

Abstract
Background and Purpose
Transient receptor potential vanilloid subtype 3 (TRPV3) is implicated in nociception and certain skin conditions. As such, it is an attractive target for pharmaceutical research. Understanding of endogenous TRPV3 function and pharmacology remains elusive as selective compounds and native preparations utilizing higher throughput methodologies are lacking. In this study, we developed medium-throughput recombinant and native cellular assays to assess the detailed pharmacological profile of human, rat and mouse TRPV3 channels.
Experimental Approach
Medium-throughput cellular assays were developed using a Ca2+-sensitive dye and a fluorescent imaging plate reader. Human and rat TRPV3 pharmacology was examined in recombinant cell lines, while the mouse 308 keratinocyte cell line was used to assess endogenous TRPV3 activity.
Key Results
A recombinant rat TRPV3 cellular assay was successfully developed after solving a discrepancy in the published rat TRPV3 protein sequence. A medium-throughput, native, mouse TRPV3 keratinocyte assay was also developed and confirmed using genetic approaches. Whereas the recombinant human and rat TRPV3 assays exhibited similar agonist and antagonist profiles, the native mouse assay showed important differences, namely, TRPV3 activity was detected only in the presence of potentiator or during agonist synergy. Furthermore, the native assay was more sensitive to block by some antagonists.
Conclusions and Implications
Our findings demonstrate similarities but also notable differences in TRPV3 pharmacology between recombinant and native systems. These findings offer insights into TRPV3 function and these assays should aid further research towards developing TRPV3 therapies.
Linked Articles
This article is part of a themed section on the pharmacology of TRP channels. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2014.171.issue-10
Keywords: TRPV3, TRP channel, FLIPR, mouse 308 keratinocytes
Introduction
A subset of the transient receptor potential (TRP) superfamily of ion channels, known as temperature-sensitive TRPs (thermoTRPs), including TRPA1, TRPV1, TRPV2, TRPV3, TRPV4 and TRPM8, activate in response to specific ranges in temperature, suggesting that they serve as temperature sensors on a molecular level (reviewed in Wu et al., 2010). However, as these channels are also activated by other diverse stimuli including chemical, osmotic, pH and mechanical challenges, their role as cellular sensors is likely much broader. Integration of these diverse stimuli appears to be a common feature of thermoTRPs. Their expression in peripheral sensory neurons and in the skin, nose and epithelia further implicates these receptors as sensors of environmental cues and in pain signalling. The role of thermoTRPs in pain signalling is the most heavily researched field to date (reviewed in (Broad et al., 2009; Moran et al., 2011).
Transient receptor potential vanilloid subtype 3 (TRPV3) is activated by innocuous temperature (31–39°C) and rather uniquely becomes sensitized with repeated heat pulses due to hysteresis of channel gating (Peier et al., 2002; Smith et al., 2002; Xu et al., 2002; Liu et al., 2011). Sensitization of the heat response can also be produced by chemical agonists (Moqrich et al., 2005). A number of non-selective, exogenous TRPV3 chemical agonists have been identified, including extracts from spices such as camphor, carvacrol, thymol and eugenol, with potencies in the high μM–low mM range (Moqrich et al., 2005; Vogt-Eisele et al., 2007). Synthetic chemical agonists include 2-aminoethoxydiphenylborate (2-APB; Chung et al., 2004a; Xu et al., 2006), its analogue diphenylborinic anhydride (DPBA; Chung et al., 2005) and terpenoid 6-tert-butyl-m-cresol (6-TBC; Vogt-Eisele et al., 2007; Supporting Information Figure S1A). Incensole acetate (IA), from Boswellia resin, is reported to be a TRPV3 agonist (Moussaieff et al., 2008), while a selective endogenous TRPV3 ligand, farnesyl pyrophosphate (FPP), has also recently been identified (Bang et al., 2010). As described for temperature, repeated bursts of chemical agonist stimulation can lead to sensitization of the receptor (Moqrich et al., 2005; Xiao et al., 2008). In addition, co-application of chemical agonists, likely acting at distinct sites on the channel, can lead to synergistic TRPV3 activation (Xu et al., 2006; Cheng et al., 2010). Activation of Gq coupled GPCRs is reported to potentiate TRPV3 activity (Xu et al., 2006), consistent with potentiation by several Gq receptor downstream signalling elements, including protein kinase C (Hu et al., 2006), Ca2+-calmodulin (Xiao et al., 2008; Phelps et al., 2010) and unsaturated fatty acids, such as arachidonic acid (AA; Supporting Information Figure S1B; Hu et al., 2006).
No selective TRPV3 antagonists are commercially available, hence a non-selective TRP channel blocker ruthenium red (RR) has been commonly employed to study TRPV3 function (Peier et al., 2002; Smith et al., 2002; Xu et al., 2002; Bang et al., 2010). Recently, however, selective TRPV3 antagonists have been disclosed by Hydra Biosciences (Chong et al., 2006; Moran et al., 2008) and Glenmark Pharmaceuticals (Lingam et al., 2009) and should prove useful tools in furthering our understanding of this channel (Supporting Information Figure S1C).
The lack of selective TRPV3 agonist and antagonist tools has hampered progress in defining the physiological roles of TRPV3 channels; however, roles in thermo-sensing, nociception and skin physiology and pathophysiology are emerging. With regards to nociception, FPP is reported to elicit pain behaviours after intraplantar injection into inflamed animals (Bang et al., 2010) and 17(R)-resolvin D, a recently identified endogenous TRPV3 antagonist, is reported to be anti-nociceptive in acute and inflammatory pain states (Bang et al., 2012). In addition, several preliminary reports elude to efficacy of selective exogenous TRPV3 antagonists in preclinical models of inflammatory and neuropathic pain (Moran et al., 2007; Lingam et al., 2009; reviewed in Joshi et al., 2010). Sanofi reports to be in Phase II with their TRPV3 antagonist SAR292833 (GRC15300) for the same indications (see Sanofi website). In contrast, TRPV3 null mice appear to have largely unaltered responses in pain models compared to control animals, showing only disruption in responses to acute noxious heat and deficits in innocuous thermal sensation (Moqrich et al., 2005). One possible complication of projecting these findings to human pain states is the variation in tissue expression profile across species. Whereas in rodents, TRPV3 appears to be mainly expressed in keratinocytes, its presence in peripheral and central nervous tissue remains contradictory (Peier et al., 2002; Bender et al., 2005; Guatteo et al., 2005; Kitahara et al., 2005; Asakawa et al., 2006; Lipski et al., 2006; Yang et al., 2006; Christoph et al., 2008). In contrast, primates and humans express TRPV3 in the CNS and dorsal root ganglia neurons as well as in keratinocytes (Smith et al., 2002; Xu et al., 2002).
In line with the high expression of TRPV3 in skin across species (Peier et al., 2002; Smith et al., 2002; Xu et al., 2002; 2006; Asakawa et al., 2006), there are several in vitro reports describing native TRPV3-mediated responses in primary keratinocytes (Chung et al., 2004b; 2005; Moqrich et al., 2005; Huang et al., 2008; Cheng et al., 2010) and in a mouse keratinocyte cell line (Hu et al., 2006; Xu et al., 2006). In mouse skin, TRPV3 promotes epidermal barrier formation and hair morphogenesis, both of which are disrupted in TRPV3 null mice (Moqrich et al., 2005; Cheng et al., 2010). TRPV3 is proposed to form a signalling complex with the EGF receptor (EGFR), whereby activation of EGFR leads to increased TRPV3 channel activity, stimulation of TGF-α release and epidermal homeostasis (Cheng et al., 2010). Adding further support to the role of TRPV3 in skin physiology and pathology, a TRPV3 gain-of-function G573S/C mutation was identified to underlie the spontaneously hairless, dermatitis phenotype of two rodent strains, DS-Nh mice and WBN/kob-HT rats, used as animal models of atopy (Asakawa et al., 2006; Yoshioka et al., 2009). Development of a TRPV3Gly573Ser transgenic mouse, recapitulated this phenotype, with significantly increased scratching behaviour (Yoshioka et al., 2009). In line with this, TRPV3 has been shown to promote the release of pro-inflammatory mediators and pruritigens, including ATP, nerve growth factor, chemokines and ILs from cultured keratinocytes (Xu et al., 2006; Huang et al., 2008; Mandadi et al., 2009; Yoshioka et al., 2009). More recently, similar mutations in TRPV3 have been identified as the cause of Olmsted syndrome in humans, a rare disorder characterized by keratoderma, alopecia and severe itching (Lai-Cheong et al., 2012; Lin et al., 2012). It is tempting to speculate that TRPV3 antagonists will be useful for the treatment of these patients and also more broadly for inflammatory skin conditions and associated itch.
In this paper, we have developed medium-throughput assays allowing detailed study of recombinant human and rat TRPV3 receptors and native TRPV3 receptors in the mouse 308 keratinocyte (m308k) cell line. Using pharmacological and genetic approaches, we were able to optimize the m308k assay and overcome the difficulty of using a complex system, expressing multiple TRP channels. Importantly, by studying the recombinant and native context, we describe important similarities and differences between the two systems.
Methods
Compounds and nomenclature
2-APB, AA, carvacrol, DPBA, eugenol, linoleic acid (LA), RR, thymol and FPP were purchased from Sigma-Aldrich (Poole, UK). Camphor was from Fluka and 6-TBC was from Acros (Loughborough, UK). IA was derived in house. TRPV3 selective antagonists were synthesized in house. Receptor nomenclature conforms to the BJP guidelines (Alexander et al., 2013).
Cell culture
Human and rat TRPV3 human embryonic kidney cells (HEK293) were cultured in DMEM (Invitrogen, Paisely, UK), 5–10% FCS (PAA), and 0.6–1 mg·mL−1 G418 (Invitrogen). m308ks were obtained from the National Cancer Institute (Bethesda, MD, USA). Unmodified and knock-down (KD) m308k cell lines were grown in Eagle's minimal essential medium (Lonza, Walkersville, MD, USA), 10% FBS, 10% lipumin (PAA), and in the case of KD cells, 4 μg·mL−1 puromycin (Sigma-Aldrich). All cells were maintained in a humidified 37°C/5% CO2 incubator. A day before conducting Ca2+-imaging experiments, cells were seeded onto 96-well, black-walled, transparent bottom, poly-D-lysine-coated fluorescent imaging plate reader (FLIPR) plates (BioCoat, Becton-Dickenson, Oxford, UK) at a density of 20 000 (m308k) or 50 000 (recombinant) cells per well.
Cell line generation
The human and rat TRPV3 monoclonal HEK293 cells were generated by Cerep SA (Poitiers, France). The coding sequences corresponding to NM_145068 (human) and AY325813.1 (rat) were synthesized chemically. A SalI restriction enzyme (RE) site and a Kozak sequence was added upstream of the ATG start codon, and a NotI RE site was engineered downstream of the stop codon. The gene was cloned into the pCI-Neo vector downstream of a cytomegalovirus (CMV) promoter between the SalI and NotI RE sites. Monoclonal cell lines were generated following plasmid transfection and G418 selection. It was noted that the rat AY325813.1 cDNA sequence differed from the rat genomic sequence. Applying GeneWise, a tool that allows for comparison between a protein and a genomic DNA sequence (Birney et al., 2004), on the rat RGSC v3.4 genome assembly (NC_005109.2), we derived a new DNA coding (NM_001025757.2) and protein (NP_001020928.2) sequence. The DNA coding sequence was codon-optimized and chemically synthesized in vitro. A HindIII RE site and a Kozak sequence was added upstream of the ATG start codon, and an XbaI RE site was placed downstream of the stop codon. The gene was inserted using the HindIII and XbaI RE sites into the pcDNA3.1 vector under control of a CMV promoter (GeneArt AG). A polyclonal rat TRPV3 HEK293 cell line was generated following transfection with Lipofectamine 2000 (Invitrogen) and selection with G418. To generate TRPV3 and control KD cell lines, m308k cells were transfected with a pLK0.1 plasmid expressing a small hairpin RNA (shRNA) against mouse TRPV3 (Thermo Scientific, Waltham, MA, USA; TRCN0000068858) or a non-target (control) shRNA (Sigma-Aldrich). After transfection with Lipofetamine 2000, polyclonal cell lines were generated following selection with puromycin.
Measurement of [Ca2+]i using FLIPR
Media was removed and cells were incubated in loading buffer (HBSS, in mM, 1.3 CaCl2, 0.5 MgCl2, 20 HEPES, 2.5 probenecid, pH 7.4) in the presence of 5 μM Fluo 4-AM/0.02% pluronic F127 (Invitrogen) for 1 h in the dark at 21°C. The solution was then replaced with assay buffer (HBSS, in mM, 0.5 CaCl2, 0.9 MgCl2, 20 HEPES, 2.5 probenecid, pH 7.4) and the plate transferred onto the FLIPR (Molecular Devices, Sunnyvale, CA, USA). Where indicated, LA or AA was added to the buffer and the cells incubated at 21°C for 10 min prior to transfer onto the FLIPR. Compound addition was automated; antagonist was added first and agonist added 3 min later (for agonist synergy experiments both agonists were added simultaneously). Fluo-4 fluorescence was excited with an argon-ion laser at 488 nm and emission was captured through a 510–570 nm bandpass interference filter. Data were captured at 1 s intervals at 0.4 s exposure. The maximum change in fluorescence (relative fluorescence units) was exported into Excel and replicates from three cell plates were combined to calculate a mean ± SEM. To obtain EC50 values, independent replicate data from n = 3 days were fitted to a four-parameter logistic curve fit model using GraphPad Prism. Statistical significance was assessed using an F-test, taking into account logEC50 values, to compare data series.
Fluorescence-based single-cell Ca2+ imaging
Transfections were performed using Lipofectamine 2000 and 150 ng plasmid alone or plasmid expressing human TRPV3, rat TRPV3 (AAQ90060.1) or rat TRPV3 (NP_001020928.2) receptor. After 24 h, cells were incubated in Hank's balanced salt solution buffer (Invitrogen) containing 4 μM Fluo-4 AM/0.1% pluronic acid (Invitrogen) for 1 h in the dark at room temperature. Cells were washed and continually perfused during the experiment with HBTS containing (in mM) 135 NaCl, 5 KCl, 1.2 MgCl2, 2.5 CaCl2, 10 HEPES, 11 glucose, pH 7.2. Dye-loaded cells were viewed using an inverted epifluorescence microscope (Axiovert, 135TV, Zeiss, Cambridge, UK). Fluo-4 fluorescence was excited at 480 ± 10 nm and emission was captured after passage through a dichroic mirror (505LP nm) and a high pass barrier filter (515LP nm). Images were processed using Imaging Workbench 5.0 software (INDEC Biosystems, Santa Clara, CA). Data were analysed by averaging individual traces collected from a large number of cells in multiple wells of the 96-well plate. Delta F/F0 values were measured by quantifying the ratio between the change in fluorescence signal intensity (delta F) and baseline fluorescence (F0).
RT-PCR
Unmodified, recombinant HEK293 and m308k cells were processed for RNA using the PARIS kit (Ambion, Paisley, UK) according to manufacturer's instructions. RNA was treated with TURBO DNase (Ambion) and quantified by spectrophotometer. Reverse transcription (RT) reactions were performed at 37°C for 1 h, and inactivated at 70°C for 5 min. The final 20 μL reaction included 300 ng RNA, 1 x reaction buffer with MgCl2, 1.25 μM random hexamers, 1.25 μM oligo-dT primers, 0.25 mM of each dNTP, 20 U RNase inhibitor, 40 U M-MulV-RT (First Strand cDNA Synthesis Kit, Fermentas, Loughborough, UK). Negative control reactions were set up in the absence of M-MulV-RT. The PCR was performed in 50 μL using a standard PCR machine and reagents from the PCR master kit (Fermentas). Reactions containing 150 ng cDNA, 1 μM of each primer and 1x Master Mix were incubated using the following cycling conditions: 94°C for 3 min, and 35 cycles at 94°C for 30 s, 58°C for 30 s and 72°C for 45 s. The primers used were rat TRPV3_F:ACGGTGGAGAACGTCTCC; rat TRPV3_R:TG TCCGTCTTATGGGCCC; human TRPV3_F:GCTGAAGAAGCGCATCTTTGCA; human TRPV3_R:TCATAGGCCTCCTCTGTGTACT; mouse TRPV3_F:ACGGTGGAGAACGTC TCC; mouse TRPV3_R:TGTCCGTCTTATGGGTCC. The PCR product was resolved on a 3% agarose gel in Tris-borate-EDTA buffer and visualized by ethidium bromide staining.
Western blot
Protein lysates were prepared from unmodified and recombinant TRPV3 HEK293 cells using the Cell Disruption Buffer supplied in the PARIS kit (Ambion) and protease inhibitor cocktail (Roche, Burgess Hill, UK). Solubilized lysates were clarified by centrifugation and the protein concentration was determined by Bradford assay (Bio-Rad, Hemel Hempstead, UK). Samples were prepared in reducing Laemmli buffer (Bio-Rad), boiled for 5 min and resolved on a 4–12% Bis-Tris NuPAGE gel (Invitrogen). The proteins were transferred onto a Hybond membrane (GE Healthcare, Amersham, UK), blocked in 5% milk (Marvel, Premier Foods, St Albans, UK) in Tris-buffered saline tween (20 mM Tris-Cl, 154 mM NaCl, 0.1% Tween-20, pH 7.4) and then incubated with anti-TRPV3 monoclonal mouse antibody (Ab) (NeuroMab, Davis, CA, USA; 1:1000 dilution) overnight at 4°C. The primary Ab was detected with goat anti-mouse-HRP Ab (Sigma-Aldrich, 1:10 000 dilution), followed by signal development with enhanced chemiluminescence reagent (Amersham, Amersham, UK) and image detection.
Determination of TRPV3 KD by RT-qPCR
RNA was isolated from the unmodified and shRNA-expressing m308k cells using the RNeasy kit (Qiagen, Crawley, UK) according to manufacturer's instructions. The RNA was treated with TURBO DNase (Ambion) and quantified by spectrophotometer. RT reactions were performed using 2 μg of RNA, 1 x reaction buffer, 2.5 mM MgCl2, 2.5 μM random hexamers, 0.25 mM of each dNTP, 40 U RNase inhibitor, 150 U MMLV-RT (Applied Biosystems, Warrington, UK) in 100 μL as described for RT-PCR. The quantitative PCR (qPCR) was performed in a 384-well plate containing 10 μL reactions in quadruplicate. Reactions contained 80–2.5 ng cDNA, 300 nM of each primer, and 1x SYBR Green Master Mix (Qiagen). The qPCR was done on an ABI Prism 7900 (Applied Biosystems) at 50°C for 2 min, 95°C for 10 min, and 40 cycles at 95°C for 15 s and 60°C for 1 min. The mouse primer sequences utilized were mTRPV3_F:ATCATCGTCTACAACACCAACAT; mTRPV3_R:AAGCAGAAGGACAAG AAGAACAT; PSMB2_F:AAATGCGGAATGGATATGAATTG; PSMB2_R:GAAGACA GTCAGCCAGGTT. The Ct values were determined automatically by the SDS 2.2 software, and the average values (Ctav) from quadruplicates were calculated. These were then averaged with values obtained from two independent qPCR experiments. Primer efficiency, determined by serial dilution of the template, was calculated to be near 100%. Changes in TRPV3 mRNA levels were measured with the ΔΔCt method, using PSMB2 (proteasome subunit beta type 2) as the housekeeping reference and the unmodified m308k cells as the calibrator.
Results
Enablement of the human TRPV3 recombinant cellular assay
A FLIPR was used to detect changes in intracellular Ca2+ in response to pharmacological treatment of Fluo-4 loaded HEK293 cells stably-expressing human TRPV3 (hTRPV3). A non-selective TRPV3 agonist DPBA elicited increases in intracellular Ca2+ in hTRPV3 cells yielding an EC50 value ∼68.5 μM (Figure 1A). At concentrations above 100 μM, non-TRPV3-mediated Ca2+ responses were observed in untransfected HEK293 cells (Figure 1B). This non-TRPV3 mediated response was not dependent on extracellular Ca2+, but was significantly inhibited (P < 0.001) by pretreatment of the cells with thapsigargin, an inhibitor of the sarcoplasmic reticulum Ca2+-ATPase pump, indicating that the Ca2+ derived from intracellular stores. 40–80 μM DPBA was selected for subsequent experiments as it selectively activated hTRPV3 mediated responses (indicated by arrow). Similar results were obtained using a second chemically related non-selective TRPV3 agonist, 2-APB (data not shown).
Figure 1.
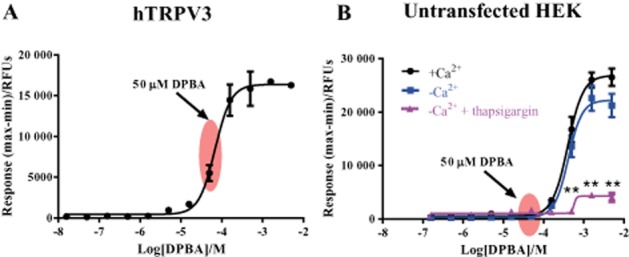
Ca2+ responses ( ) to increasing concentrations of DPBA in human recombinant TRPV3 (A) and untransfected (B) HEK293 cells. In HEK293 cells, responses were also recorded in the absence of extracellular Ca2+ (
) to increasing concentrations of DPBA in human recombinant TRPV3 (A) and untransfected (B) HEK293 cells. In HEK293 cells, responses were also recorded in the absence of extracellular Ca2+ ( ) and in the absence of extracellular Ca2+ where internal Ca2+ stores had been depleted by pre-incubation with 1 μM thapsigargin (
) and in the absence of extracellular Ca2+ where internal Ca2+ stores had been depleted by pre-incubation with 1 μM thapsigargin ( ). Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from four separate experiments. **P < 0.001 versus responses in the presence of Ca2+. RFU, relative fluorescence units.
). Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from four separate experiments. **P < 0.001 versus responses in the presence of Ca2+. RFU, relative fluorescence units.
Enablement of the rat TRPV3 recombinant cellular assay
Initially, the rat TRPV3 cDNA deriving from GenBank sequence AY325813.1 (protein id AAQ90060.1) was cloned into a mammalian expression vector and used to generate monoclonal rat TRPV3 (rTRPV3) HEK293 cell lines. Clones 47 and 66 were selected for further characterization by FLIPR, but failed to display TRPV3-specific signals in response to DPBA (Figure 2A) or 2-APB (not shown). TRPV3 mRNA and protein were readily detected in both clones by RT-PCR and Western blot, respectively (Supporting Information Figure S2A and B), indicating that the lack of rat TRPV3 function was not due to lack of expression or protein instability.
Figure 2.
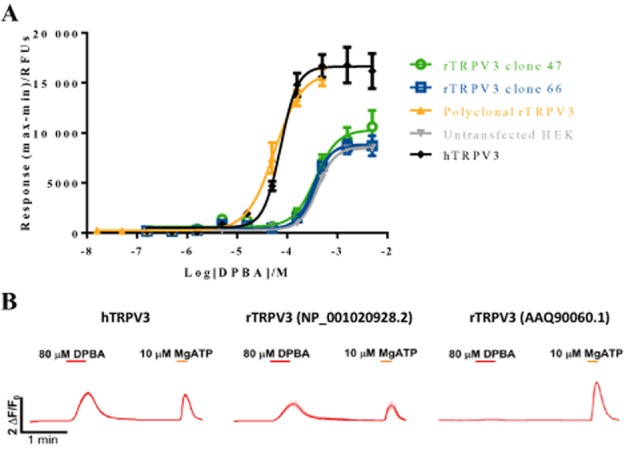
(A) Ca2+ responses to DPBA in untransfected ( ) and recombinant HEK293 cells stably expressing human TRPV3 (
) and recombinant HEK293 cells stably expressing human TRPV3 ( ), monoclonal rat TRPV3 clones 47 or clone 66 (
), monoclonal rat TRPV3 clones 47 or clone 66 ( and
and  ) or polyclonal rat TRPV3 (
) or polyclonal rat TRPV3 ( ). Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from three replicate wells. (B) Characterization of TRPV3 responses in transiently transfected HEK293 cells by fluorescence-based, intracellular Ca2+ imaging. Shown are averages from traces obtained from single-cell responses to DPBA for the various transfection conditions. ATP serves as an internal control for activation of P2 receptors leading to increases in intracellular calcium.
). Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from three replicate wells. (B) Characterization of TRPV3 responses in transiently transfected HEK293 cells by fluorescence-based, intracellular Ca2+ imaging. Shown are averages from traces obtained from single-cell responses to DPBA for the various transfection conditions. ATP serves as an internal control for activation of P2 receptors leading to increases in intracellular calcium.
Comparison with the rat genomic sequence helped identify 10 divergent amino acid residues (Supporting Information Figure S2C). A new rat TRPV3 construct was made, based on the NC_005109.2 (NP_001020928.2) sequence and tested by transient transfection in HEK293 cells. TRPV3 protein was readily detected in all orthologs by Western blot (Supporting Information Figure S2D). However, while 80 μM DPBA evoked increases in intracellular Ca2+ in cells transfected with the new rat TRPV3 construct or human TRPV3, used as a positive control (Figure 2B), the rat TRPV3 (AAQ90060.1) transfection behaved just as the plasmid alone negative control, with no responding cells. Addition of ATP, an agonist of P2-type receptors, resulted in transient increases in intracellular Ca2+, demonstrating that general Ca2+ signalling is functional under the conditions tested. Following these results, a polyclonal rat TRPV3 HEK293 cell line was generated using the new rat TRPV3 construct based on the NP001020928.2 sequence. This polyclonal rTRPV3 cell line displayed TRPV3-mediated increases in intracellular Ca2+ in response to DPBA with an EC50 value significantly different to those of the original rTRPV3 clones (P < 0.0001), and with a similar potency range as observed for human TRPV3 (Figure 2A).
Non-specific Ca2+ signalling in the m308ks
We next sought to confirm the activity of compounds in a native setting. To study the native TRPV3 receptor, we focused on the m308k cell line, which derives from immortalized cells from adult BALB/c mouse skin and expresses endogenous TRPV3, as well as TRPV1, 2 and 4 (Chung et al., 2003). Application of DPBA to m308k cells evoked increases in intracellular Ca2+; however, responses were seen only at relatively high DPBA concentrations (>50 μM) and were unaltered by removal of extracellular Ca2+ or addition of a non-selective TRP channel blocker, RR or a selective TRPV3 antagonist, 1-({[3-fluoro-5-(trifluoromethyl)pyridine-2-yl]sulfanyl}acetyl)-8-methyl-1,2,3,4-tetrahydroquinoline (FTP-THQ) (Table 1), P = 0.067 (Figure 3). Similar results were observed using a panel of other reported non-selective TRPV3 agonists, including carvacrol, camphor, thymol, eugenol, 6-TBC and 2-APB (Figure 4).
Table 1.
Summary of the pharmacological profile of FTP-THQ on a panel of thermo-TRP receptors
| hTRPV3 | hTRPV1 | hTRPV4 | hTRPM8 | hTRPA1 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | EC50 (μM) | IC50 (μM) | EC50 (μM) | IC50 (μM) | EC50 (μM) | IC50 (μM) | EC50 (μM) | IC50 (μM) | EC50 (μM) | |
| FTP-THQ | 0.135 | >60 | >36.9 | >73.5 | >30 | >60 | 2.8 | >50 | >50 | 25 |
Figure 3.
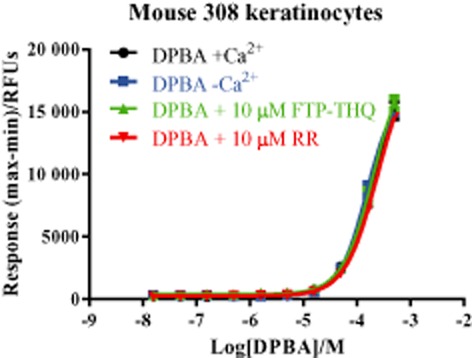
In m308ks, DPBA alone does not activate TRPV3. Mouse keratinocytes were exposed to increasing concentrations of DPBA and changes in fluorescence monitored. Responses were recorded in the presence ( ) and absence (
) and absence ( ) of extracellular Ca2+ and also after pre-incubation with 10 μM FTP-THQ (
) of extracellular Ca2+ and also after pre-incubation with 10 μM FTP-THQ ( ) or 10 μM RR (
) or 10 μM RR ( ). Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from three separate experiments. RFU, relative fluorescence units.
). Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from three separate experiments. RFU, relative fluorescence units.
Figure 4.

In m308ks, certain TRPV3 agonists synergize with DPBA to activate TRPV3. Ca2+ responses obtained in m308k cells in response to (A) carvacrol, (B) camphor, (C) thymol, (D) eugenol, (E) 6-TBC, (F) 2-APB alone ( ) or together with 50 μM DPBA (
) or together with 50 μM DPBA ( ). Responses to co-agonist application were also assessed after pre-incubation with FTP-THQ (
). Responses to co-agonist application were also assessed after pre-incubation with FTP-THQ ( ) or 10 μM RR (
) or 10 μM RR ( ). Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from three separate experiments. RFU, relative fluorescence units.
). Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from three separate experiments. RFU, relative fluorescence units.
Agonist synergy reveals TRPV3-specific activation in the m308k cells
As previous reports indicated that co-application of TRPV3 chemical agonists can increase TRPV3 mediated responses in mouse keratinocytes (Xu et al., 2006; Cheng et al., 2010), we next investigated the effects of agonist co-application in our system. As shown in Figure 4, a number of non-selective TRPV3 agonists displayed clear synergistic activation of TRPV3 on co-application with DPBA. Concentration response curves to carvacrol, camphor, thymol and 6-TBC were significantly (P < 0.001) leftward shifted in the presence of 50 μM DPBA. This leftward shift was reversed in the presence of non-selective TRP channel blocker RR (10 μM) and TRPV3 selective antagonist, FTP-THQ (10 μM), confirming it to be TRPV3 mediated. While 2-APB did not synergize with DPBA, it did synergize with carvacrol (data not shown). In contrast, eugenol failed to synergize with any agonist assessed in the current study (e.g. DPBA, Figure 4).
Linoleic and AA potentiate TRPV3 activity in m308k cells
Next we investigated the reported potentiation of TRPV3-mediated responses by unsaturated fatty acids AA and LA (Hu et al., 2006; Parnas et al., 2009). Initial experiments confirmed that concentrations of LA and AA previously reported to potentiate 2-APB, also potentiated 2-APB and DPBA-evoked responses in our system (Figure 5). At the concentrations tested, LA (40, 160 μM) and AA (10, 30 μM) alone did not significantly activate TRPV3, but did significantly potentiate DPBA-evoked responses in a concentration-dependent manner, with a more robust potentiation observed at the higher concentrations of poly-unsaturated fatty acids (PUFAs; Figure 5A). Concentration response curves to DPBA and 2-APB in m308k cells were significantly leftward shifted in the presence of LA (160 μM) and AA (30 μM; Figure 5B). This leftward shift was reversed in the presence of RR (10 μM) and FTP-THQ (10 μM), confirming it to be TRPV3 mediated. In contrast, m308k cell responses to the other TRPV3 agonists, including camphor, carvacrol, 6-tert, thymol and eugenol, were not potentiated by LA or AA (e.g. camphor, Figure 5B).
Figure 5.

Fatty acids LA and AA synergize with DPBA and 2-APB to activate TRPV3 in m308ks. (A) Effects of the fatty acids LA (40, 160 μM) and AA (10, 30 μM) alone or in combination with DPBA in m308ks. (B) Concentration response curves to DPBA, 2-APB or camphor in the absence ( ) or presence of 160 μM LA or 30 μM AA (
) or presence of 160 μM LA or 30 μM AA ( ). Responses to agonist in the presence of LA or AA were also assessed after pre-incubation with 10 μM FTP-THQ (
). Responses to agonist in the presence of LA or AA were also assessed after pre-incubation with 10 μM FTP-THQ ( ) or 10 μM RR (
) or 10 μM RR ( ). Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from a minimum of three separate experiments. *P < 0.05, ***P < 0.001 one-way anova with Tukey's post hoc test. RFU, relative fluorescence units.
). Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from a minimum of three separate experiments. *P < 0.05, ***P < 0.001 one-way anova with Tukey's post hoc test. RFU, relative fluorescence units.
KD of TRPV3 in m308k cells
To ensure that the Ca2+ signal observed in the m308k cells was indeed mediated by TRPV3, we next took a genetic approach, generating a TRPV3 KD cell line by integrating a TRPV3-specific shRNA into the m308k genome. A non-target shRNA was used to generate a control KD m308k cell line. Quantification by RT-qPCR showed a significant (P < 0.001) ∼70% reduction in TRPV3 mRNA levels in the TRPV3 KD cells compared to wild-type (WT) m308k cells (Figure 6A). The non-target control cells showed a slight increase in TRPV3 mRNA compared to WT. As described earlier, co-application of DPBA (15 nM–500 μM) with 160 μM LA (Figure 6B), 30 μM AA (Figure 6C) or 150 μM carvacrol (Figure 6D), evoked increases in intracellular Ca2+ concentration that were TRPV3 mediated in WT cells. Similar responses were also observed in the non-target control cells. In contrast, Ca2+ responses in the TRPV3 KD cells were only seen at high concentrations of DPBA, and unlike WT cells, could not be blocked with FTP-THQ.
Figure 6.
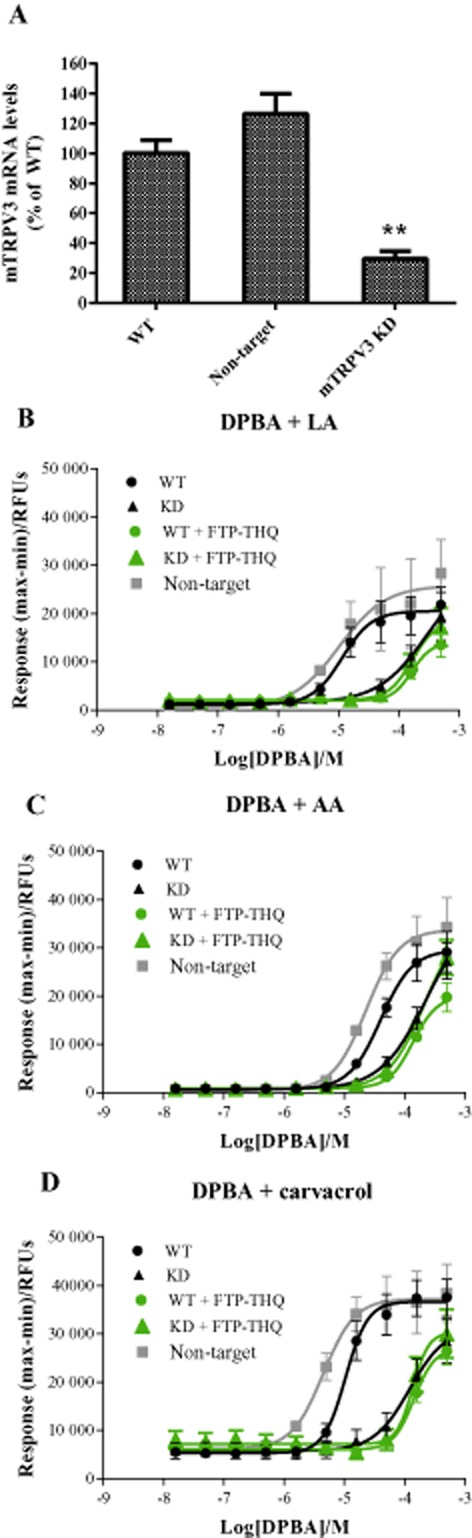
TRPV3 KD in m308k cells. (A) TRPV3 mRNA levels were measured by RT-qPCR in WT, non-target shRNA and TRPV3 KD m308k cell lines. mRNA levels are shown as a percentage of WT, set at 100%. (B) Ca2+ responses obtained in WT ( ), non-target shRNA (
), non-target shRNA ( ) and TRPV3 KD (
) and TRPV3 KD ( ) m308 keratinocytes. Cells were exposed to increasing concentrations of DPBA applied in combination with AA, LA or carvacrol and changes in intracellular Ca2+ fluorescence were monitored. Responses in WT (
) m308 keratinocytes. Cells were exposed to increasing concentrations of DPBA applied in combination with AA, LA or carvacrol and changes in intracellular Ca2+ fluorescence were monitored. Responses in WT ( ) and KD (
) and KD ( ) cells were also assessed after pre-incubation with 10 μM FTP-THQ. Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from five separate experiments. RFU, relative fluorescence units.
) cells were also assessed after pre-incubation with 10 μM FTP-THQ. Data are displayed as raw fluorescence units of the maximum minus minimum response from each experiment. Each data point represents the mean response ± SEM from five separate experiments. RFU, relative fluorescence units.
Antagonist activity across recombinant and native assays
Development of robust, medium-throughput human and rat TRPV3 recombinant assays and a mouse TRPV3 native assay allowed us to profile a number of TRPV3 antagonists and compare their activity across species (for structures, see Supporting Information Figure S1C). In human and rat TRPV3 recombinant assays, 40 μM DPBA was used to assess antagonist potency, while 50 μM DPBA in the presence of 160 μM LA was utilized as the stimulus in m308k cells. Figure 7 shows representative antagonist profiles of RR, FTP-THQ and N-[(4r)-8-chloro-3,4-dihydrospiro[chromene-2,1′-cyclobutan]-4-yl]-3-[2-(cyclopentyloxy)-3-methoxyphenyl]propanamide (CPC-MPP) in human, rat and mouse TRPV3 assays. The profiles of additional antagonists can be found in Supporting Information Figure S3A–C. Data generated with all antagonists used in this study are summarized in Table 2. Comparison of pIC50 values and % inhibition generated with a range of Hydra TRPV3 antagonists revealed a close correlation across species (rat, mouse and human) and the recombinant versus native systems. In contrast, the Glenmark compounds, CPC-MPP and Example 47, gave incomplete block, displaying significantly (P < 0.001) greater block in the native system.
Figure 7.

Inhibition of Ca2+ signalling in response to 40 μM DPBA in (A) human TRPV3, (B) rat TRPV3 and 50 μM DPBA + 160 μM LA in (C) m308ks in the presence of increasing concentrations of RR ( ), FTP-THQ (
), FTP-THQ ( ) or CPC-MPP (
) or CPC-MPP ( ). Data are displayed as percentage responses to DPBA alone. Each data point represents the mean response ± SEM from 4–6 separate experiments.
). Data are displayed as percentage responses to DPBA alone. Each data point represents the mean response ± SEM from 4–6 separate experiments.
Table 2.
Comparison of the potency and efficacy of TRPV3 antagonists on DPBA-evoked responses in cells transfected with the human or rat TRPV3 receptor and m308k cells
| Reference | Human TRPV3 | n | Rat TRPV3 | n | Mouse 308 keratinocytes | n | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| pIC50 ± SEM | % Inhibition ± SEM | pIC50 ± SEM | % Inhibition ± SEM | pIC50 ± SEM | % Inhibition ± SEM | |||||
| Ruthenium red | N/A | 6.82 ± 0.26 | 84 ± 9 | 5 | 6.93 ± 0.17 | 90 ± 7 | 4 | 6.93 ± 0.09 | 87 ± 3 | 10 |
| FTP-THQ | (Moran et al., 2008) | 6.54 ± 0.11 | 91 ± 5 | 4 | 6.73 ± 0.14 | 93 ± 7 | 3 | 6.73 ± 0.07 | 81 ± 3 | 10 |
| DCP-THQ | (Moran et al., 2008) | 6.70 ± 0.12 | 92 ± 5 | 2 | 7.04 ± 0.13 | 91 ± 5 | 2 | 7.15 ± 0.07*2006 | 87 ± 3 | 7 |
| ID101 | (Chong et al., 2006) | 6.39 ± 0.10 | 91 ± 6 | 3 | 6.02 ± 0.11 | 91 ± 9 | 3 | 6.23 ± 0.05 | 91 ± 3 | 4 |
| ID15 | (Chong et al., 2006) | 6.58 ± 0.14 | 91 ± 7 | 3 | 6.10 ± 0.09*2006 | 94 ± 6 | 3 | 6.35 ± 0.04 | 95 ± 2 | 3 |
| ID64 | (Chong et al., 2006) | 5.74 ± 0.13 | 95 ± 10 | 3 | 5.78 ± 0.14 | 89 ± 11 | 3 | 5.70 ± 0.05 | 97 ± 4 | 3 |
| ID8 | (Moran et al., 2008) | 5.94 ± 0.13 | 99 ± 11 | 3 | 6.09 ± 0.07 | 99 ± 4 | 2 | 6.35 ± 0.10 | 76 ± 4 | 5 |
| CPC-MPP | (Lingam et al., 2009) | 5.89 ± 0.27 | 28 ± 6 | 6 | 5.92 ± 0.19 | 44 ± 6 | 3 | 5.76 ± 0.11 | 67 ± 5***2006 | 7 |
| Example 47 | (Lingam et al., 2009) | 5.07 ± 1.6 | 19 ± 37 | 5 | 6.29 ± 1.29 | 17 ± 18 | 2 | 5.50 ± 0.19 | 39 ± 7 | 7 |
P < 0.05 versus hTRPV3,
P < 0.001 versus hTRPV3 – one-way anova with Tukey's post hoc test.
Mode of TRPV3 inhibition by RR and FTP-THQ in m308k cells
To compare the mechanisms by which RR and FTP-THQ inhibit the TRPV3 channel, we performed additional studies in m308k cells using agonist co-application (100 μM–7 mM camphor/50 μM DPBA) to selectively activate TRPV3 in the absence or presence of increasing antagonist concentrations (Figure 8). Increasing concentrations of RR led to step-wise decreases in the Emax of TRPV3-mediated Ca2+ responses, without affecting the EC50 value, characteristic of a non-competitive inhibitor (Figure 8A, Supporting Information Figure S4B). In contrast, application of increasing concentrations of FTP-THQ had relatively little effect on the Emax of camphor/DPBA-evoked TRPV3 Ca2+ responses, but did cause a step-wise rightward shift of the EC50. (Figure 8B). Concentrations of FTP-THQ >3 μM appeared to reach a maximal shift, indicative of an allosteric mode of binding (Figure 8B, Supporting Information Figure S4A). In line with binding to a site allosteric to the camphor binding site, FTP-THQ retained its activity as an antagonist of keratinocyte TRPV3 in the presence of 30 μM AA (pIC50 7.07 ± 0.09, 71 ± 4% block; Supporting Information Figure S3D).
Figure 8.
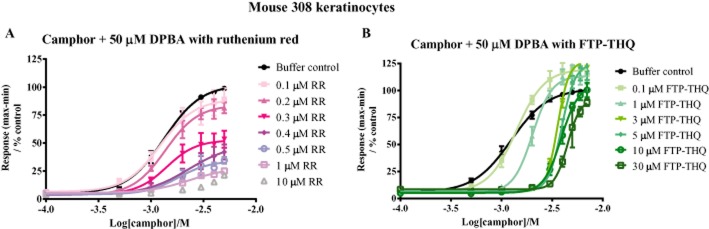
Analysis of the mechanism of inhibition of Ca2+ responses to 50 μM DPBA and camphor in m308k cells. Cells were treated with increasing concentrations of camphor in combination with 50 μM DPBA either alone ( ) or after pre-incubation with fixed concentrations of RR (A) or FTP-THQ (B). Data are displayed as percentage responses to DPBA and 5 mM camphor alone. Each data point represents the mean response ± SEM from 5–6 separate experiments.
) or after pre-incubation with fixed concentrations of RR (A) or FTP-THQ (B). Data are displayed as percentage responses to DPBA and 5 mM camphor alone. Each data point represents the mean response ± SEM from 5–6 separate experiments.
Discussion
To facilitate in vitro evaluation of TRPV3 ligands, we developed medium-throughput FLIPR assays in recombinant and native cell cultures. Using a HEK293 cell line stably expressing recombinant human TRPV3, we established optimal concentrations of DPBA and 2-APB to use for the study of TRPV3-specific activation. Higher concentrations of these agonists led to non-specific increases of intracellular Ca2+, most likely due to efflux from intracellular Ca2+ stores; therefore, caution should be used when interpreting data where such concentrations of agonists are reported.
Similar results were obtained in the recombinant rat TRPV3 assay. Development of this assay was complicated by the fidelity of the rat TRPV3 protein sequence (AAQ90060.1). Indeed, expression of this protein sequence yielded a non-functional receptor. Although other groups have reported rat TRPV3 activity in recombinant systems (De Petrocellis et al., 2012; Ortar et al., 2012), no accession number was included; therefore, it is unclear which sequence was used to generate the cell lines. Applying GeneWise on the rat reference assembly, we identified a new protein sequence (protein id NP_001020928.2) derived from NC_005109.2. This sequence differed from GenBank AAQ90060.1 by 10 amino acids; importantly, these changes were conserved between NC_005109.2 and mouse TRPV3 (Supporting Information Figure S2C). The new rat TRPV3 construct, expressing the NP_001020928.2 sequence, yielded a functional TRPV3 receptor, with a pharmacological profile closely resembling the human receptor. From the 10 amino acid residues that differ between the two protein sequences, the ones with the most radical change are found at position 466 and 641. In NP_001020928.2, both of these residues are Asp, and the negative charge is conserved in the mouse and human sequence, whereas in AAQ90060.1, those residues are both Gly. Although we cannot draw a direct conclusion that these are the changes causing loss of function, it is important to note that Asp 641 and proximal residues were previously found to be critical for TRPV3 activation (Chung et al., 2005; Grandl et al., 2008; Xiao et al., 2008).
As HEK293 cells do not express endogenous TRP channels, they allow for study of the expressed TRP channel in isolation. However, it is of value to verify the activity of compounds in native preparations before moving in vivo to assess behavioural effects. Reports of native functional TRPV3 receptors in the literature are primarily from primary rodent keratinocytes and the m308K cell line, with the latter more amenable to scaling for FLIPR type screening assays. As the mouse and rat TRPV3 sequence is 98.1% identical and behavioural assays are performed in rats and mice, a preparation expressing mouse native TRPV3 receptors served as an appropriate choice. Previous studies of TRPV3 in m308k cells utilized low-throughput electrophysiology and single-cell Ca2+ imaging experiments, with relatively limited pharmacological evaluation performed (Hu et al., 2006; Xu et al., 2006). In the present study, intracellular Ca2+ signals were seen in response to a number of non-selective TRPV3 agonists. However, these responses were present in the absence of extracellular Ca2+, and furthermore, they were not blocked by a selective TRPV3 antagonist FTP-THQ, nor with the non-selective TRP-channel inhibitor RR, proving that TRP channels were not mediating the responses. Our data indicates that the high concentrations of 2-APB and DPBA (200–300 μM) reported in the literature (Hu et al., 2004; 2006; Chung et al., 2005) are likely to have non-specific effects. The data are consistent with previous findings showing that functional TRPV3-mediated responses can be initially small and difficult to detect on addition of single-agonist applications in isolation, but increase dramatically with repeated stimulation (Xu et al., 2006). As FLIPR is a static, non-perfusion system, we were unable to readily assess sensitization after repeated drug application and focused instead on agonist co-application and fatty acid potentiation as a means to increase the magnitude of TRPV3 responses in the absence of repeat stimulation (Hu et al., 2006; Xu et al., 2006; Parnas et al., 2009). Of note, the experiments in the current study were conducted at room temperature, for technical ease, comparison with other TRP channel assays, and in line with much of the previously published in vitro TRPV3 data. However, as previous data indicate recombinant TRPV3-mediated [Ca2+]i responses to 2APB are sensitized by rising temperature (Hu et al., 2004), this would be another relevant factor to evaluate in the m308k line.
Co-application of structurally unrelated agonists has previously been reported to lead to synergistic activation of TRPV3, including in m308k cells (Xu et al., 2006; Cheng et al., 2010). Our findings confirm published work, and furthermore, reveal that in this paradigm, lower agonist concentrations are sufficient to promote selective TRPV3 activation, with the Ca2+ signals being fully inhibited by RR, FTP-THQ (a selective TRPV3 antagonist) and by KD of TRPV3. DPBA and 2-APB could synergize with terpenoids (camphor, thymol, carvacrol and 6-TBC); however, they did not synergize with each other. Given the structural similarity between 2-APB and DPBA, it is likely that the two cannot synergize because they compete for binding to the same site on TRPV3. Work by Hu et al. (Hu et al., 2009) identified two cytoplasmic residues (H426 and R696) necessary for the activation of TRPV3 by 2-APB but not by camphor. Therefore, based on structural similarity and the observation that none of the terpenoid compounds could synergize with each other, we propose that carvacrol, thymol and 6-TBC bind to the same site on TRPV3 as camphor. Eugenol, which did not synergize with any other compound, is structurally dissimilar to the other agonists tested and lacked any clear agonistic effects in the current study.
In m308k cells, LA and AA potentiated TRPV3-mediated Ca2+ responses to 2-APB, as reported previously (Hu et al., 2006; Parnas et al., 2009). Our data extends the previous findings with a more complete evaluation of the effects of PUFAs on agonist potencies, confirmation of block by antagonists (RR and FTP-THQ) and exploration of additional agonists, not all of which displayed sensitization by PUFAs. The reason for this difference is unclear, but may stem from the different binding sites for the non-2-APB type agonists on the TRPV3 channel (Hu et al., 2009). One possibility is that LA and AA binding prevents binding at the camphor-binding site, or vice versa, through competitive or allosteric means.
The high concentrations of PUFAs used in the present study to generate optimal potentiation of TPRV3 are not incompatible with PUFA effects having physiological or pathophysiological relevance. For example, Hu et al. (2006) found that 100 μM AA was required for maximal potentiation, but argued that the significant increase in TRPV3 channel activity observed at 1 μM AA was sufficient to mediate effects in vivo, such that a high level of TRPV3 activation may not be necessary. These researchers also observed that potentiation was increased by longer incubations with fatty acids.
Under the current assay conditions, the composite data confirms that we are looking at TRPV3, and not other TRP channels endogenously expressed in m308k cells (Chung et al., 2003). The evidence includes block of DPBA (or 2-APB) responses in the presence of PUFAs/co-agonists down to non-specific response levels by the selective TRPV3 receptor antagonist FTP-THQ, identical IC50 values for FTP-THQ block in m308k cells and recombinant TRPV3 cell lines, and recapitulation of the FTP-THQ findings by selective KD of TRPV3 expression. Published pharmacology data is also consistent with this explanation, specifically, AA and LA are reported to potentiate TRPV3, but not TRPV1 or TRPV2 (Hu et al., 2006; Parnas et al., 2009) and the co-agonist synergy data replicates previously reported TRPV3 findings (Xu et al., 2006). Of note, we were not able to confirm the reported activity of FPP and IA as TRPV3 agonists (Moussaieff et al., 2008; Bang et al., 2010) in the current study. Ca2+ signals evoked by FPP in m308k cells were not blocked by FTP-THQ, and were not augmented by co-agonists (DPBA and carvacrol) or potentiators (LA and AA; data not shown). IA, on the other hand, did not induce any Ca2+ responses, even at high concentrations, under any condition tested. Methodological differences (e.g. static vs. perfusion system, supplier variations) and/or technical issues working with these compounds (e.g. no independent confirmation of biological activity) cannot be discounted at this stage.
As shown in Table 2, the pIC50 values for RR, FTP-THQ, Example 47 and several other antagonists are similar across human and rat recombinant and mouse native assays. While the Hydra compounds fully blocked agonist responses across all three assays, the Glenmark TRPV3 antagonists exhibited incomplete blockade, consistent with an allosteric mechanism of action. These antagonists also showed significantly higher efficacy in blocking TRPV3 in m308k cells. Although it is possible that these differences are species specific, more likely, they are due to differences in TRPV3 protein expression levels and cell type. Although TRPV3 protein was easily detectable in recombinant cells by Western blot (Supporting Information Figure S2B), we were not able to detect it in m308k cells, indicative of lower levels of expression. Low levels of TRPV3 mRNA were, however, detected by RT-PCR (Supporting Information Figure S2A). The over-abundance of TRPV3 protein in recombinant cells may lead to non-physiological readouts or render TRPV3 insensitive to weaker effectors.
Cell type specificity is also an important factor to consider. Cultured keratinocytes, such as m308k cells, are a good cellular model to study TRPV3 function as they retain characteristics of keratinocytes in mammalian skin, and are more likely to contain the appropriate interacting factors and signalling pathways. Indeed, given the critical role now emerging for TRPV3 in skin physiology and pathology, this preparation provides an ideal model, amenable to scaling, for further TRPV3 characterization and pharmacological confirmation of potential TRPV3 therapeutic agents. Of note, AA, which we confirm as a strong potentiator of keratinocyte TRPV3 in the current study, is present at high concentrations in psoriatic dermatitis (>100 μM; Brash, 2001) indicating that TRPV3 activity may be potentiated in psoriatic epidermis. Aberrant TRPV3 activity would be anticipated to drive abnormal EGF receptor function and TGF-α release (Cheng et al., 2010) and cause release of pruritogens, promoting itch (Yoshioka et al., 2009) and abnormal epidermal homeostasis, as seen with the TRPV3 gain-of-function G573S/C mutation in rodents (Asakawa et al., 2006; Yoshioka et al., 2009) and humans (Lai-Cheong et al., 2012; Lin et al., 2012). Importantly, as our findings reveal that TRPV3 agonist-mediated activation of TRPV3 can be differentially modulated by AA interactions, TRPV3 antagonist effects might also be anticipated to vary in the presence of AA. As this appears to be a disease-relevant factor, it will be vital to confirm the efficacy of TRPV3 antagonists in the presence of AA. We were able to confirm that FTP-THQ blocked keratinocyte TRPV3 channels in both the presence and absence of AA, consistent with its action as an allosteric antagonist of the camphor-binding site.
In summary, we have developed medium-throughput recombinant and native TRPV3 assays. These methods present advantages over lower throughput electrophysiology techniques, mainly due to the quantity of data generated and should aid research towards development of TRPV3 therapeutic agents. Although recombinant systems are a good way to study receptor pharmacology in isolation, the native system is clearly a more relevant cellular model and may provide a means to study disease-relevant pharmacology.
Acknowledgments
We thank James Beck for the chemistry platform support. J. S. and L. J. B. were funded by a 1 year industrial placement at Eli Lilly.
Glossary
- 2-APB
2-aminoethoxydiphenylborate
- 6-TBC
terpenoid 6-tert-butyl-m-cresol
- AA
arachidonic acid
- DPBA
diphenylborinic anhydride
- FLIPR
fluorescent imaging plate reader
- FPP
farnesyl pyrophosphate
- HEK293
human embryonic kidney cells
- IA
incensole acetate
- LA
linoleic acid
- m308k
mouse 308 keratinocyte
- PUFA
poly-unsaturated fatty acid
- RR
ruthenium red
- RT
reverse transcription
- shRNA
small hairpin RNA
- TRP
transient receptor potential
- TRPV3
transient receptor potential vanilloid subtype 3
Conflicts of interest
The authors declare no conflicts of interest. Eli Lilly does not sell any of the compounds or devices mentioned in this article.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12303
Figure S1 Chemical structures of agonists (A), potentiators (B) and antagonists (C) used in this study.
Figure S2 Sequence and expression of human, rat and mouse TRPV3. (A) rat, human and mouse TRPV3 mRNA is detected in rat recombinant, human recombinant and m308k cells, respectively. A single band of the expected size was amplified by RT-PCR using species-specific TRPV3 primers, whereas no band was observed in HEK293 cells alone. The DNA ladder indicates size in bp. (B) TRPV3 protein is expressed in rat and human cell lines and not in untransfected HEK293 cells. TRPV3 protein was detected by Western blot using a TRPV3-specific Ab. The protein ladder indicates size in kDa. (C) Multiple sequence alignment of the human (Hs), mouse (Mm), and rat (Rn) TRPV3 protein sequences. Identical residues are highlighted in yellow. Differences between the two rat TRPV3 sequences are coloured in red. Ankyrin repeats and transmembrane regions are coloured blue and orange, respectively. The green box indicates the membrane-embedded region. (D) TRPV3 protein was detected by Western blot in HEK293 cells transiently transfected with plasmids expressing human or rat TRPV3. The rat reference sequence is indicated in brackets. The vertical line indicates splicing of the identical photo.
Figure S3 Inhibition of Ca2+ signaling in response to DPBA in human TRPV3 (A), rat TRPV3 (B) and mouse 308 keratinocytes (C) by the presence of increasing concentrations of TRPV3-selective antagonists. Data are displayed as percentage responses to DPBA alone. Each data point represents the mean response ± SEM from 4–6 separate experiments. (D) Inhibition of intracellular Ca2+ signalling by RR or FTP-THQ in the presence of DPBA and AA in m308k cells. Data are displayed as percentage responses to DPBA and AA in the absence of inhibitor. Each data point represents the mean response ± SEM from 2 separate experiments.
Figure S4 Schild plots of the mechanism of inhibition of Ca2+ responses to 50 µM DPBA and camphor in m308k cells by either (A) FTP-THQ or (B) Ruthenium Red.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1867. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakawa M, Yoshioka T, Matsutani T, Hikita I, Suzuki M, Oshima I, et al. Association of a mutation in TRPV3 with defective hair growth in rodents. J Invest Dermatol. 2006;126:2664–2672. doi: 10.1038/sj.jid.5700468. [DOI] [PubMed] [Google Scholar]
- Bang S, Yoo S, Yang TJ, Cho H, Hwang SW. Farnesyl pyrophosphate is a novel pain-producing molecule via specific activation of TRPV3. J Biol Chem. 2010;285:19362–19371. doi: 10.1074/jbc.M109.087742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang S, Yoo S, Yang T-J, Cho H, Hwang SW. 17(R)-resolvin D1 specifically inhibits transient receptor potential ion channel vanilloid 3 leading to peripheral antinociception. Br J Pharmacol. 2012;165:683–692. doi: 10.1111/j.1476-5381.2011.01568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender FL, Mederos Y, Schnitzler M, Li Y, Ji A, Weihe E, et al. The temperature-sensitive ion channel TRPV2 is endogenously expressed and functional in the primary sensory cell line F-11. Cell Physiol Biochem. 2005;15:183–194. doi: 10.1159/000083651. [DOI] [PubMed] [Google Scholar]
- Birney E, Clamp M, Durbin R. GeneWise and genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash AR. Arachidonic acid as a bioactive molecule. J Clin Invest. 2001;107:1339–1345. doi: 10.1172/JCI13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad LM, Mogg AJ, Beattie RE, Ogden AM, Blanco MJ, Bleakman D. TRP channels as emerging targets for pain therapeutics. Expert Opin Ther Targets. 2009;13:69–81. doi: 10.1517/14728220802616620. [DOI] [PubMed] [Google Scholar]
- Cheng X, Jin J, Hu L, Shen D, Dong XP, Samie MA, et al. TRP channel regulates EGFR signaling in hair morphogenesis and skin barrier formation. Cell. 2010;141:331–343. doi: 10.1016/j.cell.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JA, Fanger C, Moran MM, Underwood DJ, Zhen X, Ripka A, et al. 2006. Compounds for modulating TRPV3 function. WO2006/122156.
- Christoph T, Bahrenberg G, De Vry J, Englberger W, Erdmann VA, Frech M, et al. Investigation of TRPV1 loss-of-function phenotypes in transgenic shRNA expressing and knockout mice. Mol Cell Neurosci. 2008;37:579–589. doi: 10.1016/j.mcn.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Chung MK, Lee H, Caterina MJ. Warm temperatures activate TRPV4 in mouse 308 keratinocytes. J Biol Chem. 2003;278:32037–32046. doi: 10.1074/jbc.M303251200. [DOI] [PubMed] [Google Scholar]
- Chung MK, Lee H, Mizuno A, Suzuki M, Caterina MJ. 2-aminoethoxydiphenyl borate activates and sensitizes the heat-gated ion channel TRPV3. J Neurosci. 2004a;24:5177–5182. doi: 10.1523/JNEUROSCI.0934-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MK, Lee H, Mizuno A, Suzuki M, Caterina MJ. TRPV3 and TRPV4 mediate warmth-evoked currents in primary mouse keratinocytes. J Biol Chem. 2004b;279:21569–21575. doi: 10.1074/jbc.M401872200. [DOI] [PubMed] [Google Scholar]
- Chung MK, Guler AD, Caterina MJ. Biphasic currents evoked by chemical or thermal activation of the heat-gated ion channel, TRPV3. J Biol Chem. 2005;280:15928–15941. doi: 10.1074/jbc.M500596200. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Orlando P, Moriello AS, Aviello G, Stott C, Izzo AA, et al. Cannabinoid actions at TRPV channels: effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol (Oxf) 2012;204:255–266. doi: 10.1111/j.1748-1716.2011.02338.x. [DOI] [PubMed] [Google Scholar]
- Grandl J, Hu H, Bandell M, Bursulaya B, Schmidt M, Petrus M, et al. Pore region of TRPV3 ion channel is specifically required for heat activation. Nat Neurosci. 2008;11:1007–1013. doi: 10.1038/nn.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guatteo E, Chung KK, Bowala TK, Bernardi G, Mercuri NB, Lipski J. Temperature sensitivity of dopaminergic neurons of the substantia nigra pars compacta: involvement of transient receptor potential channels. J Neurophysiol. 2005;94:3069–3080. doi: 10.1152/jn.00066.2005. [DOI] [PubMed] [Google Scholar]
- Hu H, Grandl J, Bandell M, Petrus M, Patapoutian A. Two amino acid residues determine 2-APB sensitivity of the ion channels TRPV3 and TRPV4. Proc Natl Acad Sci U S A. 2009;106:1626–1631. doi: 10.1073/pnas.0812209106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H-Z, Gu Q, Wang C, Colton CK, Tang J, Kinoshita-Kawada M, et al. 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J Biol Chem. 2004;279:35741–35748. doi: 10.1074/jbc.M404164200. [DOI] [PubMed] [Google Scholar]
- Hu H-Z, Xiao R, Wang C, Gao N, Colton CK, Wood JD, et al. Potentiation of TRPV3 channel function by unsaturated fatty acids. J Cell Physiol. 2006;208:201–212. doi: 10.1002/jcp.20648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Lee H, Chung MK, Park U, Yu YY, Bradshaw HB, et al. Overexpressed transient receptor potential vanilloid 3 ion channels in skin keratinocytes modulate pain sensitivity via prostaglandin E2. J Neurosci. 2008;28:13727–13737. doi: 10.1523/JNEUROSCI.5741-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NK, Maharaj N, Thomas A. The TRPV3 receptor as a pain target: a therapeutic promise or just some more new biology? Open Drug Discov J. 2010;2:89–97. [Google Scholar]
- Kitahara T, Li HS, Balaban CD. Changes in transient receptor potential cation channel superfamily V (TRPV) mRNA expression in the mouse inner ear ganglia after kanamycin challenge. Hear Res. 2005;201:132–144. doi: 10.1016/j.heares.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Lai-Cheong JE, Sethuraman G, Ramam M, Stone K, Simpson MA, McGrath JA. Recurrent heterozygous missense mutation, p.Gly573Ser, in the TRPV3 gene in an Indian boy with sporadic Olmsted syndrome. Br J Dermatol. 2012;167:440–442. doi: 10.1111/j.1365-2133.2012.11115.x. [DOI] [PubMed] [Google Scholar]
- Lin Z, Chen Q, Lee M, Cao X, Zhang J, Ma D, et al. Exome sequencing reveals mutations in TRPV3 as a cause of Olmsted syndrome. Am J Hum Genet. 2012;90:558–564. doi: 10.1016/j.ajhg.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingam VSP, Thomas A, Gharat LA, Ukirde DV, Phatangare SK, Minde AS, et al. 2009. Chromane derivatives as TRPV3 modulators. WO2009/084034.
- Lipski J, Park TI, Li D, Lee SC, Trevarton AJ, Chung KK, et al. Involvement of TRP-like channels in the acute ischemic response of hippocampal CA1 neurons in brain slices. Brain Res. 2006;1077:187–199. doi: 10.1016/j.brainres.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Liu B, Yao J, Zhu MX, Qin F. Hysteresis of gating underlines sensitization of TRPV3 channels. J Gen Physiol. 2011;138:509–520. doi: 10.1085/jgp.201110689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandadi S, Sokabe T, Shibasaki K, Katanosaka K, Mizuno A, Moqrich A, et al. TRPV3 in keratinocytes transmits temperature information to sensory neurons via ATP. Pflugers Arch. 2009;458:1093–1102. doi: 10.1007/s00424-009-0703-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moqrich A, Hwang SW, Earley TJ, Petrus MJ, Murray AN, Spencer KS, et al. Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science. 2005;307:1468–1472. doi: 10.1126/science.1108609. [DOI] [PubMed] [Google Scholar]
- Moran MM, Wei D, Zhen X, Mandel-Brehm J, Witek J, Yaksh T, et al. Potent and Selective Antagonists Validate TRPV3 as a Target for Analgesic Therapeutics. San Diego, CA: Society for Neuroscience; 2007. Online Program No. 143.5. 2007 Neuroscience Meeting Planner. [Google Scholar]
- Moran MM, Chong JA, Fanger C, Ripka A, Larsen GR, Zhen X, et al. 2008. Compounds for modulating TRPV3 function. WO2008/033564.
- Moran MM, McAlexander MA, Biro T, Szallasi A. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov. 2011;10:601–620. doi: 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- Moussaieff A, Rimmerman N, Bregman T, Straiker A, Felder CC, Shoham S, et al. Incensole acetate, an incense component, elicits psychoactivity by activating TRPV3 channels in the brain. FASEB J. 2008;22:3024–3034. doi: 10.1096/fj.07-101865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortar G, Morera L, Moriello AS, Morera E, Nalli M, Di Marzo V, et al. Modulation of thermo-transient receptor potential (thermo-TRP) channels by thymol-based compounds. Bioorg Med Chem Lett. 2012;22:3535–3539. doi: 10.1016/j.bmcl.2012.03.055. [DOI] [PubMed] [Google Scholar]
- Parnas M, Peters M, Minke B. Linoleic acid inhibits TRP channels with intrinsic voltage sensitivity: implications on the mechanism of linoleic acid action. Channels (Austin) 2009;3:164–166. doi: 10.4161/chan.3.3.8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peier AM, Reeve AJ, Andersson DA, Moqrich A, Earley TJ, Hergarden AC, et al. A heat-sensitive TRP channel expressed in keratinocytes. Science. 2002;296:2046–2049. doi: 10.1126/science.1073140. [DOI] [PubMed] [Google Scholar]
- Phelps CB, Wang RR, Choo SS, Gaudet R. Differential regulation of TRPV1, TRPV3, and TRPV4 sensitivity through a conserved binding site on the ankyrin repeat domain. J Biol Chem. 2010;285:731–740. doi: 10.1074/jbc.M109.052548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GD, Gunthorpe MJ, Kelsell RE, Hayes PD, Reilly P, Facer P, et al. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature. 2002;418:186–190. doi: 10.1038/nature00894. [DOI] [PubMed] [Google Scholar]
- Vogt-Eisele AK, Weber K, Sherkheli MA, Vielhaber G, Panten J, Gisselmann G, et al. Monoterpenoid agonists of TRPV3. Br J Pharmacol. 2007;151:530–540. doi: 10.1038/sj.bjp.0707245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev. 2010;62:381–404. doi: 10.1124/pr.110.002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R, Tang J, Wang C, Colton CK, Tian J, Zhu MX. Calcium plays a central role in the sensitization of TRPV3 channel to repetitive stimulations. J Biol Chem. 2008;283:6162–6174. doi: 10.1074/jbc.M706535200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Ramsey IS, Kotecha SA, Moran MM, Chong JA, Lawson D, et al. TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature. 2002;418:181–186. doi: 10.1038/nature00882. [DOI] [PubMed] [Google Scholar]
- Xu H, Delling M, Jun JC, Clapham DE. Oregano, thyme and clove-derived flavors and skin sensitizers activate specific TRP channels. Nat Neurosci. 2006;9:628–635. doi: 10.1038/nn1692. [DOI] [PubMed] [Google Scholar]
- Yang XR, Lin MJ, McIntosh LS, Sham JS. Functional expression of transient receptor potential melastatin-and vanilloid-related channels in pulmonary arterial and aortic smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1267–L1276. doi: 10.1152/ajplung.00515.2005. [DOI] [PubMed] [Google Scholar]
- Yoshioka T, Imura K, Asakawa M, Suzuki M, Oshima I, Hirasawa T, et al. Impact of the Gly573Ser substitution in TRPV3 on the development of allergic and pruritic dermatitis in mice. J Invest Dermatol. 2009;129:714–722. doi: 10.1038/jid.2008.245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Chemical structures of agonists (A), potentiators (B) and antagonists (C) used in this study.
Figure S2 Sequence and expression of human, rat and mouse TRPV3. (A) rat, human and mouse TRPV3 mRNA is detected in rat recombinant, human recombinant and m308k cells, respectively. A single band of the expected size was amplified by RT-PCR using species-specific TRPV3 primers, whereas no band was observed in HEK293 cells alone. The DNA ladder indicates size in bp. (B) TRPV3 protein is expressed in rat and human cell lines and not in untransfected HEK293 cells. TRPV3 protein was detected by Western blot using a TRPV3-specific Ab. The protein ladder indicates size in kDa. (C) Multiple sequence alignment of the human (Hs), mouse (Mm), and rat (Rn) TRPV3 protein sequences. Identical residues are highlighted in yellow. Differences between the two rat TRPV3 sequences are coloured in red. Ankyrin repeats and transmembrane regions are coloured blue and orange, respectively. The green box indicates the membrane-embedded region. (D) TRPV3 protein was detected by Western blot in HEK293 cells transiently transfected with plasmids expressing human or rat TRPV3. The rat reference sequence is indicated in brackets. The vertical line indicates splicing of the identical photo.
Figure S3 Inhibition of Ca2+ signaling in response to DPBA in human TRPV3 (A), rat TRPV3 (B) and mouse 308 keratinocytes (C) by the presence of increasing concentrations of TRPV3-selective antagonists. Data are displayed as percentage responses to DPBA alone. Each data point represents the mean response ± SEM from 4–6 separate experiments. (D) Inhibition of intracellular Ca2+ signalling by RR or FTP-THQ in the presence of DPBA and AA in m308k cells. Data are displayed as percentage responses to DPBA and AA in the absence of inhibitor. Each data point represents the mean response ± SEM from 2 separate experiments.
Figure S4 Schild plots of the mechanism of inhibition of Ca2+ responses to 50 µM DPBA and camphor in m308k cells by either (A) FTP-THQ or (B) Ruthenium Red.