

Abstract
Background and Purpose
The retention of plasma low-density lipoprotein (LDL) particles in subendothelial space following transcytosis across the endothelium is the initial step of atherosclerosis. Whether or not C-reactive protein (CRP) can directly affect the transcytosis of LDL is not clear. Here we have examined the effect of CRP on transcytosis of LDL across endothelial cells and have explored the underlying mechanisms.
Experimental Approach
Effects of CRP on transcytosis of FITC-labelled LDL were examined with human umbilical vein endothelial cells and venous rings in vitro and, in vivo, ApoE-/- mice. Laser scanning confocal microscopy, immunohistochemistry and Oil Red O staining were used to assay LDL.
Key Results
CRP increased transcytosis of LDL. An NADPH oxidase inhibitor, diphenylene iodonium, and the reducing agent, dithiothreitol partly or completely blocked CRP-stimulated increase of LDL transcytosis. The PKC inhibitor, bisindolylmaleimide I and the Src kinase inhibitor, PP2, blocked the trafficking of the molecules responsible for transcytosis. Confocal imaging analysis revealed that CRP stimulated LDL uptake by endothelial cells and vessel walls. In ApoE-/- mice, CRP significantly promoted early changes of atherosclerosis, which were blocked by inhibitors of transcytosis.
Conclusions and Implications
CRP promoted atherosclerosis by directly increasing the transcytosis of LDL across endothelial cells and increasing LDL retention in vascular walls. These actions of CRP were associated with generation of reactive oxygen species, activation of PKC and Src, and translocation of caveolar or soluble forms of the N-ethylmaleimide-sensitive factor attachment protein.
Keywords: LDL, transcytosis, C-reactive protein, endothelial cells, atherosclerosis, caveolae, PKC, Src, ROS
Introduction
Currently, more and more pieces of evidence have demonstrated that the subendothelial retention of apolipoprotein B100 (apoB100)-containing lipoproteins, such as low-density lipoprotein (LDL), is the initial step of atherogenesis, and is usually termed the ‘response to retention hypothesis’ (Williams and Tabas, 1995; Lusis, 2000; Tabas et al., 2007; Fan et al., 2010; Libby et al., 2011; Weber and Noels, 2011; Didangelos et al., 2012; Fogelstrand and Boren, 2012). As the diameter of the LDL particles (20–30 nm) is much larger than the gap junctions (3–6 nm) between the vascular endothelial cells, the only pathway for the LDL particles to traffic across the intact endothelial barrier is through the transporting process called transcytosis (Quest et al., 2004; Frank et al., 2008a, 2008b; Sun et al., 2010; Sowa, 2012), the transport of macromolecular cargo from one side (e.g. within the lumen) of polar cells to the other (e.g. basolateral side) within membrane-bounded carriers. Converging lines of evidence have pointed to a possible link between LDL transcytosis across endothelial cells and the initiation of atherosclerosis (Quest et al., 2004; Frank et al., 2008a, 2008b; Sun et al., 2010; Sowa, 2012).
Meanwhile, over the past couple of decades, evidence has accumulated suggesting that inflammation is critically involved in atherosclerosis (AAAS, 2012). In an effort to better identify patients at high risk for cardiovascular problems, inflammatory markers have received much more attention than before. Many reports have suggested that high levels of C-reactive protein (CRP) correlate tightly with an increased risk of atherosclerosis. Administration of 1, 6-bis (phosphocholine)-hexane, a specific low MW inhibitor of CRP to rats undergoing acute myocardial infarction, blocked the increase in infarct size and cardiac dysfunction produced by injection of human CRP (Pepys et al., 2006). The JUPITER (Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin) trial found that statins (rosuvastatin) reduced heart attacks and strokes in people with elevated CRP levels, but without hyperlipidemia (Ridker et al., 2008). A recent study reported that measurement of the CRP level in subjects with intermediate risk could help prevent one additional cardiovascular event. The hazard ratio for cardiovascular events to CRP levels is close to those for hyperlipidemia and high blood pressure (Kaptoge et al., 2012; 2013).
Although there is still some controversy about the role of CRP in cardiovascular disease (Elliott et al., 2009; Manace and Babyatsky, 2012), no study has denied that CRP serves as a biomarker to predict future cardiovascular events. The predictive value of CRP appears to be based on its ability to incite endothelial dysfunction. CRP stimulates production of reactive oxygen species (ROS) and NF-κB signalling in endothelial cells, up-regulates a range of adhesion molecules and chemokines, as well as displaying a pro-inflammatory phenotype. However, whether or not CRP is able to directly exert a pro-atherogenic effect through the up-regulation of the transcytosis of LDL across the endothelial barrier and thus to accelerate LDL retention in vascular walls, is not clear.
In the present study, we first established an in vitro model to assay the transcytosis of LDL across endothelial cells. In this model, we found that CRP indeed increased LDL transcytosis across endothelial cells and accelerated the retention of LDL in the subendothelial space of the wall of human umbilical vein. Following this, we further explored the underlying molecular mechanisms and found that CRP stimulated ROS signalling, the activation of PKC and Src kinase (nomenclature follows Alexander et al., 2013), as well as the translocation of transcytosis-related proteins into membrane raft domains, which were critically involved in this reinforcement of atherogenesis.
Methods
This study was approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China) and conducted in accordance with the Declaration of Helsinki (2008) and all applicable national and local regulations. All subjects provided written informed consent prior to the initiation of the study. All animals were kept in accordance with standard animal care requirements and maintained in a 21 ± 2°C room with a 12 h light/dark cycle.
Isolation and culture of HUVECs
Isolation of HUVECs was performed using methods that have been were described earlier (Weymann et al., 2013). The HUVECs were cultured in ECM (ScienCell, Carlsbad, CA, USA) with 10% FBS, 100 U·mL−1 penicillin, 100 U·mL−1 streptomycin (ScienCell) and 30 μg·mL−1 endothelial cell growth supplement (ScienCell) at 37°C with 5% CO2. Cells were passaged when 80%∼90% confluent and were used between passages 3 and 9.
LDL labelling
LDL was labelled using FITC (Biosharp, Hefei, China) as described previously (De Cicco et al., 2012). All procedures were performed in the dark. LDL (2mg; the Institute of Clinical Pharmacology of Sun Yat-Sen University) and FITC (120 μg) were mixed and incubated at 37°C for 2 h. Unbound FITC was removed by dialysis against PBS for 72 h at 4°C. After the measurement of protein with the BCA assay kit (Thermo Scientific, Rockford, IL, USA), FITC-LDL was then stored at 4°C for later use.
Establishment of the model to study LDL transcytosis
Figure 1 summarises the preparation of the model used to measure LDL transcytosis. In brief, HUVECs were seeded (∼4 × 104 cells per insert) on polyester membranes in transwells (6.5 mm diameter, 0.4 μm pore size; Costar, New York, NY, USA). The integrity of the cell monolayer was tested by a method described previously (Tuma and Hubbard, 2003; Cankova et al., 2011). Two inserts of cell monolayers with equal integrity were used for each sample: the non-competitive insert and the competitive insert. The non-competitive insert was incubated with FITC-LDL for 3 h to determine the total amount of transendothelial LDL passing across the endothelial cell monolayer. Paracellular transport was determined, in the competitive insert, by incubating the cells with FITC-LDL (50 μg·mL−1) and a sixfold excess of unlabelled LDL. The choice of sixfold was based on the finding that LDL at a concentration of more than 350 μg·mL−1 was toxic to endothelial cells (data not shown). Samples were then collected from the outer chambers and further dialysed against PBS to remove the free FITC, due to possible degradation or metabolism in the cell. The FITC fluorescent intensity was measured via a fluorescence spectrophotometer (Infinite F200PRO; Tecan, Männedorf, Switzerland) with excitation and emission wavelengths of 490 nm and 520 nm respectively. Background fluorescence was determined by measuring the serum-free endothelial cell medium (ECM). This background was then subtracted from the fluorescence of each sample containing FITC-LDL. The amount of LDL that was carried by transcytosis was the difference in FITC fluorescent intensity between the non-competitive insert and the competitive insert.
Figure 1.
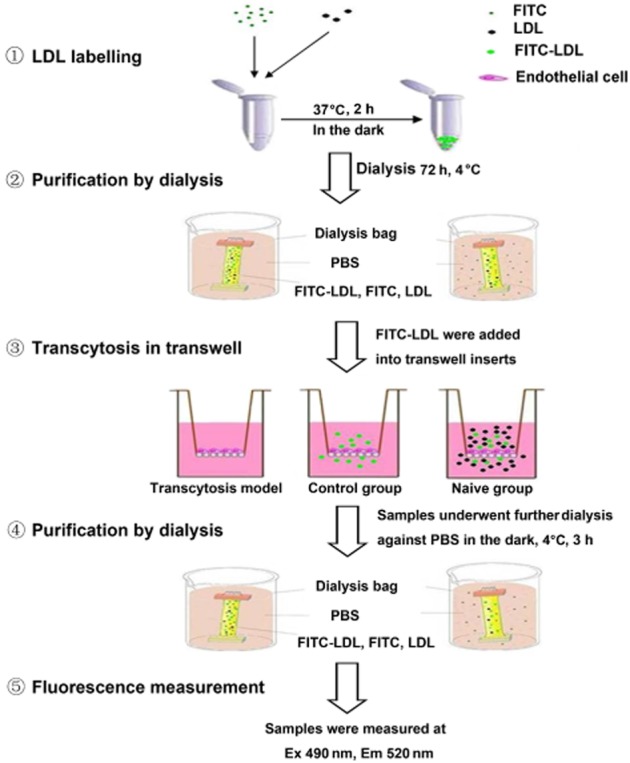
Schematic diagram of the model of LDL transcytosis. Passage of FITC-LDL through inserts with HUVECs monolayers was carried out at 37°C. LDL was added to the upper side of the inserts (50 μg·mL−1 or 100 μg·mL−1 FITC-LDL to all inserts and an additional sixfold excess of unlabelled LDL to naive inserts) for 3 h. Samples were collected from the outer chambers and further dialysed against PBS at 4°C. The FITC fluorescent intensity was assessed by fluorescence spectrophotometry.
Measurements of intracellular ROS levels
For measuring the intracellular levels of ROS, HUVECs were incubated with 3 μmol·L−1 2′,7′-dichlorofluorescein (DCF-DA; Applygen Technologies Inc, Beijing, China) at room temperature in the dark for 30 min as described previously (Jin et al., 2007), and then the medium with DCF was removed. The cells were pretreated with 10 μmol·L−1 diphenylene iodonium (DPI; Sigma), or 30 μmol·L−1 dithiothreitol (DTT) (MDBio Inc, Piscataway, NJ, USA) for further 30 min, followed by exposure to 20 μg·mL−1 CRP (C4063; Sigma-Aldrich, St. Louis, MO, USA) for 30 min; this preparation of CRP is isolated from human plasma and verified to be pentameric (Zwaka et al., 2001). Using afluorescence spectrophotometer with excitation and emission wavelengths of 490 nm and 520 nm, respectively, the fluorescence intensity of the ROS-reactive dichlorofluorescin was dynamically monitored.
PKC and Src kinase activity assay
PKC and Src kinase activity were measured by using the PKC kinase assay kit (Genmed Scientifics Inc, Arlington, MA, USA) and the Src kinase assay kit (Genmed Scientifics Inc), respectively, according to the manufacturer's instructions (Yao et al., 2011).
Isolation of caveolin-1-enriched membrane raft fractions
The most important steps of transcytosis are the endocytosis and exocytosis of the transporting cargo. As caveolae are abundant in endothelial cells, these two processes mainly take place in caveolin-1 (Cav-1)-enriched raft domains of the cell membrane. Cav-1-enriched membrane fractions were isolated with the detergent-free (Na2CO3) sucrose density gradient fractionation method as described previously with minor modifications (Meye et al., 2007; Jin et al., 2008a, 2008b; Wang et al., 2011). In brief, cells were lysed on ice for 30 min with 2 mL 500 mmol·L−1 Na2CO3 (pH 11) containing protease inhibitor cocktail (Amresco, Solon, OH, USA). The extract was collected with a cell scraper and homogenized with 15 strokes through a 25-gauge needle followed by sonication (three 15 s bursts). The total concentration of protein in the homogenate was determined and equal amounts of protein, between the control and experimental samples, were adjusted to a final volume of 2 mL with MBS (0.15 mol·L−1 NaCl, 25 mmol·L−1 2-(N-morpholino) ethanesulfonic acid, pH 6.5). By the addition of 2 mL of 90% sucrose prepared in MBS, the extract was adjusted to 45% sucrose and layered at the bottom of an ultracentrifuge tube, followed by 4 mL of 35% sucrose, 4 mL of 5% sucrose in the MBS buffer containing 250 mmol·L−1 Na2CO3. The tubes were then centrifuged at 26 808× g for 18 h at 4°C in a SW 41 rotor (Beckman Instruments, Fullerton, CA, USA). After that, a total of 12 fractions per 1 mL were collected carefully from top to bottom. Each fraction was precipitated with 10% cold trichloroacetic acid and washed with cold acetone. Precipitates were then dissolved in an SDS-PAGE lysis buffer for Western blot analysis.
Western blotting
Resuspended proteins were separated by the SDS-PAGE gel and transferred to a PVDF membrane. The membranes were probed with primary antibodies against Cav-1 (1:8000, a lipid rafts (LRs) marker protein; Cell Signaling, Danvers, MA, USA), Cavin-1 (1:500, Anbo), N-ethylmaleimide (NEM)-sensitive factor (NSF, 1:300; Proteintech, Chicago, IL, USA), α-soluble NSF attachment protein (SNAP, 1:2000; Abcam, Cambridge, UK), VAMP3/cellubrevin (1:300, Proteintech), syntaxin 4 (STX4, 1:300; Proteintech) and dynamin 2 (DNM2, 1:300; Proteintech). The immunoreactive bands were visualized by the ECL (Thermo Scientific) Western blot detection system.
Confocal imaging analysis of the intermediate status of LDL during transcytosis by quantification of LDL particles in cultured HUVECs
To determine the LDL uptake in HUVECs, cells were first incubated with 50 μg·mL−1 FITC-LDL for 24 h and then treated with DPI, DTT, bisindolylmaleimide I (BIM I; 5 μmol·L−1), PP2 (5 μmol·L−1), 3 mmol·L−1 methyl-β-cyclodextrin (MβCD; Sigma-Aldrich) or 10 μmol·L−1 NEM (Sigma-Aldrich) followed by CRP in the dark for 3 h at 37°C. Images were obtained with a confocal laser scan microscopy (Olympus FV500, Center Valley, PA, USA) using a 40× objective. For fluorescence imaging, wavelengths of 490 nm and 520 nm were used for excitation and emission respectively. The fluorescence images were analysed using the Image J software (Santa Clara, CA, USA). The individual microscopic field was randomly selected to include at least 15 cells and the numbers of cells were counted. The integrated fluorescence intensities were measured. The fluorescence intensities were normalized to the number of cells (Wang et al., 2007).
Confocal imaging analysis of LDL retention in isolated umbilical venous wall
To determine the LDL retention in human umbilical venous walls, rings of human umbilical vein were suspended in serum-free ECM medium, bubbled with 95% O2 and 5% CO2, and incubated with FITC-LDL, and/or CRP, DPI, DTT, BIM I, PP2, MβCD, NEM for 3 h in 37°C. Then frozen tissue sections of 10 μm in thickness were cut consecutively in a cryostat (Leica CM1900, Wetzlar, Germany) and further stained with DAPI. For each optical section, the region above the basilar membrane was defined as the region of interest (ROI). The fluorescent area and the area of ROI were quantified using Image J. For the fluorescence quantification, we used a weighted protocol described previously (Devlin et al., 2008). The weighted analysis was performed by first determining the area of fluorescence within the ROI of each optical section for three fluorescence intensity value ranges: 86–123, 124–161 and 162–200. These three area measurements were then multiplied by 1, 2 or 3, respectively, to give greater weight to areas of highest intensities. These weighted values were then summed for each optical section and divided by the area of ROI.
Experiments in ApoE-/- mice
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 49 animals were used in the experiments described here. Male homozygous ApoE-/- mice (C57BL/6J genetic background) were purchased from Beijing HFK Bio-Technology at 7 weeks of age. The mice were fed a standard diet for 2 weeks and then followed by a ‘Western diet’ (21% fat, 0.15% cholesterol) for 8 weeks. ApoE-/- mice (n = 7) were randomly assigned to seven treatment groups: group 1 received 0.9% saline as control; group 2 received human CRP 2.5 mg·kg−1 per week s.c.; in addition to human CRP, groups 3–7 were treated with DTT 63 mg·kg−1 per day, BIM I 1 mg·kg−1 per 3 days i.p., PP2 0.48 mg·kg−1 per 3 days i.p., MβCD 100 mg·kg−1 per day i.p., NEM 3.5 mg·kg−1 per week i.v. respectively. After treatment for 8 weeks, mice were killed by cervical dislocation. Serial cross sections (8 μm) of the heart throughout the entire aortic valve area were cut in a cryostat (Leica CM1900) and the atherosclerotic lesions were stained with Oil Red O. Plaque size was quantified using the Image J pro plus software as described previously (Missiou et al., 2010; Matsubara et al., 2012). The sections stained with CD154 (1:50; Proteintech) were examined under light microscopy at a magnification of ×400 with a semi-quantitative scoring system (0 to 4) by a method described previously (Schwedler et al., 2005). At least five animals from each experimental condition were included. All analyses were carried out without knowledge of the treatments.
Data analysis
All data are expressed as the mean ± SEM from at least three separate experiments. Unpaired Student t-tests with Bonferroni correction were used to analyse individual group statistical comparisons, and one-way ANOVA with post hoc testing was used to evaluate multiple group comparisons. Statistical significance is defined as P < 0.05.
Results
Establishment of the LDL transcytosis model and the assay of LDL transcytosis
In this study, we developed a new in vitro model of transcytosis across endothelial cell monolayers (Figure 1) to examine the mechanisms underlying the transcytosis of LDL.
As shown in Figure 2A, the total transport and the paracellular transport of FITC-LDL (50 μg·mL−1 ) across the monolayer of HUVECs was measured. The paracellular transport was determined with HUVECs adding the FITC-LDL and a sixfold excess of unlabelled LDL. Similarly, in Figure 2B the total and paracellular transport of LDL are shown, after the addition of 100 μg·mL−1 FITC-LDL into the inner inserts. The contribution of transcytosis to the total LDL transported was calculated by subtracting the paracellular transport from the total transport. These data were obtained at two time points (Figure 2C) and LDL transcytosis increased with prolonged incubation with LDL (24 h vs. 3 h). Furthermore, we also found that LDL transcytosis was significantly higher with 100 μg·mL−1 LDL compared to that with 50 μg·mL−1 LDL (Figure 2D).
Figure 2.
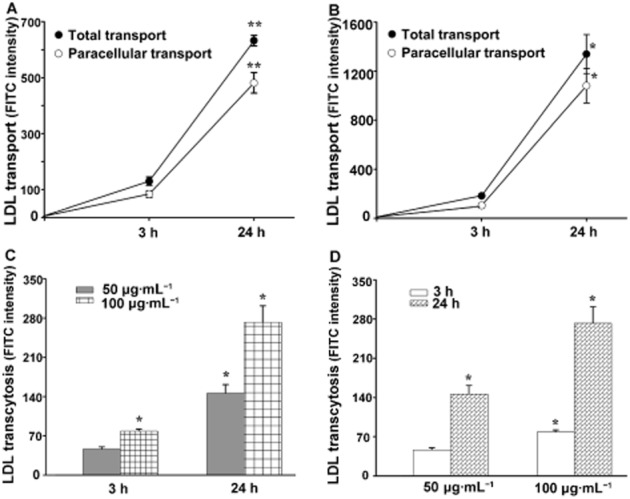
Analysis of LDL transcytosis in an in vitro model. Receptor-mediated transport (LDL transcytosis) was calculated by subtracting the FITC intensity obtained in the presence of native LDL (paracellular transport, Figure 2A, B) from that obtained in the absence of native LDL (total transport, Figure 2A, B) and are summarized in Figure 2C, D respectively. A,B: After incubation with 50 μg·mL−1 (A) or 100 μg·mL−1 (B) FITC-LDL for the indicated amount of time, the total transport and paracellular transport were measured. **P < 0.01 versus 3 h group; *P < 0.05 versus 3 h group, n = 4; (C) Time-dependent transcytosis of 50 μg·mL−1 and 100 μg·mL−1 FITC-LDL. *P < 0.05 versus 3 h 50 μg·mL−1 group, n = 4. (D) Concentration-dependent transcytosis of FITC-LDL at 3 h and 24 h. *P < 0.05 versus 3 h 50 μg·mL−1 group, n = 4.
ROS are involved in CRP-induced LDL transcytosis
We then tested the effects of adding CRP to our model of LDL transcytosis. The LDL transcytosis was normalized to that obtained after incubating 50 μg·mL−1 FITC-LDL for 3 h. As shown in Figure 3A, exposure to CRP for 3 h significantly increased LDL transcytosis and, in the same model, CRP increased intracellular ROS levels (Figure 3B), with peak production at about 5 min after the addition of CRP.
Figure 3.
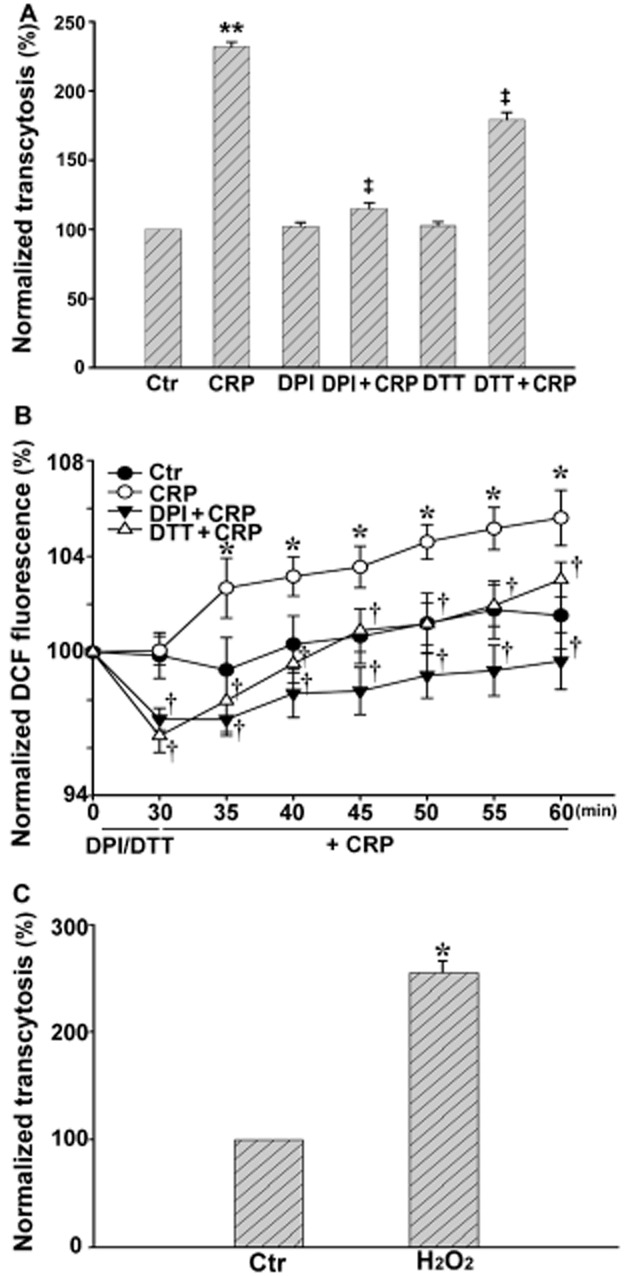
Effect of CRP on LDL transcytosis and ROS production in HUVECs. HUVECs were pretreated with 10 μmol·L−1 DPI, or 30 μmol·L−1 DTT for 30 min, and then exposed to 20 μg·mL−1 CRP or 0.1 μmol·L−1 H2O2 for 30 min. The intracellular ROS level was detected with DCF-DA. (A) LDL transcytosis in the absence or presence of CRP, DPI + CRP or DTT + CRP. **P < 0.01 versus Ctr; ‡P < 0.01 versus CRP; n = 4. (B) Production of ROS in the absence or presence of CRP, DPI + CRP or DTT + CRP. *P < 0.05 versus Ctr; †P < 0.05 versus CRP; n = 4. (C) LDL transcytosis with or without H2O2 (0.1 μmol·L−1). *P < 0.05 versus Ctr, n = 4.
To further clarify the role of ROS in CRP-induced LDL transcytosis, we pretreated HUVECs with an NADPH oxidase inhibitor, DPI or with the reducing agent DTT for 30 min, then followed that with CRP stimulation for 30 min. Pretreatment with DPI or DTT largely attenuated CRP-induced LDL transcytosis, whereas DPI or DTT alone had no effect on the transcytosis in basal conditions (Figure 3A). DPI or DTT both markedly decreased the level of intracellular ROS induced by CRP (Figure 3B). As shown in Figure 3C, 3 h incubation with 0.1 μmol·L−1 H2O2 significantly increased LDL transcytosis, which was very similar to the effect of CRP.
CRP stimulation increased PKC and Src kinase activity
In our model, CRP up-regulated PKC and Src activity (Figure 4) and this up-regulation was inhibited by the PKC inhibitor BIM I or the Src inhibitor PP2. As summarized in Figure 4, the CRP-increased PKC and Src kinase activities were also diminished in the presence of DPI or DTT.
Figure 4.
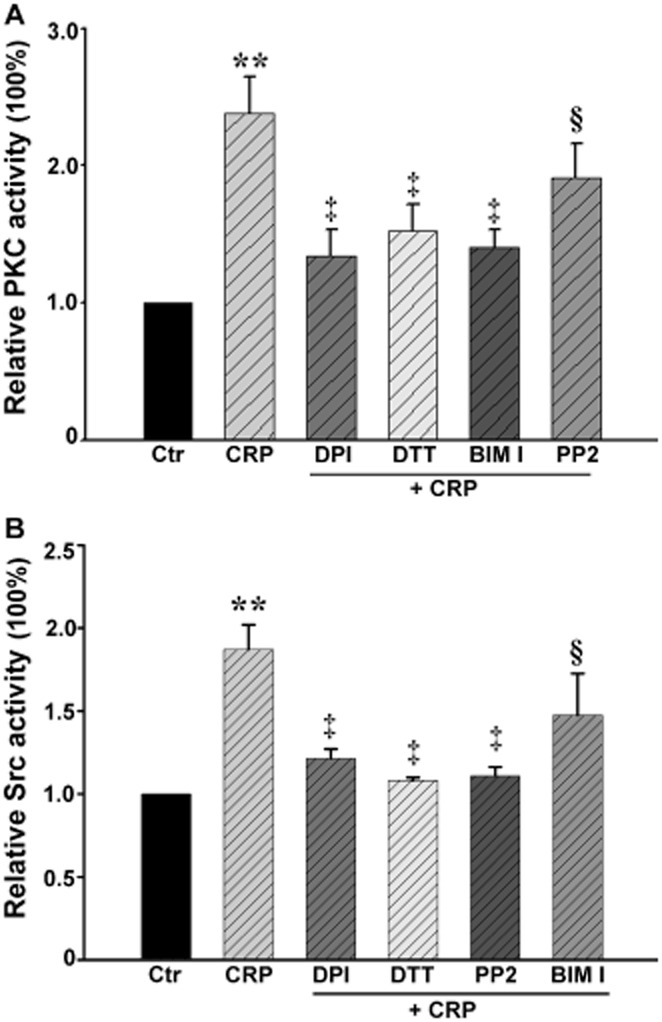
Effect of CRP on the activity of PKC and Src kinase in HUVECs. HUVECs were pretreated with 10 μmol·L−1 DPI, or 30 μmol·L−1 DTT for 30 min, and then exposed to 20 μg·mL−1 CRP for 5 min. (A) PKC activity in the absence or presence of CRP, DPI + CRP, DTT + CRP, BIM I + CRP or PP2 + CRP. **P < 0.01 versus Ctr; ‡P < 0.01 versus CRP; §P > 0.05 versus CRP; n = 4. (B) Src kinase activity in the absence or presence of CRP, DPI + CRP, DTT + CRP, BIM I + CRP or PP2 + CRP. **P < 0.01 versus Ctr; ‡P < 0.01 versus CRP; §P > 0.05 versus CRP; n = 4.
CRP stimulated the expression of molecules involved in endothelial transcytosis in Cav-1-enriched membrane raft domains
Cav-1 is a well-documented marker of LRs. We isolated Cav-1-enriched membrane fractions by using the detergent-free, sucrose density gradient fractionation method (Meye et al., 2007). The upper 12 fractions (1 mL of each) were analysed by Western blotting and showed that Cav-1 was only detected in the floating low-density fractions (fractions 6 and 7) (Figure 5A). Analysis of these fractions (Figure 5B) showed that the expression of proteins involved in LDL endocytosis (Cav-1, cavin-1 and DNM2) was increased after incubating with CRP for 3 h. Similarly, the proteins known to be involved in exocytosis (NSF, α-SNAP, VAMP 3 and STX4) were also up-regulated by CRP.
Figure 5.
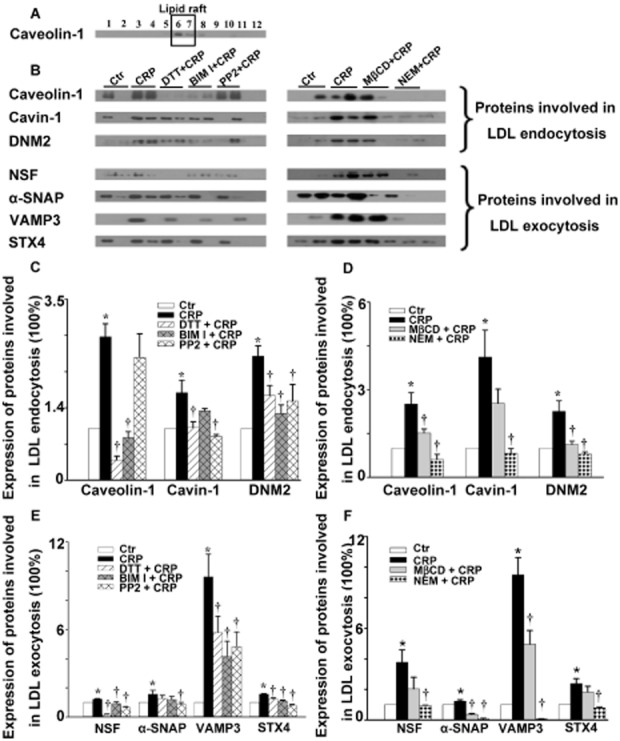
Effect of CRP on the expression of proteins involved in LDL endocytosis and exocytosis in lipid rafts in HUVECs. HUVECs were first incubated with 10 μmol·L−1 DPI, 30 μmol·L−1 DTT, 3 mmol·L−1 MβCD, 10 μmol·L−1 NEM for 30 min or 5 μmol·L−1 BIM I, 5 μmol·L−1 PP2 for 1 h, followed by 20 μg·mL−1 CRP exposure for 3 h. (A) Representative Western blot showing the subcellular localization of caveolin-1 without CRP stimulation. (B) Representative Western blot showing the expression of proteins involved in LDL endocytosis and exocytosis in lipid rafts in the absence or presence of CRP, DTT + CRP, BIM I + CRP, PP2 + CRP, MβCD + CRP or NEM +CRP. (C–F) Summary bar graph showing the expression of proteins involved in LDL endocytosis (C–D) and exocytosis (E–F) in lipid rafts in the absence or presence of CRP, DTT + CRP, BIM I + CRP, PP2 + CRP, MβCD + CRP or NEM + CRP respectively. *P < 0.05 versus Ctr; †P < 0.05 versus CRP; n = 4.
We then measured the effects of a range of inhibitors on the levels of these proteins in our HUVEC model. Cells were incubated with DTT, MβCD, NEM for 30 min or BIM I, PP2 for 1 h, followed by CRP stimulation for 3 h. As shown in Figure 5C–E, DTT blocked the CRP-induced up-regulation of Cav-1, cavin-1, DNM2, NSF, VAMP 3 and STX4, but not that of α-SNAP. The PKC inhibitor BIM I attenuated the up-regulation of all proteins except for cavin-1 and α-SNAP. PP2 significantly diminished the up-regulation of all proteins except for Cav-1. MβCD (Figure 5D–F) diminished the up-regulation of all proteins except for cavin-1, NSF and STX4. However, increases in all proteins after CRP were markedly inhibited by NEM.
CRP stimulation increased the uptake of FITC-LDL in HUVECs
After incubation with FITC-LDL, the HUVECs were found to be full of small, individual, discrete vesicles throughout the cells. The fluorescent intensity in each individual cell reflected the extent of LDL uptake. As shown in Figure 6, CRP exposure significantly increased the fluorescent intensity, which represented an increase in LDL uptake. In contrast, pretreatment with DPI, DTT, BIM I, PP2, MβCD or NEM significantly diminished CRP-stimulated internalization of FITC-LDL.
Figure 6.
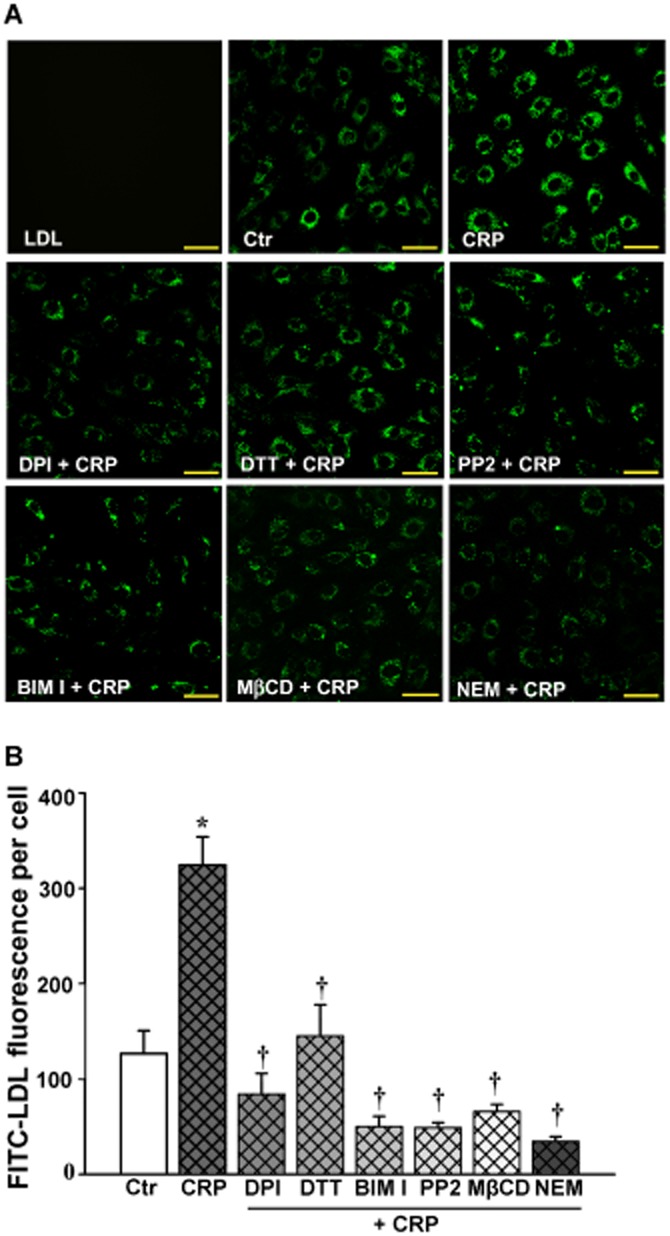
Confocal analysis of FITC-LDL uptake in HUVECs. HUVECs were first incubated with 50 μg·mL−1 FITC-LDL for 24 h, and then incubated with 10 μmol·L−1 DPI, 30 μmol·L−1 DTT, 3 mmol·L−1 MβCD, 10 μmol·L−1 NEM for 30 min or 5 μmol·L−1 BIM I, 5 μmol·L−1 PP2 for 1 h, followed by 20 μg·mL−1 CRP exposure for 3 h. (A) Confocal microscopy images of FITC-LDL uptake stimulated by CRP alone or after pretreatment with DPI, DTT, BIM I, PP2, MβCD or NEM in HUVECs. Scale bars = 50 μm. (B) Quantification summary of FITC-LDL uptake in HUVECs. *P < 0.05 versus Ctr; †P < 0.05 versus CRP; n = 4.
CRP stimulation increased the retention of LDL in human umbilical venous walls
In the preparation of rings of human umbilical vein incubated with FITC-LDL, more FITC-LDL accumulated in the region above the basilar membrane after CRP stimulation (Figure 7A). Pre-treatment with DPI, DTT, BIM I, PP2, MβCD or NEM significantly blocked CRP-stimulated FITC-LDL accumulation. Quantification of these images showed (Figure 7B) that accumulation of FITC-LDL in the presence of CRP was about threefold higher than that in the absence of CRP. DPI, DTT, BIM I or PP2, MβCD or NEM treatment resulted in about 30% decrease of FITC-LDL accumulation compared to that in CRP-treated groups.
Figure 7.
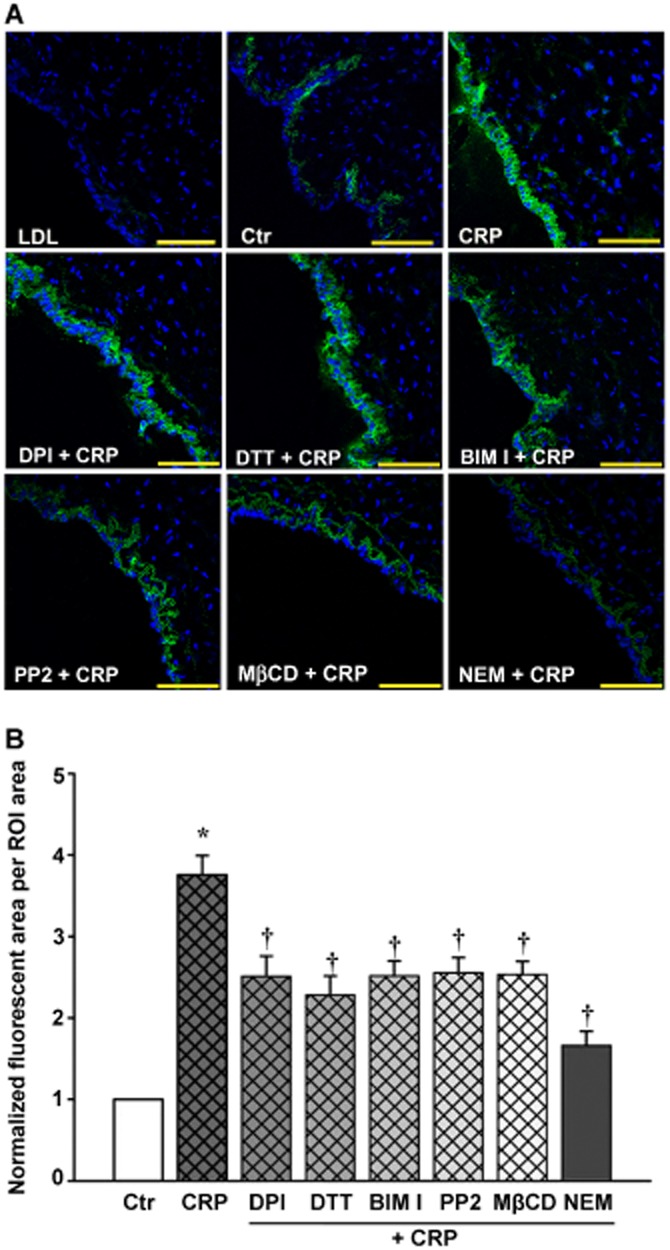
Confocal analysis of FITC-LDL retention in human umbilical venous wall. The human umbilical venous rings were incubated with 50 μg·mL−1 FITC-LDL, and/or 20 μg·mL−1 CRP, 10 μmol·L−1 DPI, 30 μmol·L−1 DTT, 5 μmol·L−1 BIM I, 5 μmol·L−1 PP2, 3 mmol·L−1 MβCD, 10 μmol·L−1 NEM for 3 h. (A) Confocal microscopic images of FITC-LDL retention stimulated by CRP alone or with DPI, DTT, BIM I, PP2, MβCD or NEM in human umbilical venous rings. Scale bars = 100 μm. (B) Quantitative summary of FITC-LDL retention in vessels. *P < 0.05 versus Ctr; †P < 0.05 versus CRP; n = 4.
CRP promoted atherosclerotic lesion formation in ApoE-/- mice
The experimental protocols used in the ApoE-/- mice are outlined in Figure 8A. As shown in Figure 8B and summarized in Figure 8C, in samples of the aortic root taken from these animals at the end of the 15 week protocol, CRP promoted formation of atherosclerotic lesions, characterized by the lipid deposition (Oil Red O staining positive) in the subendothelial space. As shown in Figure 8D and E, the aortic plaques in ApoE-/- mice also showed strong CD154 staining, which is used as a marker of arterial inflammation and correlates with early atherosclerosis (Schwedler, 2005). It was noted that the immunostaining for CD154 showed an increased signal in the endothelium of plaque lesions of CRP-treated ApoE-/- mice compared with other groups.
Figure 8.
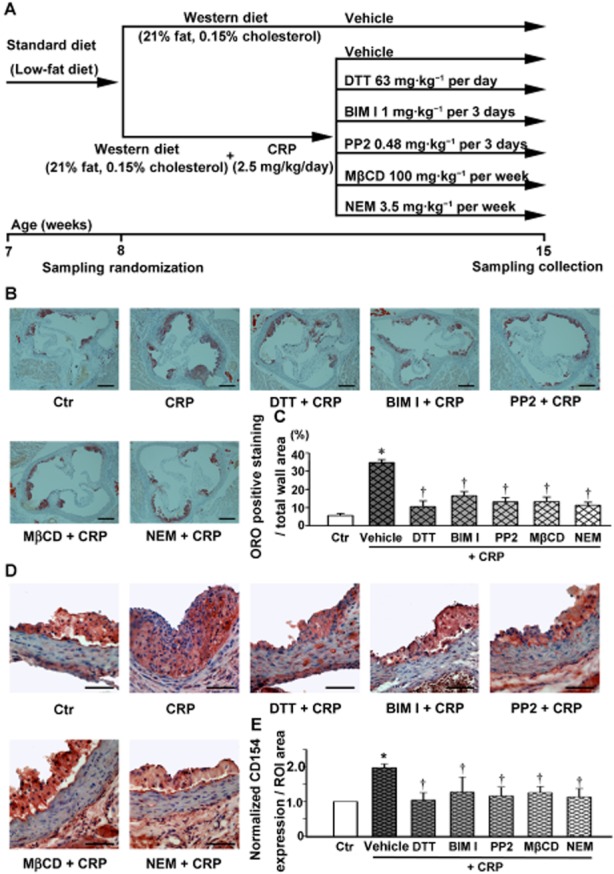
CRP increases atherosclerotic lesion formation, CD154 staining within plaques in ApoE-/- mice. (A) The treatment protocols used for the analysis of the effects of CRP. (B) Oil Red O–stained aortic root sections. Scale bars = 600 μm. (C) Quantitative summary of the percentage of the area of atherosclerotic lesion in the aortic root. *P < 0.05 versus Ctr; †P < 0.05 versus CRP; n = 7. ORO: Oil Red O. (D) Immunostaining for CD154 in aortic root sections. Scale bars = 200 μm. (E) Quantitative summary of the expression of CD154. *P < 0.05 versus Ctr; †P < 0.05 versus CRP; n = 7.
Discussion
Currently, it is generally accepted that the subendothelial retention of LDL, particularly oxidized LDL, is the key initiating event in early atherosclerosis (Libby et al., 2011; Weber and Noels, 2011). Importantly, LDLs are lipoprotein particles 20–30 nm in diameter and would not be able to cross the intact endothelium via a paracellular pathway, but rather via the transcellular process of transcytosis.
First of all, we established an in vitro model to assay LDL transcytosis. Although LDL particles cannot pass through the intact endothelial barrier via paracellular gap junctions in vivo, it is possible for these particles to pass through the in vitro cultured endothelial cells via paracellular spaces because the conditions of in vitro cultures are very different from those in vivo. Endothelial cells are enriched in LDL receptors and the transcytosis of LDL across these cells is mainly through a receptor-dependent pathway (Frank et al., 2008b). Unlike the paracellular pathway, the receptor-mediated transcytosis is saturable, which means this pathway can be competitively inhibited by excessive ligand occupation. However, the paracellular transport of FITC-LDL remains constant. By subtracting the fluorescent intensity in competitive conditions from that in non-competitive conditions, the contribution of paracellular transport of FITC-LDL can be excluded and the receptor-mediated transcytosis can be assessed.
This newly developed method for analyzing transcytosis is easy to perform, highly sensitive and avoids the use of radioactive compounds (Fry et al., 1992; Stary et al., 1994; Rosengren et al., 2002). By using this transcytosis model, we first characterized the concentration and time-dependent characteristics of LDL transcytosis. In our model, the higher concentration of LDL induced a higher transcytosis of LDL across the endothelial cell monolayer and this higher transcytosis leads to higher accumulation of LDL in the subendothelial space, which in turn leads to a higher risk of atherogenesis. Although this is seemingly a very obvious conclusion, the present study provides the first direct experimental evidence supporting the assumption that hypercholesterolemia contributes to higher risks of atherosclerosis, based on the ‘response to retention’ hypothesis (Tabas et al., 2007).
With our in vitro transcytosis model, we investigated the effects of CRP on the transcytosis of LDL across endothelial cell monolayers and found that CRP increased this transcytosis. To our knowledge, this is the first report that CRP directly enhances the transport of LDL through transcytosis in endothelial cells. Such an effect might underlie the predictive value of CRP in cardiovascular diseases.
We then analysed the CRP-induced increase in transcytosis. ROS have been emerging as essential intracellular second messengers and are also known to increase endothelial permeability (Sun et al., 2009). CRP did indeed stimulate ROS production in endothelial cells and both DPI and DTT substantially reduced the CRP-stimulated up-regulation of LDL transcytosis. Moreover, a low concentration of H2O2 (0.1 μmol·L−1) also increased LDL transcytosis in vitro, mimicking the action of CRP, further supporting the involvement of ROS signalling in the CRP-stimulated increase of LDL transcytosis. As LDL transcytosis is a significant component of the initiation of atherosclerosis, these findings strongly indicate the involvement of oxidative stress in atherosclerosis and could therefore explain the usefulness of antioxidants in the prevention or treatment of atherosclerosis, as described in numerous earlier reports (Curtiss, 2009).
One of the most important downstream events of ROS signalling is its effect on PKC and Src kinase, as recently demonstrated (Knock and Ward, 2011). In our study, we found that CRP activated PKC and Src, consistent with previous reports (Kawanami et al., 2006; Sundgren et al., 2011). BIM I was able to inhibit PKC activity but did not affect the activation of Src. In contrast, PP2 did not interfere with the activation of PKC, but inhibited Src kinase activity almost completely. These results further confirmed that BIM I and PP2 are selective inhibitors of PKC and Src respectively. Meanwhile, CRP-induced PKC and Src activation were also completely abolished by DPI and DTT, further supporting that activation of PKC and Src are downstream events of ROS signalling in endothelial cells.
Currently, there are two main processes known to be involved in the transcytosis of LDL: caveolae-mediated endocytosis and SNAP receptor (SNARE)-mediated exocytosis (Predescu et al., 2007). Caveolae are a particular type of LR and have been described as 50–100 nm, flask-shaped, non-clathrin-coated invaginations of the plasma membrane. As the population of clathrin-coated pits and vesicles is relatively small in endothelial cells, caveolae play a major role in the continuous exchange of molecules across the endothelium (Predescu et al., 2007). Caveolae exhibit their highest frequency in endothelial cells (∼10 000/cell) (Simionescu et al., 2009). The major structural component of caveolae is Cav-1, an integral membrane protein (20–22 kDa) with amino and carboxyl ends exposed to the cytoplasmic aspect of the membrane.
Recently, the discovery of a class of caveolae regulatory proteins, the cavins (also known as polymerase I and transcript release factors; PTRF), revealed new perspectives for the characterization of the caveolar coat. Expression of caveolin proteins is reduced in the absence of functional cavin-1 (Liu and Pilch, 2007; Liu et al., 2008; Hill et al., 2008). Of note, oxidative stress can up-regulate cavin-1 expression and increase the number of caveolae, as well as promoting the interaction between cavin-1 and Cav-1 (Volonte and Galbiati, 2011).
PKC is able to phosphorylate Cav-1 at Ser37 (Lisanti et al., 1995) and Src phosphorylates Cav-1 at Tyr14 (Joshi et al., 2012). As shown in our study, CRP exposure significantly increased the expression of Cav-1 and cavin-1 in membrane raft domains and the process of the caveolae/Cav-1/cavin-1 system participating in the transcellular transport of LDL was regulated by PKC or Src kinases.
In fact, dynamin, which mainly localizes at the neck of caveolae serves as the GTPase mediating the fission of caveolae (Schnitzer, 2001). Three distinct dynamin genes have been identified in mammals and DNM2 is found in all tissues. Dynamin is a downstream target for Src (Ahn, 2002) and PKC (Powell et al., 2000), and has been reported to activate signalling events associated with caveolae internalization. Our study confirmed that the translocation of DNM2 is also dependent on the activation of PKC and Src kinases.
The ‘SNARE hypothesis’ describes the mechanism by which the SNAREs promote membrane docking and fusion through interactions with an ATPase, NSF and its ‘receptor’, SNAP (Sollner et al., 1993). NSF has been implicated as an important regulator of transcytosis in endothelial cells. NSF and its receptor SNAPs (α-, β- and γ-SNAP), which recruit NSF and activate its ATPase activity, are also reported to be strongly associated with the assembly–disassembly cycle of the SNARE complex (Predescu et al., 2007), which can facilitate cargo release. In mammals, α-SNAP is the most abundant and ubiquitous isoform of the SNAPs.
SNAREs are divided into v-SNAREs [vesicle-associated membrane protein (VAMP)] and t-SNAREs (syntaxin), with their targeting receptors located on the vesicles and the target membrane respectively. VAMPs (also called synaptobrevins) are a family of integral membrane, including VAMP-1, VAMP-2 and VAMP-3 (cellubrevin). VAMP-3 has a broad tissue distribution that includes endothelial cells. There are two families of t-SNARE proteins: the syntaxins and the SNAP-25 family. To date, at least 18 syntaxin family members have been identified in mammals, of which five (STX 1A, 1B, 2–4) are plasma membrane-specific isoforms. Most importantly, both STX4 and SNAP-23 are restricted to the basolateral plasma membrane of endothelial cells (Tuma and Hubbard, 2003). SNARE proteins are also regulated by PKC and Src kinase. In our study, CRP may increase the transcytosis of LDL through facilitating the formation of both caveolin-1/cavin-1/DNM2 complex and the SNARE complex.
An intermediate status of LDL transcytosis is that LDL particles are taken up by endothelial cells and still reside in the cell cytosol. We conducted confocal microscopic analysis and found that CRP stimulated a significant increase of LDL uptake by endothelial cells and vessel walls. In contrast, inhibitors significantly diminished CRP-stimulated LDL endocytosis and subendothelial retention (mainly located above the internal elastic lamina of vessel wall).
It is noted that the endothelial cells and vessels used in the present study were isolated from the human umbilical veins, not arteries. This was based on the fact that the umbilical vein carries oxygenated arterial blood but not deoxygenated venous blood. Another reason for using human umbilical veins is that they are most easily available and are considered to be very frequently used human vessels for the study of cardiovascular diseases.
In ApoE-/- mice, CRP treatment significantly promoted the development of atherosclerosis. These results were consistent with many previous studies, which demonstrate that treaetment with native pentameric CRP accelerates the progression of atherosclerosis in ApoE-/- mice (Paul et al., 2004; Schwedler et al., 2005). Also, ROS and PKC or Src kinase-stimulated transcytosis of LDL played an essential role in CRP-related early atherogenesis.
CRP is thought to assist in complement binding to foreign or damaged cells and enhancing phagocytosis by macrophages. Although the role of CRP in atherosclerosis (Singh et al., 2008; Elliott et al., 2009; Ortiz et al., 2009; Manace and Babyatsky, 2012) is controversial, the present study demonstrates that a direct effect of CRP on human vasculature is to increase the endocytosis and exocytosis of LDL, namely the transcytosis of LDL across endothelial cells and to accelerate LDL retention in human vascular walls, which may represent an initiating mechanism for atherosclerosis.
In conclusion, the present study demonstrates for the first time that the transcytosis of LDL across endothelial cells was concentration-dependent in our model and that CRP increased LDL transcytosis across endothelial cells and therefore promotes LDL retention in human vascular walls. This CRP-stimulated LDL retention in vessel walls appears to be a novel and key step in initiating atherosclerosis. Mechanistically, it appears to be associated with the production of ROS, activation of PKC and Src,and involve the caveolae and SNARE signalling pathway in endothelial cells, which is summarized in Figure 9. Hopefully, these findings will be able to provide new insights into the pathogenesis of atherosclerosis, as well as some novel strategies for the prevention or treatment of this widespread disease.
Figure 9.
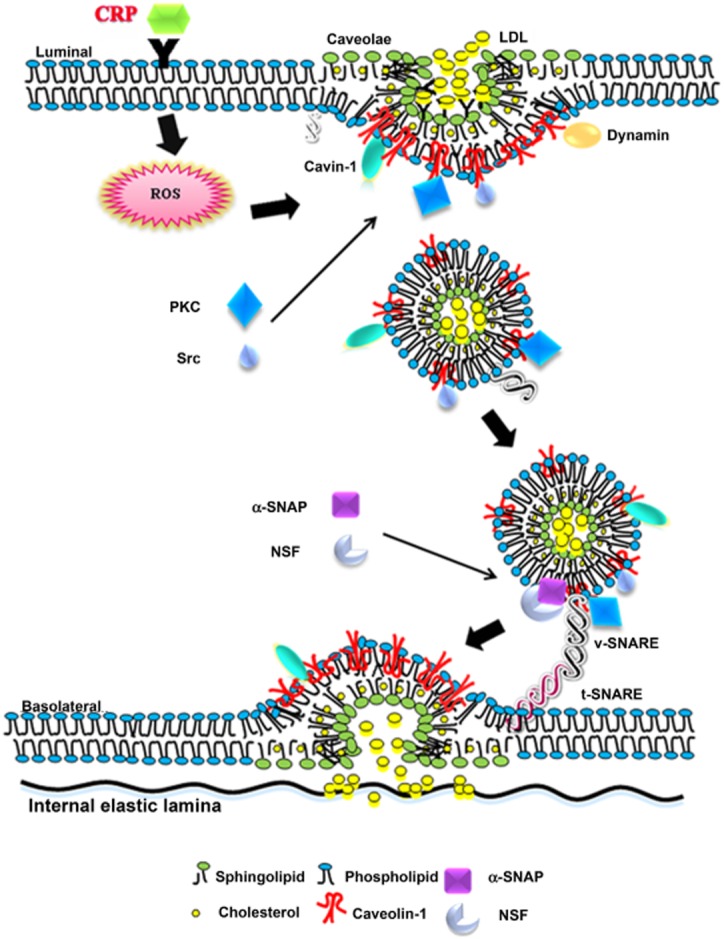
Proposed model of CRP-induced LDL transcytosis in endothelial cells.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81072634, 81000080, 81070190), grants from the Ministry of Education of China (NCET-10-0409, 2011TS069, 20101561) as well as from the National Science and Technology Major Projects (2013zx09103-001-020, 2011zx09102-004-001).
Glossary
- BIM I
bisindolylmaleimide I
- Cav-1
caveolin-1
- CRP
C-reactive protein
- DCF-DA
2′, 7′-dichlorofluorescein
- DPI
diphenylene iodonium
- LDL
low-density lipoprotein
- LR
lipid raft
- MβCD
methyl-β-cyclodextrin
- NEM
N-ethylmaleimide
- NSF
N-ethylmaleimide-sensitive factor
- ROS
reactive oxygen species
- SNAP
soluble NSF attachment protein
- SNARE
SNAP receptor
Conflicts of interest
None.
References
- AAAS. Massive trials to test inflammation hypothesis. Science. 2012;337:1158. doi: 10.1126/science.337.6099.1158. [DOI] [PubMed] [Google Scholar]
- Ahn S. Src-dependent tyrosine phosphorylation regulates dynamin self-assembly and ligand-induced endocytosis of the epidermal growth factor receptor. J Biol Chem. 2002;277:26642–26651. doi: 10.1074/jbc.M201499200. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. B J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cankova Z, Huang JD, Kruth HS, Johnson M. Passage of low-density lipoproteins through Bruch's membrane and choroid. Exp Eye Res. 2011;93:947–955. doi: 10.1016/j.exer.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtiss LK. Reversing atherosclerosis? N Engl J Med. 2009;360:1144–1146. doi: 10.1056/NEJMcibr0810383. [DOI] [PubMed] [Google Scholar]
- De Cicco NN, Pereira MG, Correa JR, Andrade-Neto VV, Saraiva FB, Chagas-Lima AC, et al. LDL uptake by Leishmania amazonensis: involvement of membrane lipid microdomains. Exp Parasitol. 2012;130:330–340. doi: 10.1016/j.exppara.2012.02.014. [DOI] [PubMed] [Google Scholar]
- Devlin CM, Leventhal AR, Kuriakose G, Schuchman EH, Williams KJ, Tabas I. Acid sphingomyelinase promotes lipoprotein retention within early atheromata and accelerates lesion progression. Arterioscler Thromb Vasc Biol. 2008;28:1723–1730. doi: 10.1161/ATVBAHA.108.173344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didangelos A, Mayr U, Monaco C, Mayr M. Novel role of ADAMTS-5 protein in proteoglycan turnover and lipoprotein retention in atherosclerosis. J Biol Chem. 2012;287:19341–19345. doi: 10.1074/jbc.C112.350785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott P, Chambers JC, Zhang W, Clarke R, Hopewell JC, Peden JF, et al. Genetic Loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302:37–48. doi: 10.1001/jama.2009.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Shi F, Liu J, Dong J, Bui HH, Peake DA, et al. Selective reduction in the sphingomyelin content of atherogenic lipoproteins inhibits their retention in murine aortas and the subsequent development of atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:2114–2120. doi: 10.1161/ATVBAHA.110.213363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogelstrand P, Boren J. Retention of atherogenic lipoproteins in the artery wall and its role in atherogenesis. Nutr Metab Cardiovasc Dis. 2012;22:1–7. doi: 10.1016/j.numecd.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Frank PG, Pavlides S, Cheung MW-C, Daumer K, Lisanti MP. Role of caveolin-1 in the regulation of lipoprotein metabolism. Am J Physiol Cell Physiol. 2008a;295:C242–C248. doi: 10.1152/ajpcell.00185.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank PG, Pavlides S, Lisanti MP. Caveolae and transcytosis in endothelial cells: role in atherosclerosis. Cell Tissue Res. 2008b;335:41–47. doi: 10.1007/s00441-008-0659-8. [DOI] [PubMed] [Google Scholar]
- Fry DL, Haupt MW, Pap JM. Effect of endothelial integrity, transmural pressure, and time on the intimal-medial uptake of serum 125I-albumin and 125I-LDL in an in vitro porcine arterial organ-support system. Arterioscler Thromb Vasc Biol. 1992;12:1313–1328. doi: 10.1161/01.atv.12.11.1313. [DOI] [PubMed] [Google Scholar]
- Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell. 2008;132:113–124. doi: 10.1016/j.cell.2007.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Yi F, Li P-L. Contribution of lysosomal vesicles to the formation of lipid raft redox signaling platforms in endothelial cells. Antioxid Redox Signal. 2007;9:1417–1426. doi: 10.1089/ars.2007.1660. [DOI] [PubMed] [Google Scholar]
- Jin S, Yi F, Zhang F, Poklis JL, Li PL. Lysosomal targeting and trafficking of acid sphingomyelinase to lipid raft platforms in coronary endothelial cells. Arterioscler Thromb Vasc Biol. 2008a;28:2056–2062. doi: 10.1161/ATVBAHA.108.172478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Zhang Y, Yi F, Li PL. Critical role of lipid raft redox signaling platforms in endostatin-induced coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2008b;28:485–490. doi: 10.1161/ATVBAHA.107.159772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi B, Bastiani M, Strugnell SS, Boscher C, Parton RG, Nabi IR. Phosphocaveolin-1 is a mechanotransducer that induces caveola biogenesis via Egr1 transcriptional regulation. J Cell Biol. 2012;199:425–435. doi: 10.1083/jcb.201207089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaptoge S, Di Angelantonio E, Pennells L, Wood AM, White IR, Gao P, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367:1310–1320. doi: 10.1056/NEJMoa1107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaptoge S, Thompson SG, Danesh J. C-reactive protein, fibrinogen, and cardiovascular risk. N Engl J Med. 2013;368:85–86. doi: 10.1056/NEJMc1213688. [DOI] [PubMed] [Google Scholar]
- Kawanami D, Maemura K, Takeda N, Harada T, Nojiri T, Saito T, et al. C-reactive protein induces VCAM-1 gene expression through NF-κB activation in vascular endothelial cells. Atherosclerosis. 2006;185:39–46. doi: 10.1016/j.atherosclerosis.2005.01.057. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knock GA, Ward JPT. Redox regulation of protein kinases as a modulator of vascular function. Antioxid Redox Signal. 2011;15:1531–1547. doi: 10.1089/ars.2010.3614. [DOI] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- Lisanti MP, Schereg PE, Tang Z, Kiibleg E, Koleske AJ, Sargiacomo M. Caveolae and human disease: functional roles in transcytosis, potocytosis, signalling and cell polarity. Dev Biol. 1995;6:47–58. [Google Scholar]
- Liu L, Pilch PF. A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J Biol Chem. 2007;283:4314–4322. doi: 10.1074/jbc.M707890200. [DOI] [PubMed] [Google Scholar]
- Liu L, Brown D, McKee M, LeBrasseur NK, Yang D, Albrecht KH, et al. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 2008;8:310–317. doi: 10.1016/j.cmet.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manace LC, Babyatsky MW. Putting genome analysis to good use: lessons from C-reactive protein and cardiovascular disease. Cleve Clin J Med. 2012;79:182–191. doi: 10.3949/ccjm.79a.09169. [DOI] [PubMed] [Google Scholar]
- Matsubara J, Sugiyama S, Sugamura K, Nakamura T, Fujiwara Y, Akiyama E, et al. A dipeptidyl peptidase-4 inhibitor, des-fluoro-sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. J Am Coll Cardiol. 2012;59:265–276. doi: 10.1016/j.jacc.2011.07.053. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meye C, Schumann J, Wagner A, Gross P. Effects of homocysteine on the levels of caveolin-1 and eNOS in caveolae of human coronary artery endothelial cells. Atherosclerosis. 2007;190:256–263. doi: 10.1016/j.atherosclerosis.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Missiou A, Kostlin N, Varo N, Rudolf P, Aichele P, Ernst S, et al. Tumor necrosis factor receptor-associated factor 1 (TRAF1) deficiency attenuates atherosclerosis in mice by impairing monocyte recruitment to the vessel wall. Circulation. 2010;121:2033–2044. doi: 10.1161/CIRCULATIONAHA.109.895037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz MA, Campana GL, Woods JR, Boguslawski G, Sosa MJ, Walker CL, et al. Continuously-infused human C-reactive protein is neither proatherosclerotic nor proinflammatory in apolipoprotein E-deficient mice. Exp Biol Med (Maywood) 2009;234:624–631. doi: 10.3181/0812-RM-347. [DOI] [PubMed] [Google Scholar]
- Paul A, Ko KW, Li L, Yechoor V, McCrory MA, Szalai AJ, et al. C-reactive protein accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:647–655. doi: 10.1161/01.CIR.0000114526.50618.24. [DOI] [PubMed] [Google Scholar]
- Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature. 2006;440:1217–1221. doi: 10.1038/nature04672. [DOI] [PubMed] [Google Scholar]
- Powell KA, Valova VA, Malladi CS, Jensen ON, Larsen MR, Robinson PJ. Phosphorylation of dynamin I on Ser-795 by protein kinase C blocks its association with phospholipids. J Biol Chem. 2000;275:11610–11617. doi: 10.1074/jbc.275.16.11610. [DOI] [PubMed] [Google Scholar]
- Predescu SA, Predescu DN, Malik AB. Molecular determinants of endothelial transcytosis and their role in endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2007;293:L823–L842. doi: 10.1152/ajplung.00436.2006. [DOI] [PubMed] [Google Scholar]
- Quest AFG, Leyton L, Párraga M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem Cell Biol. 2004;82:129–144. doi: 10.1139/o03-071. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- Rosengren B-I, Rayyes OA, Rippe B. Transendothelial transport of low-density lipoprotein and albumin across the rat peritoneum in vivo: effects of the transcytosis inhibitors NEM and filipin. J Vasc Res. 2002;39:230–237. doi: 10.1159/000063688. [DOI] [PubMed] [Google Scholar]
- Schnitzer JE. Caveolae: from basic trafficking mechanisms to targeting transcytosis for tissue-specific drug and gene delivery in vivo. Adv Drug Deliv Rev. 2001;49:265–280. doi: 10.1016/s0169-409x(01)00141-7. [DOI] [PubMed] [Google Scholar]
- Schwedler SB. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2005;112:1016–1023. doi: 10.1161/CIRCULATIONAHA.105.556530. [DOI] [PubMed] [Google Scholar]
- Schwedler SB, Amann K, Wernicke K, Krebs A, Nauck M, Wanner C, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2005;112:1016–1023. doi: 10.1161/CIRCULATIONAHA.105.556530. [DOI] [PubMed] [Google Scholar]
- Simionescu M, Popov D, Sima A. Endothelial transcytosis in health and disease. Cell Tissue Res. 2009;335:27–40. doi: 10.1007/s00441-008-0688-3. [DOI] [PubMed] [Google Scholar]
- Singh SK, Suresh MV, Prayther DC, Moorman JP, Rusinol AE, Agrawal A. C-reactive protein-bound enzymatically modified low-density lipoprotein does not transform macrophages into foam cells. J Immunol. 2008;180:4316–4322. doi: 10.4049/jimmunol.180.6.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- Sowa G. Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front Physiol. 2012;2:1–13. doi: 10.3389/fphys.2011.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W, Rosenfeld ME, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb Vasc Biol. 1994;14:840–856. doi: 10.1161/01.atv.14.5.840. [DOI] [PubMed] [Google Scholar]
- Sun S, Zu X, Tuo Q, Chen L, Lei X, Li K, et al. Caveolae and caveolin-1 mediate endocytosis and transcytosis of oxidized low density lipoprotein in endothelial cells. Acta Pharmacol Sin. 2010;31:1336–1342. doi: 10.1038/aps.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Hu G, Zhang X, Minshall RD. Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ Res. 2009;105:676–685. doi: 10.1161/CIRCRESAHA.109.201673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundgren NC, Zhu W, Yuhanna IS, Chambliss KL, Ahmed M, Tanigaki K, et al. Coupling of Fcγ receptor I to Fcγ receptor IIb by SRC kinase mediates C-reactive protein impairment of endothelial function. Circ Res. 2011;109:1132–1140. doi: 10.1161/CIRCRESAHA.111.254573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- Tuma PL, Hubbard AL. Transcytosis: crossing cellular barriers. Physiol Rev. 2003;83:871–932. doi: 10.1152/physrev.00001.2003. [DOI] [PubMed] [Google Scholar]
- Volonte D, Galbiati F. Polymerase I and transcript release factor (PTRF)/cavin-1 is a novel regulator of stress-induced premature senescence. J Biol Chem. 2011;286:28657–28661. doi: 10.1074/jbc.C111.235119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang AX, Liu Z, Barrett EJ. Insulin signaling stimulates insulin transport by bovine aortic endothelial cells. Diabetes. 2007;57:540–547. doi: 10.2337/db07-0967. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhen H, Yao W, Bian F, Zhou F, Mao X, et al. Lipid raft-dependent activation of dual oxidase 1/H2O2/NF-κB pathway in bronchial epithelial cells. Am J Physiol Cell Physiol. 2011;301:C171–C180. doi: 10.1152/ajpcell.00363.2010. [DOI] [PubMed] [Google Scholar]
- Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- Weymann A, Schmack B, Okada T, Radovits T, Strabu B, Karck M, et al. Reendothelialization of human heart valve neoscaffolds using umbilical cord-derived endothelial cells. Circ J. 2013;77:207–216. doi: 10.1253/circj.cj-12-0540. [DOI] [PubMed] [Google Scholar]
- Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao XQ, Zhang XX, Yin YY, Liu B, Luo DJ, Liu D, et al. Glycogen synthase kinase-3β regulates Tyr307 phosphorylation of protein phosphatase-2A via protein tyrosine phosphatase 1B but not Src. Biochem J. 2011;437:335–344. doi: 10.1042/BJ20110347. [DOI] [PubMed] [Google Scholar]
- Zwaka TP, Hombach V, Torzewski J. C-reactive protein-mediated low density lipoprotein uptake by macrophages: implications for atherosclerosis. Circulation. 2001;103:1194–1197. doi: 10.1161/01.cir.103.9.1194. [DOI] [PubMed] [Google Scholar]