

Abstract
Hypogammaglobulinaemias are the most common primary immunodeficiency diseases. This group of diseases is very heterogeneous, and little is known about these diseases in children. In the Pediatric Predominantly Antibody Deficiencies (PedPAD) study, we analysed data from the European Society for Immunodeficiencies (ESID) online database to gain more insight into the characteristics of children with hypogammaglobulinaemia; 46 centres in 18 different countries agreed to participate. Data from 2076 of the 3191 children who were registered at the time of data extraction with a diagnosis of hypogammaglobulinaemia (this excludes agammaglobulinaemia and defects in class-switch recombination) were available for analysis. The data set showed several limitations. Because of country-related differences in diagnostic criteria used for the classification of different types of primary hypogammaglobulinaemia, further analysis of the data was performed in the combined data set. The most striking observation is the strong majority of male patients in the group of children with primary hypogammaglobulinaemia (n = 1292, 63%). This male predominance was observed in each of the 18 countries involved. The boys were younger at diagnosis (mean age males 5·3 years; mean age females 5·8 years). Moreover, one or more complications were more frequently reported in boys (12%) compared to girls (5%). The male predominance suggests that patients with an undetected or unknown X-linked genetic cause are included in this group of children registered as primary hypogammaglobulinaemia.
Keywords: child, hypogammaglobulinaemia, predominantly antibody deficiencies, primary immunodeficiency
Introduction
Primary immunodeficiencies (PID) are inherited defects of the immune system, with predominantly antibody deficiencies (PAD) being the largest subgroup. The hallmark of PAD is a marked reduction or absence of immunoglobulins, with an increased susceptibility to mainly bacterial infections that typically involve the upper and lower respiratory tracts. PADs can be divided into agammaglobulinaemias, defects of class-switch recombination and hypogammaglobulinaemias. ‘Hypogammaglobulinaemia’ is by far the most common entity, comprising nearly half of all PID diagnoses [1]. For most diseases in the first two subgroups, the genetic defects have been described and their effects on B cell differentiation studied [2]. The hypogammaglobulinaemia subgroup, however, is extremely heterogeneous, and much less is known about these diseases. Among the hypogammaglobulinaemias, common variable immunodeficiency disorders (CVID) is the most commonly reported diagnosis in registries, followed by immunoglobulin (Ig)G subclass deficiency [1].
Because PIDs are relatively rare disorders, international collaboration is necessary to study the overall clinical characteristics of these diseases. Since 2004, the European Society for Immunodeficiencies (ESID) has been running an online database for primary immunodeficiencies: the ESID Online Database. This database registers demographical, clinical and laboratory characteristics of patients with PID [3]. At present, more than 18 000 cases from 119 centres in Europe are registered [4]. Biennial reports from the ESID Online Database have been published, but none of these reports has focused specifically on children [1,5,6]. Also, in other available literature, most published data on hypogammaglobulinaemia refer to adult patients.
Children with hypogammaglobulinaemia constitute a special group due to their maturing immune system. Some antibody deficiencies disappear over time and seem to be a physiological variation, whereas other antibody deficiencies remain and manifestations of more severe immunodeficiency will develop. Currently, it is impossible to predict which child will recover and which one will suffer from more or less severe disease in the future. Moreover, data from adults cannot be simply extrapolated to children. Separate studies have to be performed. We used the data from the ESID Online Database to gain more insight into the characteristics of children with hypogammaglobulinaemia.
Materials and methods
The objective of this study was to describe numbers, geographical distribution, underlying diseases, age and laboratory values at presentation and diagnosis, the diagnostic delay and follow-up data in children with hypogammaglobulinaemia (this excludes agammaglobulinaemia and defects in class-switch recombination) registered in the ESID Online Database.
The system and structure of the ESID Online Database has been described previously [3,5]. The data of the ESID Online Database are stored on secure servers of the IT Centre of the University Hospital in Freiburg, Germany. Categorization is based on the classification defined by the International Union of Immunological Societies (IUIS) [7]. There is a set of identical fields for all patients, which comprises diagnosis, gender, age of patient at diagnosis, information regarding whether it is a sporadic or familial case, current medication, adverse effects of treatment, basic laboratory values and genetic mutation data. These fields form the ‘core data set’. Patients give informed consent before their data are entered into the database. The aim of the database is long-term documentation of a patient; documentation is requested at least once a year for each patient. The date of a patient's attendance at the clinic is recorded and data associated with this patient visit, such as a change in the treatment regimen, can be documented. Some centres and national networks maintain local databases for their PID patients. Their data are transferred electronically to the ESID database at regular intervals. The database has an inbuilt automatic quality assurance system, including field type, range and plausibility checks. In addition, data sets are checked regularly for plausibility and completeness by the database administration.
For this study, all centres that registered children with hypogammaglobulinaemia (age <19 years) were approached; 46 centres from 18 countries agreed to participate. Only data from patients reported by these collaborating centres were included in our study.
Standard statistical analyses were applied to investigate associations between categorical variables [χ2 analysis and Fisher's exact test (Monte Carlo; 10 000 samples) when expected cell values were lower than 5] and differences between groups [the t-test (comparing two groups) and analysis of variance (comparing more than two groups)]. This means that the data can be summarized as frequencies in cross-tabulations, e.g. by cross-tabulating country by gender, consanguinity cases (yes/no) or familial cases (yes/no). When age variables were involved (age of onset, presentation or diagnosis, all continuous variables) differences between groups were tested by using the t-test (comparing two groups) and analysis of variance (comparing more than two groups). The statistical software package used was IBM spss statistics version 20.
Results
The data for 2076 of 3191 children registered within the ‘Hypogammaglobulinaemias’ registry at the time of data extraction were available for analysis. These data were collected in 46 centres in 18 different countries; some countries only reported one or a few diagnoses (Supporting information, Table S1). Estonia (20), Turkey (14), the Netherlands (eight), Spain (four) and Greece (four) reported most affected children per 1 million inhabitants (Fig. 1). In absolute numbers, Turkey reported most children (968), followed by Germany (257) and Spain (182).
Figure 1.
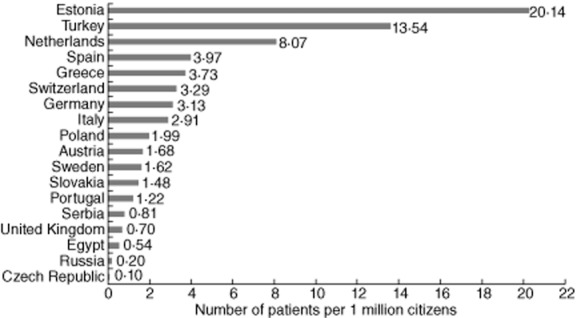
Number of paediatric patients per 1 million inhabitants registered in the European Society for Immunodeficiencies (ESID) online database per country participating in the Pediatric Predominantly Antibody Deficiencies (PedPAD) study.
Unfortunately, the data set showed several limitations, making interpretation of the data challenging. Obvious mistakes were excluded from further analysis (e.g. date of diagnosis or laboratory value before date of birth or clinical presentation), as well as laboratory data without dates or units. Many entries were incomplete (Table 1); children frequencies in the statistical analyses may vary because of differences in data available in subsets of data. Immunoglobulin (Ig)G substitution was not documented clearly in most cases, therefore no analysis of reported IgG levels was performed (IgG substitution was reported for 1152 episodes in 598 patients; 813 intravenously, 331 subcutaneously, two intramuscularly administered and six by unknown route).
Table 1.
Available information in the Hypogammaglobulinaemias registry of the European Society for Immunodeficiencies (ESID) online database.
| Data regarding: | Number of patients with available data | Percentage of total group |
|---|---|---|
| Infections | 414 | 20% |
| Autoimmunity | 85 | 4% |
| Other concomitant diseases | 276 | 13% |
| Therapy | 1312 | 63% |
| Immunizations | 16 | 0·8% |
| Blood count | 1830 | 88% |
| Immunoglobulins | 1872 | 90% |
| IgG subclasses | 890 | 43% |
| White blood cells | 1333 | 64% |
| T and B cells | 827 | 40% |
| Additional laboratory values | 319 | 15% |
| Total | 2076 | na |
n.a. = not applicable.
Furthermore, when comparing immunoglobulin levels and diagnoses, we noticed that different criteria for registering under a certain diagnosis were used in different countries. The current ESID diagnostic criteria (http://www.esid.org) were not always followed. Because reliable immunoglobulin levels at diagnosis were lacking in many patients, diagnosis could not be checked in every patient [4]. Therefore, further analysis of the data was performed within the total group of children with primary hypogammaglobulinaemia, which includes the subregistries common variable immunodeficiency (CVID), deficiency of specific IgG, IgA with IgG subclass deficiency, isolated IgG subclass deficiency, other hypogammaglobulinaemias, other immunoglobulin gene deletions, selective IgA deficiency, selective IgM deficiency and transient hypogammaglobulinaemia of infancy (THI) of the registry (Table 2, n = 2052).
Table 2.
Different diagnoses reported in the 2076 children from the Hypogammaglobulinaemias registry.
| Diagnosis | Number of patients reported | Percentage of total hypogammaglobulinaemia group | Percentage of boys |
|---|---|---|---|
| Common variable immunodeficiency (CVID) | 469 | 23% | 64% |
| Deficiency of specific IgG | 39 | 2% | 56% |
| IgA with IgG subclass deficiency | 12 | 0·6% | 67% |
| Isolated IgG subclass deficiency | 301 | 15% | 59% |
| Other hypogammaglobulinaemias | 188 | 9% | 61% |
| Other immunoglobulin gene deletions | 1 | 0·1% | 100% |
| Secondary hypogammaglobulinaemia | 8 | 0·4% | Unknown* |
| Secondary selective IgA deficiency | 15 | 0·7% | Unknown* |
| Selective IgA deficiency | 513 | 25% | 58% |
| Selective IgM deficiency | 41 | 2% | 89% |
| Thymoma with immunodeficiency | 1 | 0·1% | Unknown* |
| Transient hypogammaglobulinaemia of infancy | 488 | 24% | 69% |
| Total hypogammaglobulinaemia group | 2076 | 100% | 63% |
Only the diagnoses in italics were included in the group of ‘total primary hypogammaglobulinaemia’ that were used for further analysis.
Percentage cannot be calculated due to lack of data. Ig = immunoglobulin.
General characteristics
The mean age at onset (defined as first symptoms) was 2·6 years [standard deviation (s.d.) 3·15, median 1·0 years, range 0–18 years, n = 1833]. The mean age at diagnosis was 5·5 years (s.d. 3·98, median 5·0 years, range 0–19 years, n = 1508), resulting in a mean diagnostic delay of 2·6 years (s.d. 2·87, median 2·0 years, range 0–17 years, n = 1508). No information regarding survival was available in 594 patients (30%); 1416 patients (69%) were reported as alive at the last update (9 October 2012). Although there were statistically significant differences in age at onset and age at diagnosis between countries, these were somewhat small (effect sizes of less than 0·10). In the sample of 1508 patients for whom the relevant age data was present, six countries had a small sample of 10 patients or fewer. We therefore excluded the Czech Republic (one), Austria (three), Portugal (eight), Serbia (five), Slovakia (two) and Sweden (nine) from this analysis. An analysis of variance was applied, giving a significant effect for age at diagnosis (F(1,11) = 2·217, P = 0·012, partial ε2 = 0·016) and age at presentation (F(1,11) = 3·818, P = 0·000, partial ε2 = 0·028). Switzerland (5·17, n = 18) had a mean age at presentation later than 4 years. Russia (7·4, n = 18) had a mean age at diagnosis later than 7 years. No relevant subgroups were detected in a post-hoc analysis.
Consanguinity was present in 133 patients (6%), absent in 1590 patients (78%) and not reported in 329 (16%). The consanguinity differences between the countries were significant (Fisher's exact test = 120·68, df = 17, P = 0·000). Egypt (17 of 37 patients, 47%) and Serbia (six of six, 100%) reported by far the largest percentage of consanguineous cases, but several other countries also showed higher percentages: Turkey (87 of 938 patients, 9%) Russia (three of 15 patients, 20%), Sweden (three of 14 patients, 21%) and Austria (one of six patients, 17%). Consanguinity was not associated with gender (χ2(1, n = 1723) = 1·236, P = 0·266), age at onset (t(1409) = −1·056, P = 0·291) or age at diagnosis (t(1409) = −1·079, P = 0·281).
Familial cases were present in 198 patients (10%), absent in 1500 patients (73%) and not reported in 354 patients (17%). The frequencies of familial cases between the countries differed significantly (Fisher's exact test = 99·93, P = 0·000). High numbers (more than 20%) were reported by the Netherlands (13 of 34, 26%), Serbia (six of six, 100%) and United Kingdom (six of 24, 25%). Familial cases were not associated with gender (χ2(1, n = 1698) = 0·008, P = 0·928), age at onset (t(1380) = −1·520, P = 0·129) or age at diagnosis (t(1380) = 0·483, P = 0·630). There were 58 family cases (28 families) reported with available information of the family member. If we look at the 19 families in which the child who was presented secondly was diagnosed after the first presented child, there was a significant decrease in diagnostic delay for the second child (one-sample t-test, test value 0: t(18) = −3·144, P = 0·006).
A total of 297 patients reported consanguinity and/or familial cases. There was no association with gender (χ2(1, n = 1763) = 0·592, P = 0·442), age at onset (t(1434) = 1·653, P = 0·096) or age at diagnosis (t(1434) = 0·948, P = 0·344. Egypt (17 of 36, 47%) and Serbia (six of six, 100%) reported significantly higher numbers than other countries.
A gene mutation was reported in only 12 patients [seven transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), four cluster of differentiation (CD)19, one 11q23 mutation].
Male patients are in the majority [63%; χ2 based on an equal distribution of males and females, χ2(1, n = 2052) = 137·93, P = 0·000]. This male predominance was present in each of the 18 countries. There was no specific association between country and gender (Fisher's exact test, P = 0·446). Furthermore, male patients tend to be younger at onset (mean age male patients 2·7 years; mean age female patients 3·1 years; t(1509) = 1·83, P = 0·068) and differ significantly at diagnosis (mean age male patients 5·3 years; mean age female patients 5·8 years; t(1509) = 2·38, P = 0·012).
Laboratory characteristics
IgA levels were available in 1469 patients; IgM levels were available in 1477 patients. We categorized them as low, normal or high for age [8]; 643 (44%), 796 (54%) and 30 (2%) patients showed low, normal and high IgA levels at presentation, and 107 (7%), 1313 (89%) and 57 (4%) patients showed low, normal and high IgM levels at presentation, respectively (Fig. 2). IgA and IgM levels were not associated with gender (IgA: χ2(2, n = 1469) = 0·354, P = 0·831; IgM: χ2(2, n = 1477) = 3·38, P = 0·187) or consanguinity (IgA: χ2(2, n = 1299) = 0·679, P = 0·712; IgM: χ2(2, n = 1304) = 0·498, P = 0·780). Country was associated with IgA levels (Fisher's exact test (n = 1469) = 206·10, P = 0·000) and with IgM levels (Fisher's exact test (n = 1477) = 138·08, P = 0·000). Low IgA levels were reported frequently (more than 70%) in children from the Czech Republic (one of one patient, 100%), Egypt (26 of 34 patients, 77%), Estonia (21 of 27, patients 78%), Greece (25 of 35 patients, 72%), Poland (37 of 50 patients, 74%), Russia (15 of 21 patients, 71%), Slovakia (seven of seven patients, 100%), Spain (52 of 57 patients, 91%) and Sweden (six of eight patients, 75%). Low IgM levels were reported frequently (more than 15%) in the Czech Republic (one of one patient, 100%), Egypt (eight of 34 patients, 24%), Germany (23 of 27 patients, 18%), Russia (nine of 22 patients, 41%), Slovakia (three of seven patients, 43%) and the United Kingdom (eight of 23 patients, 35%). The 57 patients with reported high IgM levels were distributed equally among the countries.
Figure 2.
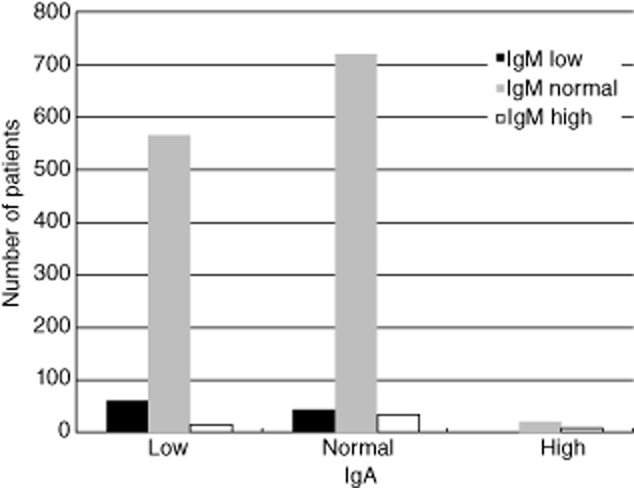
Immunoglobulin (Ig)A and IgM levels at presentation of the children in the ‘total primary hypogammaglobulinaemia’ group. Patients are divided into different groups based on their levels of IgA and IgM at presentation: IgA low (left), IgA normal (middle), IgA high (right), IgM low (black), IgM normal (grey) and IgM high (white).
Blood counts (n = 1247) showed 38 patients with severe neutropenia (<0·5 × 10e9/l). Absolute numbers of B lymphocytes (defined as CD19+ lymphocytes) were reported in 780 patients, 23 without units. Fifty-two of 757 patients (7%) show low absolute B cell numbers when compared to age-matched reference values (range −0·004 to −0·18 × 10e9/l below age-matched reference value) [9]). Five patients with absent B lymphocytes were reported. Absolute T lymphocyte numbers (defined as CD3+ lymphocytes) were reported in 792 patients, 23 without units. Forty-five of 769 patients (6%) show low absolute T cell numbers when compared to age-matched reference values (range −0·02 to −0·85 × 10e9/l below age-matched reference values) [9]. Absent T lymphocytes were not reported.
Clinical phenotypes
We divided the hypogammaglobulinaemia children with available data on complications (n = 195 from seven countries) into the different clinical phenotypes of CVID as described by Chapel in 2008 [10]: autoimmunity (80), polyclonal lymphocytic infiltration (31), enteropathy (seven) and lymphoid malignancy (three). Additionally, the ESID Online Database contained two more relevant subgroups; ‘granulomatous infiltration’ (nine) and ‘pulmonary disease’ (107). It was not possible to define patients ‘without complications’, because of the incompleteness of the data. Some patients (42) fitted several clinical phenotypes.
No differences between countries in clinical phenotypes were observed [Fisher's exact test (n = 237) = 35·99, P = 0·720], nor between males and females (χ2(5, n = 237) = 2·576, P = 0·781). There was no association between clinical phenotype and gender [Fisher's exact test (n = 237) = 2·701, P = 0·750]. The clinical phenotypes were reported for 195 patients, 41 females (5% of the 760 females) and 154 males (12% of the 1292 males). Males were shown to be over-represented in the clinical phenotypes, over and above their over-representation in the total sample (χ2(1, n = 2052) = 23·688, P = 0·000; odds ratio for gender = 2·373). This supports the hypothesis that male patients suffer more complications, and thus have more severe disease. However, there was no specific clinical phenotype that contained more boys.
Follow-up
Not all centres reported follow-up data for (all) their patients; 1546 infectious episodes were reported in 413 patients. Czech Republic, Italy, Portugal, Serbia and the United Kingdom reported no follow-up data on infectious episodes. Most infections were localized in the respiratory tract: upper respiratory tract infection (156), pharyngitis (283), bronchitis (183), otitis media (150) and pneumonia (283). In addition, 1227 episodes of anti-microbial treatment were reported in 601 patients: antibiotics (1150), anti-fungals (28), anti-malarials (one), anti-mycobacterials (12), anti-parasitics (four) and anti-virals (32).
A total of 132 episodes of autoimmune disease were reported in 80 patients. The most frequently reported autoimmune diseases were: thyroiditis (16), coeliac disease (11), idiopathic thrombocytopenic purpura (20), insulin-dependent diabetes mellitus (14) and haemolytic anaemia (25). No differences between male and female patients were found in reported autoimmune disease (χ2(1, n = 2052) = 2·264, P = 0·132). However, country was associated with the frequency of autoimmune disease (Fisher's exact test (n = 2052) = 14·945, P = 0·000). Autoimmune diseases were reported frequently (more than 10%) in Austria (two of 14, 14%), Estonia (six of 27, 27%), Greece (13 of 32, 31%), Poland (nine of 76, 12%) and Sweden (four of 15, 27%). Age was available for only 39 patients with autoimmune disease. Mean age at onset in patients with reported autoimmune disease was 2·0 years and mean age at diagnosis was 5·5 years. The diagnostic delay was longer (2·6 years) in patients with autoimmune disease (t(1506) = −2·707, P = 0·007).
Three cases of lymphoma were reported during follow-up, one each in Germany, Turkey and Spain; these were all male patients. One case of acute lymphoblastic leukaemia was reported in a Dutch girl, and one case of a benign neoplasm of bone and articular cartilage was reported in a Spanish girl. Other concomitant diseases were reported in 605 patients, ranging from possibly related issues such as hepatosplenomegaly and Down syndrome to unrelated issues such as concussion and dizziness. Only ‘asthma’ was reported in more than 1% of the total group (n = 76, 4%). It is not clear whether this is truly asthma, or a misdiagnosis of airway disease caused by the immunodeficiency.
Discussion
Hypogammaglobulinaemia in childhood is the laboratory manifestation of an extremely heterogeneous group of diseases; solid criteria for the diagnosis are difficult to define. The objective of this study was to obtain insight into the general, clinical and laboratory characteristics of children with hypogammaglobulinaemia registered in the ESID Online Database and to determine whether or not different clinical phenotypes could be defined. Analysis of the data was performed in the combined data set, because of country-related differences in diagnostic criteria used for classification of different types of primary hypogammaglobulinaemia. Although this hampers comparison with separate diagnostic groups, the overall data are still very useful. No such large data set in children with hypogammaglobulinaemia has been reported so far.
Strikingly, male patients are clearly in the majority (63%), whereas the diseases in the subregistry ‘Hypogammaglobulinaemias’ are not known to be inherited by the X-linked route. This male predominance was seen in every country. Furthermore, their younger age at onset and at diagnosis suggests more severe disease. This is supported by the occurrence of more complications in boys (12%) compared to girls (5%). Although not previously discussed explicitly, this predominance of males can be found in previously published papers [1,10,11]. In the study by Chapel et al., in which CVID patients are classified into different clinical phenotypes, 58% of patients were males (mean age 49·4 years) [10]. Also, in the latest update from the ESID Registry Working Party, 60·8% of all registered patients were male, and 64·6% in the group of 15 years or younger [1]. A recent update from the German registry for PID also shows a higher percentage of boys: 57·2% male in the overall group and twice as many boys in children below 12 years of age [11]. They attributed this male predominance to known X-linked diseases such as X-linked agammaglobulinaemia (XLA) and Wiskott–Aldrich syndrome (WASP). This can be an explanation for the male predominance overall, but it cannot explain the higher percentage of boys in the group of children with primary hypogammaglobulinaemia, and suggests that this group also contains patients with diseases inherited by the X-linked route. This can be due to either insufficient diagnostic procedures or atypical presentation (i.e. a known X-linked PID diagnosis has been missed in both cases), or to as yet unknown X-linked diseases that cause primary hypogammaglobulinaemia. Little is known as yet about the genetic defects in primary hypogammaglobulinaemias. Genetic defects in CVID patients have been discovered in past years [e.g. inducible co-stimulator (ICOS), TACI, B cell activator of the tumour necrosis factor-α family receptor (BAFF-R), CD19 complex and CD20], but in more than 95% of CVID patients the genetic background is as yet unknown [2]. A search for known and unknown X-linked genetic defects in boys with primary hypogammaglobulinaemia could be very interesting. Focusing on boys with early-onset hypogammaglobulinaemia and complications will probably provide the highest yield. At least, they need to be evaluated for B lymphocyte numbers and known X-linked disease, such as a mutation in the Bruton's agammaglobulinaemia tyrosine kinase gene.
A recent study from Driessen et al. showed that patients with idiopathic primary hypogammaglobulinaemia (IPH), defined as patients with hypogammaglobulinaemia who do not fulfil the diagnostic criteria for CVID, suffered from the same infectious complications as CVID patients (i.e. respiratory infections, bronchiectasis) but did not show the non-infectious complications (autoimmune cytopaenia, polyclonal lymphocytic proliferation and persistent enteropathy) [12], which have been linked to increased mortality [10,13]. In our study, autoimmune disease, polyclonal lymphocytic proliferation and enteropathy were reported in a minority of the total group of hypogammaglobulinaemias (4%). Our data are compatible with the above interpretation, demonstrating a subgroup of patients with early onset of immune dysregulation.
Unfortunately, the current ESID Online Database has several limitations, making interpretation of the data challenging; many data had to be excluded, and the determination of different clinical or laboratory phenotypes was not possible. Currently, the ESID Registry Working Party is reorganizing the database. There will be three levels of documentation. Level 1 will consist of a baseline form at initial registration and a short yearly follow-up form; this level will offer good insight into the prevalence of PID. Level 2 will consist of a diagnosis form and a current visit form, which is specific for each IUIS category; this level will provide additional information on individual diseases. Temporary level 3 forms can be used for collecting in-depth information in dedicated studies. Because the quality of the data is dependent upon the users who enter the data, more complex automated checks based on diagnostic criteria, training sessions with a uniform protocol containing clear definitions of diseases and the meaning of data fields and increased local as well as central monitoring are being considered. National registries benefited from employing data entry clerks, especially in the reporting rate [11,14]. With the new database, more research on children with hypogammaglobulinaemia will be possible in the future; the level 3 option will be particularly suited for this purpose.
In conclusion, this study gives a first overview of the general characteristics of children with primary hypogammaglobulinaemia as registered in the current ESID Online Database. Determining different clinical or laboratory phenotypes was not possible because of the limitations in the collected data. The male predominance in the group of children with hypogammaglobulinaemias suggests that X-linked diseases are ‘hidden’ in this group. This finding warrants further exploration.
Acknowledgments
This study was approved and supported by the ESID Registry Working Party (Chair Professor Dr S. Ehl). We thank all patients and their parents for their consent to be registered in the ESID Online Database.
Disclosure
There are no conflicts of interest.
Author contributions
E. J. H. S. and E. d. V. designed the study and wrote the manuscript. E. J. H. S. and R. W. N. M. v. H. performed the statistical analysis of the data. B. G. represents the ESID Registry Working Party and extracted the data for analysis. All the authors in the PedPAD consortium were responsible for the data entry in the ESID Online Database for their centre. R. W. N. M. v. H., B. G. and all the authors in the PedPAD consortium contributed to the final version of the manuscript.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Number of reported patients for each diagnosis in the different countries in the European Society for Immunodeficiencies (ESID) online database.
References
- 1.Gathmann B, Grimbacher B, Beaute J, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol. 2009;157(Suppl. 1):3–11. doi: 10.1111/j.1365-2249.2009.03954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Burg M, van Zelm MC, Driessen GJ, van Dongen JJ. New frontiers of primary antibody deficiencies. Cell Mol Life Sci. 2012;69:59–73. doi: 10.1007/s00018-011-0836-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guzman D, Veit D, Knerr V, et al. The ESID Online Database network. Bioinformatics. 2007;23:654–655. doi: 10.1093/bioinformatics/btl675. [DOI] [PubMed] [Google Scholar]
- 4. Diagnostic criteria PID. Available at: http://www.esid.org (accessed 24 February 2014)
- 5.Eades-Perner AM, Gathmann B, Knerr V, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2004–06. Clin Exp Immunol. 2007;147:306–312. doi: 10.1111/j.1365-2249.2006.03292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gathmann B, Binder N, Ehl S, Kindle G. The European internet-based patient and research database for primary immunodeficiencies: update 2011. Clin Exp Immunol. 2012;167:479–491. doi: 10.1111/j.1365-2249.2011.04542.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol. 2011;2:54. doi: 10.3389/fimmu.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Vries E. Patient-centred screening for primary immunodeficiency, a multi-stage diagnostic protocol designed for non-immunologists: 2011 update. Clin Exp Immunol. 2012;167:108–119. doi: 10.1111/j.1365-2249.2011.04461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schatorje EJ, Gemen EF, Driessen GJ, et al. Age-matched reference values for B-lymphocyte subpopulations and CVID classifications in children. Scand J Immunol. 2011;74:502–510. doi: 10.1111/j.1365-3083.2011.02609.x. [DOI] [PubMed] [Google Scholar]
- 10.Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–286. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 11.Gathmann B, Goldacker S, Klima M, et al. The German national registry for primary immunodeficiencies (PID) Clin Exp Immunol. 2013;173:372–380. doi: 10.1111/cei.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Driessen GJ, Dalm VA, van Hagen PM, et al. Common variable immunodeficiency and idiopathic primary hypogammaglobulinemia: two different conditions within the same disease spectrum. Haematologica. 2013;98:1617–1623. doi: 10.3324/haematol.2013.085076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapel H, Lucas M, Patel S, et al. Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Allergy Clin Immunol. 2012;130:1197–8 e9. doi: 10.1016/j.jaci.2012.05.046. [DOI] [PubMed] [Google Scholar]
- 14.Le Centre de Référence Déficits Immunitaires Héréditaires (CEREDIH) The French national registry of primary immunodeficiency diseases. Clin Immunol. 2010;135:264–272. doi: 10.1016/j.clim.2010.02.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Number of reported patients for each diagnosis in the different countries in the European Society for Immunodeficiencies (ESID) online database.