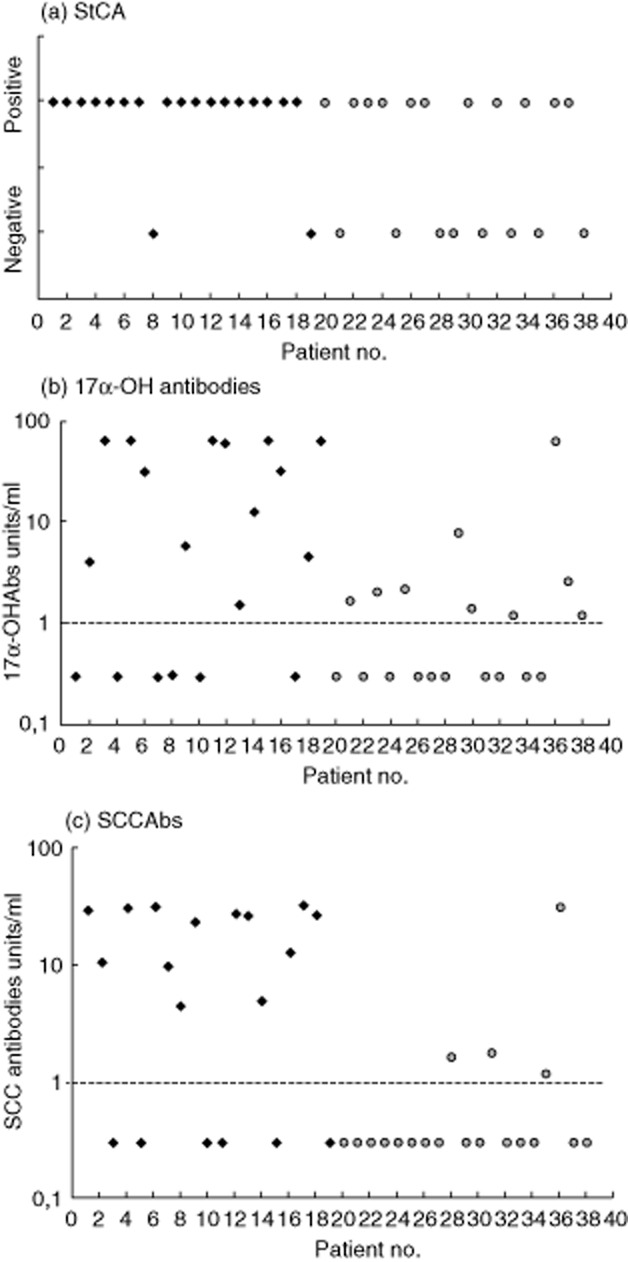
Autoantibodies in 38 steroid–enzyme autoantibodies (SEAbs)(+) patients with autoimmune Addison's disease (AAD) with autoimmune polyendocrine syndrome type 1 (APS-1) (diamond) numbers 1–19, with non-APS-1 (circle) numbers 20–38 (with APS-2 numbers 20–36, a patient with APS-4 number 37 and a patient with isolated AAD number 38). All patients were positive for adrenal cortex autoantibody (ACA) and/or 21-hydroxylase autoantibodies (OHAbs). (a) StCA expressed as positive or negative for the antibody. (b) 17αOHAbs values ≤1 unit/ml were considered negative (shown with dashed line). (c) Side chain cleavage enzyme autoantibody (SCCAb) values ≤1 unit/ml were considered negative (shown with dashed line).
Patient no. 10 (Fig. 2) presented with seizures at 2 years of age, and was diagnosed with chronic hypoparathyroidism together with diffuse vitiligo at the age of 3 years. One year later he developed chronic mucocutaneous candidiasis at nail and oral localizations. At the age of 6 years, keratoconjunctivitis and nail pitting were diagnosed. ACA were found positive when the patient was aged 8 years, and 2 years later he presented with overt AAD. He was also affected by gastrointestinal dysfunction, alopecia, essential hypertension, nephrocalcinosis and severe renal insufficiency. Autoimmune regulator (AIRE) gene analysis showed a homozygous R257X mutation. The patient went through normal puberty and never had mumps. At 17 years of age, when first assessed for SEAbs, he was found positive for StCA while 17α-OHAbs and SCCAbs were negative. He was also positive for autoantibodies to interferon-ω (IFNωAbs), tryptophan hydroxylase (TPHAbs) and aromatic-L-amino acid decarboxylase (AADC), while antibodies to NACHT leucine-rich-repeat protein 5 were negative. At that time FSH was 1·4 U/l, LH 9·96 U/l, TT 1·09 nmol/l, SHGB 25·1 nmol/l, AMH 21 ng/ml and I-B 383 pg/ml. At 17 years he developed renal failure due to nephrocalcinosis and went on dialysis. Four months later during a hypertensive crisis he suffered a cerebral haemorrhage and died. The post-mortem histological examination of the testes revealed a reduction of the volume, absence of spermatozoa, few Leydig cells and lymphocytic infiltration.
Overall, in 136 of 154 patients (88·3%), there was agreement between the presence/absence of StCA and 17α-OHAbs and/or SCCAbs; however, 18 of 154 (11·7%) samples were discordant (eight positive for StCA and negative for 17α-OHAbs or SCCAbs and 10 positive for 17α-OHAbs and/or SCCAbs and negative for StCA).
Gonadal function
The mean gonadal hormone concentrations of FSH, LH, TT, SHBG, AMH, I-B in the 85 males with AAD [12 aged < 15 years, six SEAbs(+) and six SEAbs(–); 41 aged from 15–40 years, 17 SEAbs(+) and 24 SEAbs(–) and 32 > 40 years of age, seven SEAbs(+) and 25 SEAbs(–)] were not significantly different versus age-matched controls.
We compared the serum reproductive hormone levels in 11 SEAbs(+) patients with a high titre of 17α-OHAbs (30·8 U/ml) and/or SCCAbs (19·9 U/ml) (aged > 15 years), with those in 14 SEA(+) patients with a low titre of 17α-OHAbs (5·2 U/ml) and/or SCCAbs (1·8 U/ml) (aged > 15 years). There was no difference between the two groups.
Follow-up
Twenty-six patients [10 SEAbs(+) and 16 SEAbs(–)] were included in the follow-up. During the follow-up, the behaviour of SEAbs and gonadal function were re-evaluated. Eight of 10 SEAbs(+) patients maintained the immunological pattern during follow-up; two patients initially positive for three autoantibodies maintained positivity for two autoantibodies. During follow-up three of 16 initially SEAbs(–) patients seroconverted for StCA after 2, 7 and 8 years, while 13 patients remained negative for all the SEAbs.
Nine of 10 persistently SEAbs(+) patients were affected by APS-1 and one was affected by APS-4. The three initially SEAbs(–) patients who seroconverted to StCA positivity were affected by APS-2.
Seven of 13 initially and persistently SEAbs(–) patients had APS-2, four had isolated AAD, one had APS-4 and one APS-1.
Four of 10 SEAbs(+) patients at the start of follow-up were prepubertal; two patients were followed from age 8 to 21 and 23 years, respectively, and the other two from age 9 to 28 and 19 years, respectively. All four underwent normal puberty with reproductive hormone levels in the normal range at the end of follow-up (mean levels of FSH 1 ± 0·42 U/l, LH 1·1 ± 1·9 U/l, TT 0·98 ± 1·5 nmol/l, SHBG 48 ± 16 nmol/l, AMH 18·9 ± 2·8 ng/ml, I-B 197·6 ± 174·8 pg/ml). Six of 10 SEAbs (+) patients were adult at entry to the study and were followed-up for a mean period of 7·3 years. All these patients showed normal gonadal function at entry and at the end of follow-up. The mean reproductive hormone levels at the end of follow-up were 6·1 ± 6·7 U/l for FSH, 4·63 ± 2·3 U/l for LH, 17·3 ± 8·6 nmol/l for TT, 45 ± 16·9 nmol/l SHBG, 9·7 ± 6·6 ng/ml for AMH and 198·6 ± 135·5 pg/ml for I-B.
The three patients who seroconverted from SEAbs(–) to SEAbs(+) were followed-up for a mean period of 6·3 years, and they had normal gonadal function at the beginning of the follow-up and at the end of observation. At the beginning of the follow-up the mean concentrations of FSH were 5·5 ± 4·4 U/l, LH 5·3 ± 1·1 U/l, TT 21·2 ± 8·9 nmol/l, SHBG 44·6 ± 21·3 nmol/l, AMH 4·5 ± 2 ng/ml and I-B 172·9 ± 141·4 pg/ml. At the end of follow-up the mean levels were FSH 7·53 ± 2·9 U/l, LH 5·5 ± 0·81 U/l, TT 16·4 ± 5 nmol/l, SHBG 30·9 ± 12·8 nmol/l, AMH 2·7 ± 0·7 ng/ml and I-B 72·8 ± 40·3 pg/ml. The differences were not significant (P > 0·05).
Thirteen persistently SEAbs(–) males were followed-up for a mean period of 4·8 years; there were 12 adults and one prepubertal patient. All the patients showed normal gonadal function during the follow-up. Mean hormone levels at the end of follow-up were FSH 5·6 ± 3·1 U/l, LH 6·2 ± 3·8 U/l, TT 11·75 ± 7·25 nmol/l, SHBG 38·9 ± 28·9 nmol/l, AMH 8·27 ± 6·7 ng/ml and I-B 142·5 ± 108 pg/ml.
Serum hormone concentrations between SEAbs(+) and SEAbs(–) patients were not different (P > 0·05). Four SEAbs(+) and eight SEAbs(–) patients reported fathering children.
None of the 13 SEAbs(+) males with AAD and none of the 13 persistently SEAbs(–) with AAD who were followed-up for a mean of 7·6 years (range 1–19) developed hypergonadotrophic hypogonadism.
Discussion
StCA were first described in two males with AAD without gonadal dysfunction [13]. Subsequently, the prevalence of StCA or 17α-OHAbs and SCCAbs, in particular, was studied with a limited number of male patients with AAD [1,11,12,14]. However, gonadal function was not assessed systematically in these studies. For example, case reports in the late 1970s described a 16-year-old male with AAD who was positive for StCA and presented with gynaecomastia, left testicular atrophy and absence of spermatogonia [11] and another StCA-positive male with AAD with testicular atrophy [16]. A different report described 11 males with APS-1 (not tested for SEAbs), of whom three patients showed undefined signs of hypogonadism [17]. In a further study, seven males with gonadal dysfunction were described: three had azoospermia, three had hyperpogonadotrophic hypogonadism and one had hypogonadotrophic hypogonadism; however; none were tested for SEAbs [18]. In a further study, all seven males with APS-1 who were positive for SCCAbs had normal testicular function [14], while in another study the only patient with APS-1 tested and positive for all three SEAbs was affected by hypergonadotrophic hypogonadism [15]. Yet another report described two males with APS-1 and hypogonadotrophic hypogonadism who had detectable SEAbs in addition to antibodies to pituitary (against LH-, GHRH-and GH-secreting cells), suggesting that in these patients autoimmune hypophysitis was responsible for gonadal dysfunction [12,19]. To date, there are no comprehensive data on the relationship between SEAbs positivity and gonadal function in males with AAD.
This is in contrast to the well-documented correlation between the presence of SEAbs and hypergonadotrophic hypogonadism characterized by lymphocytic oophoritis in females with AAD [8]. Furthermore, SEAbs in females with AAD are markers of future POF [7,9,10].
This is the first study in a large cohort of males with AAD on the prevalence of all three SEAbs (StCA, 17α-OHAbs and SCCAbs) with systematical evaluation of gonadal function and analysis of change in positivity for SEAbs and gonadal function over time.
In our study the prevalence of one or more SEAbs in AAD males reached 66·8% in APS-1 and 19% in non APS-1 patients. Furthermore, the mean serum concentrations of 17α-OHAbs and SCCAbs were significantly higher in the SEAbs-positive APS-1 than in non-APS-1 males (P < 0·05). There was a good correlation between StCA and 17α-OHAbs and/or SCCAbs in AAD males, as demonstrated previously in AAD females [7]. This confirms that the major autoantigens recognized by StCA are 17αOH and SCC in both female and male patients with AAD. The discrepancies may be related to the differences in the methods used to measure antibodies. For example, IPAs are likely to be more sensitive than the IICF technique, and this could account for some samples that tested positive for 17α-OHAbs and/or SCCAbs and negative for StCA. In the case of samples positive for StCA while negative for 17α-OHAbs and/or SCCAbs, it is possible that the positive IICF signal was due to the presence of as yet unidentified autoantigens (different from 17α-OH or SCC).
We investigated gonadal function in a cohort of 85 males with AAD, assessing the patients in three different age groups (<15 years, 15–40 years and >40 years) at entry to the study. We did not find differences in mean reproductive hormone concentrations between SEAbs(+) and SEAbs(–) patients in these age-related groups. Furthermore, none of the patients had hypergonadotrophic hypogonadism and their reproductive hormone levels were all normal compared to age-matched normal controls. Our study showed that not only was Leydig cell function normal, but Sertoli cell function was also normal, as reflected by AMH and I-B levels, and this would exclude potential hypogonadism in childhood [20].
Although clinical symptoms and serum concentrations of reproductive hormones are helpful in the assessment of the gonadal function in SEAsb(+) patients, insight into the histology of the gonads in these patients is very limited. In this study a post-mortem sample of testicular tissue from a StCA(+) APS-1 patient (no. 10, Fig. 2) was available for examination. Gonadal function tests in this patient showed low levels of TT with inappropriately normal levels of FSH and LH, whereas SHBG, AMH and I-B were normal. Histology of the testicular tissue showed the absence of spermatozoa and no signs of lymphocytic infiltration. This presentation would be consistent with primary hypogonadism associated with partial pituitary dysfunction. Another SEAbs(+) patient (no. 16, Fig. 2) presented clinical symptoms of gonadal dysfunction and oligospermia, while reproductive hormone levels were within the normal range, with the exception of slightly elevated FSH. These observations suggest that the serum markers of gonadal function and of autoimmunity may not always reflect the actual status of the gonads themselves.
Previous reports on the follow-up of StCA(+) males described only eight, two and two patients, respectively, in three different studies, and none of them developed gonadal dysfunction during a mean period of 5·7 years of observation [8–10].
In this study a group of 26 males was followed-up for a mean period of 7·6 years. All the 10 SEAbs(+) males maintained autoantibody positivity, while three of 16 SEAbs(–) became positive for StCA. Four SEAbs(+) and one SEAbs(–) patients were prepubertal at entry to follow-up; however, all went through normal puberty.
Of 26 adult SEAbs(+) and SEAbs(–) patients, none developed gonadal dysfunction at the end of follow-up. In particular, the mean gonadal hormone levels in SEAbs(+) and SEAbs(–) patients were not different and none of the SEAbs(+) or SEAbs(–) males with AAD developed hypergonadotrophic hypogonadism. These observations suggest that the presence of SEAbs has no apparent effect on gonadal development in the pubertal period, and furthermore does not appear to affect testicular function in adult males.
Our study suggests that although there is evidence for autoimmune reactivity against the gonadal antigens in patients with AAD, SEAbs do not appear to be markers of an autoimmune testis disease. However, the testis can be considered as an immunoprivileged site due to the presence of the effective blood–testis barrier. In addition, Leydig cells have been shown to secrete high doses of testosterone, and local macrophages, peritubular cells and Sertoli cells produce immunosuppressive factors (transforming growth factor-β, granulocyte–macrophage colony-stimulating factor, pro-opiomelanocortin, α-endorphin, met-encephalin, insulin growth factor). Furthermore, the number of resident T cells in the testis is low, while regulatory T cells (CD4+CD25+) are present [21]. These observations could provide an explanation while, irrespective of the presence of circulating autoimmune markers, there is no evidence of autoimmune testicular failure in SEAbs(+) male patients with AAD.
This is in direct contrast to women with AAD, in whom SEAbs are useful diagnostic and predictive markers of POF.
Acknowledgments
This study was supported in part from the European Union 7th Framework Programme, the EurAdrenal project Grant Agreement no. 2008-201167 and in part from a 60% grant of the University of Padova.
Disclosure
B. R. S., S. C. and J. F. are employed by RSR Ltd. RSR Ltd is a developer of medical diagnostics including kits for measuring 21-OHAbs and IFNωAbs.
References
- 1.Erichsen MM, Løvås K, Skinningsrud B, et al. Clinical, immunological and genetic features of autoimmune primary adrenal insufficiency: observations from a Norwegian registry. J Clin Endocrinol Metab. 2009;94:4882–4900. doi: 10.1210/jc.2009-1368. [DOI] [PubMed] [Google Scholar]
- 2.Betterle C, Dal Prà C, Mantero F, Zanchetta R. Autoimmune adrenal insufficiency and autoimmune syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev. 2002;23:327–364. doi: 10.1210/edrv.23.3.0466. [DOI] [PubMed] [Google Scholar]
- 3.Falorni A, Brozzetti A, Aglietti MC, et al. Progressive decline of residual follicle pool after clinical diagnosis of autoimmune ovarian insufficiency. Clin Endocrinol (Oxf) 2012;77:453–458. doi: 10.1111/j.1365-2265.2012.04387.x. [DOI] [PubMed] [Google Scholar]
- 4.Sotsiou F, Bottazzo GF, Doniach D. Immunofluorescence studies on autoantibodies to steroid-producing cells, and to germline cells in endocrine disease and infertility. Clin Exp Immunol. 1980;39:97–111. [PMC free article] [PubMed] [Google Scholar]
- 5.Chen S, Sawicka J, Betterle C, et al. Autoantibodies to steroidogenic enzymes in autoimmune polyglandular syndrome, Addison's disease and premature ovarian failure. J Clin Endocrinol Metab. 1996;81:1871–1876. doi: 10.1210/jcem.81.5.8626850. [DOI] [PubMed] [Google Scholar]
- 6.Betterle C, Greggio NA, Volpato M. Autoimmune polyglandular syndrome type 1. J Clin Endocrinol Metab. 1998;83:1049–1055. doi: 10.1210/jcem.83.4.4682. [DOI] [PubMed] [Google Scholar]
- 7.Reato G, Morlin L, Chen S, et al. Premature ovarian failure in patients with autoimmune Addison's disease: clinical features and immunological evaluation. J Clin Endocrinol Metab. 2011;96:1255–1261. doi: 10.1210/jc.2011-0414. [DOI] [PubMed] [Google Scholar]
- 8.Hoek A, Schoemaker J, Drekhage HA. Premature ovarian failure and ovarian autoimmunity. Endocr Rev. 1997;18:107–134. doi: 10.1210/edrv.18.1.0291. [DOI] [PubMed] [Google Scholar]
- 9.Ahonen P, Miettinen A, Perheentupa J. Adrenal and steroidal cell antibodies in patients with autoimmune polyglandular disease type I and risk of adrenocortical and ovarian failure. J Clin Endocrinol Metab. 1987;64:494–500. doi: 10.1210/jcem-64-3-494. [DOI] [PubMed] [Google Scholar]
- 10.Betterle C, Rossi A, Dalla Pria S, et al. Premature ovarian failure: autoimmunity and natural history. Clin Endocrinol (Oxf) 1993;39:35–43. doi: 10.1111/j.1365-2265.1993.tb01748.x. [DOI] [PubMed] [Google Scholar]
- 11.Irvine WJ, Barnes EW. Addison's disease, ovarian failure and hypoparatiroidism. J Clin Endocrinol Metab. 1975;4:379–433. [Google Scholar]
- 12.Meloni A, Willcox N, Meager A, et al. Autoimmune polyendocrine syndrome type 1: an extensive longitudinal study in Sardinian patients. J Clin Endocrinol Metab. 2012;97:1114–1124. doi: 10.1210/jc.2011-2461. [DOI] [PubMed] [Google Scholar]
- 13.Anderson JR, Goudie RB, Gray K, Stuart-Smith DA. Immunological features of idiopathic Addison's disease: an antibody to cell producing steroid hormones. Clin Exp Immunol. 1968;3:107–117. [PMC free article] [PubMed] [Google Scholar]
- 14.Wolff AS, Erichsen MM, Meager A, et al. Autoimmune polyendocrine syndrome type 1 in Norway: phenotypic variation, autoantibodies, and novel mutations in the autoimmune regulator gene. J Clin Endocrinol Metab. 2007;92:595–603. doi: 10.1210/jc.2006-1873. [DOI] [PubMed] [Google Scholar]
- 15.Magitta NF, Pura M, Bøe Wolff AS, et al. Autoimmune polyendocrine syndrome type I in Slovakia: relevance of screening patients with autoimmune Addison's disease. Eur J Endocrinol. 2008;158:705–709. doi: 10.1530/EJE-07-0843. [DOI] [PubMed] [Google Scholar]
- 16.Colls J, Betterle C, Volpato M, Rees Smith B, Furmaniak J. A new immunoprecipitation assay for autoantibodies to steroid 21-hydroxylase in Addison's disease. Clin Chem. 1995;41:375–380. [PubMed] [Google Scholar]
- 17.McNatty KP, Short RV. The cytotoxic effect of serum from patients with Addison's disease and autoimmune ovarian failure on human granulose cells in culture. Clin Exp Immunol. 1975;22:378–384. [PMC free article] [PubMed] [Google Scholar]
- 18.Zlotogora J, Shapiro MS. Polyglandular syndrome type I among Iranian Jews. J Med Genet. 1992;29:824–826. doi: 10.1136/jmg.29.11.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perheentupa J. Autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy. J Clin Endocrinol Metab. 2006;91:2843–2850. doi: 10.1210/jc.2005-2611. [DOI] [PubMed] [Google Scholar]
- 20.Barkan AL, Kelch RP, Marshall JC. Isolated gonadotrope failure in the polyglandular autoimmune syndrome. N Engl J Med. 1985;312:1535–1540. doi: 10.1056/NEJM198506133122402. [DOI] [PubMed] [Google Scholar]
- 21.Rey RA, Grinspon RP, Gottlieb S, et al. Male hypogonadism: an extended classification based on a developmental, endocrine physiology-based approach. Andrology. 2013;1:3–16. doi: 10.1111/j.2047-2927.2012.00008.x. [DOI] [PubMed] [Google Scholar]
- 22.Jacobo P, Guazzone AV, Theas MS, Lustig L. Testicular autoimmunity. Autoimmun Rev. 2011;10:201–204. doi: 10.1016/j.autrev.2010.09.026. [DOI] [PubMed] [Google Scholar]