

Abstract
Background and Purpose
Aleglitazar is a dual PPARα/γ agonist but little is known about its effects on vascular function and atherogenesis. Hence, we characterized its effects on circulating angiogenic cells (CAC), neoangiogenesis, endothelial function, arteriogenesis and atherosclerosis in mice.
Experimental Approach
C57Bl/6 wild-type (WT, normal chow), endothelial NOS (eNOS)−/− (normal chow) and ApoE−/− (Western-type diet) mice were treated with aleglitazar (10 mg·kg−1·day−1, i.p.) or vehicle.
Key Results
Aleglitazar enhanced expression of PPARα and PPARγ target genes, normalized glucose tolerance and potently reduced hepatic fat in ApoE−/− mice. In WT mice, but not in eNOS−/−, aleglitazar up-regulated Sca-1/VEGFR2-positive CAC in the blood and bone marrow and up-regulated diLDL/lectin-positive CAC. Aleglitazar augmented CAC migration and enhanced neoangiogenesis. In ApoE−/− mice, aleglitazar up-regulated CAC number and function, reduced markers of vascular inflammation and potently improved perfusion restoration after hindlimb ischaemia and aortic endothelium-dependent vasodilatation. This was associated with markedly reduced formation of atherosclerotic plaques. In human cultured CAC from healthy donors and patients with coronary artery disease with or without diabetes mellitus, aleglitazar increased migration and colony-forming units in a concentration-dependent manner. Furthermore, oxidative stress-induced CAC apoptosis and expression of p53 were reduced, while telomerase activity and expression of phospho-eNOS and phospho-Akt were elevated. Comparative agonist and inhibitor experiments revealed that aleglitazar's effects on CAC migration and colony-forming units were mediated by both PPARα and PPARγ signalling and required Akt.
Conclusions and Implications
Aleglitazar augments the number, function and survival of CAC, which correlates with improved vascular function, enhanced arteriogenesis and prevention of atherosclerosis in mice.
Keywords: PPARα/γ agonist, endothelial progenitor cells, circulating angiogenic cells, endothelial function, arteriogenesis, endothelial NO synthase, atherosclerosis, aleglitazar
Introduction
PPARα and γ are nuclear receptors (see Alexander et al., 2013) that regulate the transcription of genes important for fatty acid utilization, carbohydrate metabolism and inflammation, especially in the pathogenesis of obesity, hypercholesterolaemia, insulin resistance and atherosclerosis (Brown and Plutzky, 2007). PPARα agonists such as fibrates lower plasma triglycerides and increase high-density lipoprotein cholesterol. PPARγ agonists such as thiazolidinediones improve insulin sensitivity. Simultaneous activation of both PPARα and PPARγ may offer the opportunity to combine these favourable metabolic benefits (Benardeau et al., 2009; Charbonnel, 2009; Henry et al., 2011; Cavender and Lincoff, 2010; Sanwald-Ducray et al., 2010; Herz et al., 2011). Receptor binding and transcriptional transactivation assays have shown that the dual PPARα/γ agonist aleglitazar is a high-affinity agonist of both PPARα and PPARγ (Benardeau et al., 2009; Dietz et al., 2012). However, very little is known regarding the effect of aleglitazar on vascular function and atherogenesis.
Circulating endothelial progenitor cells (EPC) originate from the bone marrow and are recruited to sites of ischaemia and endothelial damage, where they mediate re-endothelialization and neovascularization by incorporating into the endothelium and/or by mediating paracrine effects (Asahara et al., 1997; Dimmeler, 2010; Fadini et al., 2012). EPC enhance angiogenesis, promote vascular repair and improve endothelial function (Hill et al., 2003; Werner et al., 2003; Urbich et al., 2005; Gensch et al., 2007; Foteinos et al., 2008; Fadini et al., 2012). There is no single universal definition of EPC (Fadini et al., 2012), but in a large prospective clinical study CD34/KDR-positive cells were shown to predict cardiovascular events (Werner et al., 2005). Therefore, Sca-1/VEGFR2-positive cells, representing the murine equivalent of human CD34/KDR positive cells, are well accepted as biomarkers of cardiovascular risk. Vascular risk factors, inflammation and especially type 2 diabetes, down-regulate EPC numbers and hamper their function (Vasa et al., 2001; Hill et al., 2003; Dimmeler, 2010; Jarajapu and Grant, 2010; Hamed et al., 2011), while the reduction of lipids induced by statins or physical activity is able to increase EPC numbers and improve their function (Dimmeler et al., 2001; Walter et al., 2002; Laufs et al., 2004; Mayr et al., 2011).
Increasing understanding has led to the need for increasingly complex current definitions of ‘EPC’. As recently reviewed (Fadini et al., 2012), circulating angiogenic cells (CAC; previously termed ‘early EPC’ or ‘monocytic EPC’) and outgrowth EPC (endothelial colony-forming cells) have to be distinguished. CAC emerge from short-term culture and may predominantly enhance vessel formation and, not necessarily related to endothelial commitment, angiogenesis. They mainly originate from myeloid haematopoietic cells and share features with immune cells, particularly monocytes and macrophages. According to this current concept, in our study we predominantly investigated the CAC subpopulation of EPC.
The PPARγ agonist pioglitazone up-regulates the number and function of CAC (Pistrosch et al., 2005; Gensch et al., 2007; Sorrentino et al., 2007; Werner et al., 2007; 2011). The role of PPARα signalling in the function of CAC has not been studied in detail; the only available report suggests that PPARα is required for CAC-mediated neovascularization in mice (Benameur et al., 2010).
The vascular effects of PPARα/γ agonist aleglitazar, which has not been tested in mice, are unknown. The aim of this study was to characterize the effects of the PPARα/γ agonist on CAC as a cellular marker for vascular health, on endothelial function, arteriogenesis and atherogenesis. The observed effects were confirmed in studies on human CAC.
Methods
Animals and aleglitazar treatment
Animal experiments were approved by the animal welfare authority of the Universität des Saarlandes and complied with national guidelines (directive 63/2010 of the European Parliament) as well as the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010) and procedures were as humane as possible. Mice were housed in a 22°C room with a 12 h light/dark cycle under standard conditions with food and water available ad libitum. Aleglitazar ((2S)-2-methoxy-3-[4-[2-(5-methyl-2-phenyl-4-oxazolyl)ethoxy]-7-benzothiophenyl]propanoic acid) was provided by F. Hoffmann-La Roche, Basel, Switzerland. Twelve-week-old male C57Bl/6 wild-type (WT) and endothelial NOS (eNOS) knockout (B6.129/P2-Nos3) mice of the same age and gender (Charles River Laboratories, Sulzfeld, Germany) were treated for 21 days with aleglitazar (10 mg·kg−1 body weight) or vehicle (5% DMSO in 0.9% saline) by i.p. injection. Ten-week-old male apolipoprotein E-deficient (ApoE−/−) mice (C57Bl/6 background; bred at our own animal facilities) were placed on an atherogenic Western-type diet (WTD) containing 21% fat, 19.5% casein and 1.25% cholesterol (Ssniff, Soest, Germany) and were treated with aleglitazar (10 mg·kg−1·day−1, i.p. injection) or vehicle for 6 or 8 weeks. Untreated WT mice were used as control groups where indicated. Heart rate and BP were measured by a computerized tail-cuff system (BP-2000, Visitech Systems, Apex, NC, USA) in conscious animals. Mice were trained for 3 consecutive days in the pre-warmed tail-cuff device to become accustomed to the procedure, followed by measurements of heart rate and BP at the indicated time-points. During each procedure, 30 sequential measurements were obtained at minute-by-minute intervals and averaged for each individual animal. The mean values of all analyses were used for comparisons. Body weight was documented weekly in the indicated groups and organ weights were measured immediately after the animals had been killed.
Mice were killed by i.p. injection of undiluted ketamine (1 g·kg−1) and xylazine (0.1g·kg−1). Anaesthesia was performed with a 1:10 dilution of ketamine and xylazine, and carprofen (5 mg·kg−1 s.c.) was given for post-operative analgesia.
Peritoneal glucose tolerance tests (GTT)
Glucose tolerance tests were performed in WT and ApoE−/− mice after 8 h fasting, as reported previously by i.p. injection of a glucose bolus (1.5 mg·g−1 body weight; Lenski et al., 2011). Blood samples were obtained from the tail vein at fasting and after 20, 30, 60, 90, 120 and 150 min after glucose administration. Blood glucose concentrations were measured using the glucose oxidase method with OneTouch test strips (Accu-Chek Sensor, Roche, Mannheim, Germany).
Adiponectin
Venous blood was obtained before and after treatment with aleglitazar or vehicle in WT mice (aleglitazar 10 mg·kg−1·day−1 or vehicle for 3 weeks, n = 4) and ApoE−/− mice (aleglitazar 10 mg·kg−1·day−1 or vehicle for 6 weeks, n = 4). Adiponectin concentrations were measured in diluted serum samples using the mouse adiponectin/Acrp30 Quantikine elisa (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. A standard curve was produced using recombinant mouse adiponectin (10 ng·mL−1 to 0.16 ng·mL−1).
Quantification of EPC by flow cytometry
EPC are characterized by the expression of surface markers such as Sca-1 and vascular endothelial growth factor receptor2 (VEGFR2; corresponding to human CD34 and KDR respectively). Mouse blood and bone marrow were processed for FACS analysis as previously described (Gensch et al., 2007; Werner et al., 2008; Pöss et al., 2010). The viable lymphocyte population was incubated with Sca-1-fluorescein isothiocyanate (FITC; E13–161.7, BD Pharmingen, Heidelberg, Germany) and VEGFR2 (Flk-1) (Avas12α1, BD Pharmingen) antibodies conjugated with the corresponding phycoerythrin (PE)-labelled secondary antibody (Sigma-Aldrich, Taufkirchen, Germany). Isotype-identical antibodies (IgG2a κ FITC and PE, BD Pharmingen) served as controls in every experiment (Becton-Dickinson, Heidelberg, Germany). FACS analysis was performed immediately after staining using a FACSCalibur instrument (Becton-Dickinson) and Cell Quest software version 6.0 (BD Biosciences, Heidelberg, Germany).
Isolation, culture and assessment of CAC numbers and function
To select for EPC, mononuclear cells (MNC) were isolated from spleen homogenates from WT, ApoE−/− and eNOS−/− mice by Ficoll density gradient centrifugation (Biocoll Separating Solution; Biochrom, Berlin, Germany) and cultured on fibronectin-coated culture dishes in endothelial basal medium (EBM; Lonza, Wuppertal, Germany) with supplements (1 μg·mL−1 hydrocortisone, 3 μg·mL−1 bovine brain extract, 30 μg·mL−1 gentamicin, 50 μg·mL−1 amphotericin B, 10 μg·mL−1 human endothelial growth factor and 20% fetal calf serum; Gensch et al., 2007; Pöss et al., 2010; Werner et al., 2011). In experiments using human CAC, 80 mL of fresh peripheral blood were drawn into tubes containing sodium citrate from healthy volunteers or patients with coronary artery disease (CAD) after obtaining informed consent. MNC were isolated using the Ficoll method and cultured on fibronectin-coated dishes in EBM with supplements as previously described (Werner et al., 2005; 2007) and treated as described below. Experiments with human blood samples conformed to the principles outlined in the Declaration of Helsinki.
To identify CAC, 4 × 106 MNC per animal were plated on fibronectin-coated 24-well plates in triplicate wells. After 4 days in culture, the cells were washed with PBS and CAC were identified by the uptake of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine-labelled acetylated low density lipoprotein (DiLDL, 2.4 μg·mL−1; CellSystems, St. Katharinen, Germany) and binding to FITC-labelled Ulex europaeus agglutinin I (lectin, 10 μg·mL−1; Sigma-Aldrich). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Linaris, Dossenheim, Germany). Four random fields of cells were counted for each well using a Nikon DS-Ri1 digital camera (Nikon, Düsseldorf, Germany) mounted on a Nikon Eclipse E600 fluorescence microscope at 100 × magnification. Both total cell number and number of diLDL-lectin-double-positive CAC were determined using NIS 3.0 BR software (Nikon) by an investigator blinded to the treatment group.
The migratory capacity of CAC derived from WT, ApoE−/− and eNOS−/− mice and human blood was determined in modified Boyden chamber assays (Vasa et al., 2001). For each animal or blood sample, 4 × 106 MNC were plated on fibronectin-coated six-well culture dishes and cultured for 4 days. Culture medium was removed, cells were harvested and suspended in EBM without supplements and counted under a light microscope, followed by transfer of 1 × 105 cells to a migration chamber (HTS Fluoroblock, 8 μm pore size, BD Biosciences). The chambers were then placed in a 24-well plate containing EBM without supplements and 100 ng·mL−1 SDF-1 to induce migration (R&D Systems). After incubation at 37°C for 24 h, the filters were carefully washed, cells fixed and incubated with diLDL as described above. DiLDL-positive CAC that had migrated to the lower surface of the filter in response to SDF-1 were quantified using fluorescence microscopy (400 × magnification) by an observer blinded to the study. For migration experiments performed with human CAC, treatment with aleglitazar (1 nmol·L−1–100 nmol·L−1; each in DMSO), pioglitazone (10 μmol·L−1; Cayman Chemicals, Ann Arbor, MI, USA), GW9662 (1 μmol·L−1; Cayman Chemicals), fenofibric acid (150 μmol·L−1; Sigma-Aldrich), the NOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME, Sigma-Aldrich, 1 mM·L−1), the phosphoinositide 3-kinase inhibitor LY294002 (Calbiochem, Darmstadt, Germany, 10 μM·L−1) or the direct Akt inhibitor 1L-6-hydroxymethyl-chiro-inositol2-[(R)-2-O-methyl-3-ooctadecylcarbonate] (Calbiochem, 10 μM·L−1) was started after 4 days in culture and lasted for 24 h before the migration assay was initiated.
Colony-forming capability is a key measure of CAC proliferative capacity (Hill et al., 2003). After Ficoll isolation from human blood as described above, 5 × 106 MNC were cultured in fibronectin-coated dishes in EBM with supplements. After 48 h, the non-adherent cells in the supernatant were collected and counted. One million cells were transferred to fibronectin-coated 24-well plates and cultured in EBM with supplements for 5 more days. Colonies, defined as clusters of more than 15 viable cells, were counted under a light microscope (Olympus CK2, Hamburg, Germany, 100 × magnification) by an observer blinded to the study. For experiments with human CAC, treatment with aleglitazar (10 pmol·L−1–10 μmol·L−1; each in DMSO), pioglitazone (10 μmol·L−1; Cayman Chemicals), GW9662 (1 μmol·L−1; Cayman Chemicals), fenofibric acid (150 μmol·L−1; Sigma-Aldrich) or the above-mentioned signal transduction inhibitors was performed once after reseeding the cells and lasted until counting of the colonies. In a control experiment, CAC colonies were counterstained with DiLDL and lectin to confirm the endothelial phenotype of the cells (data not shown). Furthermore, Ki67 staining (rabbit NCL-Ki67p, Leica Microsystems, Newcastle Upon Tyre, UK) using a FITC-conjugated donkey anti-rabbit secondary antibody (Dianova, Hamburg, Germany) was performed to assess proliferating cells within colonies.
Measurement of CAC apoptosis by AnnexinV FACS
Flow cytometric assessment of AnnexinV/propidium iodide staining was employed to quantify apoptosis in cultured human CAC exposed to oxidative stress. Following Ficoll isolation, 4 × 106 MNC were cultured in six-well plates in EBM for 4 days followed by treatment with aleglitazar (10 nmol·L−1) alone or in combination with hydrogen peroxide (H2O2, 500 μmol·L−1) for 24 h. Cells were washed with PBS and CAC were detached by gentle trypsinization, suspended in binding buffer (10 mmol·L−1 HEPES/NaOH, 140 mmol·L−1 NaCl, 2.5 mmol·L−1 CaCl2, pH 7.4) and subjected to FACS analysis. Assays were performed using FITC-conjugated anti-AnnexinV (BD Pharmingen), propidium iodide (Sigma-Aldrich), on a FACSCalibur instrument with Cell Quest Pro software version 6.0 (Becton-Dickinson) to detect fluorescence, forward scatter and side scatter. All measurements were made in duplicate.
Disc neoangiogenesis
We assessed angiogenesis in vivo by implanting sterilized polyvinyl alcohol sponge discs covered with a cell-impermeable nitrocellulose filter (Biorad, München, Germany) that permits capillaries to grow only through the rim of the disc. Discs were implanted s.c. in anaesthetized WT mice (n = 6, two discs per mouse). After 14 days, space-filling fluorescent microspheres (0.2 μm; Invitrogen, Karlsruhe, Germany) were injected into the left ventricle to deliver them to the microvasculature as previously described (Laufs et al., 2004; Gertz et al., 2012). The in-growth of new vessels was assessed using quantitative fluorescence microscopy with NIS 3.0 BR software. The extent of vascularization was calculated as the vascularized disc area divided by the total disc area × 100.
Aortic ring preparation and tension recording
Endothelial function was measured in aortic rings from normal WT mice on standard diet and from ApoE−/− mice after 6 weeks on the WTD with and without aleglitazar treatment. Vasodilatation and vasoconstriction of isolated aortic ring preparations were determined as described previously (Werner et al., 2009). After excision of the descending thoracic aorta all adventitial fat was removed and it was immersed in Tyrode's solution (NaCl 118 mmol·L−1, CaCl2 2.5 mmol·L−1, KCl 4.73 mmol·L−1, MgCl2 1.2 mmol·L−1, KH2PO4 1.2 mmol·L−1, NaHCO3 25 mmol·L−1, NaEDTA 26 μmol·L−1, D-(+) glucose 5.5 mmol·L−1; pH 7.4). For each animal, four 3 mm rings were mounted in organ bath chambers filled with Tyrode's solution (37°C, continuously aerated with 95% O2 and 5% CO2) and were attached to a force transducer for recording isometric tension. The aortic rings were gradually stretched to a resting tension of 10 mN, which was maintained throughout the experiment, and were allowed to equilibrate for a further 30 min. Contraction of the rings was induced with the α1-adrenoceptor agonist phenylephrine-HCl (5 μmol·L−1). Once a steady state of contraction was reached, drugs were added in increasing concentrations. Cumulative concentration-response curves for carbachol (carbamylcholine chloride, 1 nmol·L−1–100 μmol·L−1) to measure endothelium-dependent relaxation, and glyceryl trinitrate (1 nmol·L−1–10 μmol·L−1) as an NO donor, to assess endothelial-independent relaxation, were obtained. Carbachol-induced relaxation was abolished by adding L-NAME (1 μmol·L−1). Aortic rings, which relaxed <10% in response to carbachol, were excluded from the analysis due to a possible loss of the endothelium during preparation. The results are presented as % of the maximal phenylephrine-induced relaxation versus the log of the concentration.
Oil red O staining of aortic and hepatic sections
The extent of atherosclerotic plaques present in the aortic root of ApoE−/− mice after 6 weeks or 8 weeks on a WTD with or without aleglitazar treatment was quantified as described previously (Custodis et al., 2008). In brief, hearts containing the ascending aorta were embedded in Tissue Tek OCT embedding medium (VWR, Darmstadt, Germany), snap-frozen and sectioned on a Leica cryostat (10 μm), starting at the apex and progressing through the aortic valve area into the ascending aorta. Sections were fixed with 3.7% formaldehyde and stained with oil red O. Lipid-staining area and total area of each section was determined in a minimum of 25 sequential sections per animal. Atherosclerosis data are expressed as plaque area as % of total aortic sinus area.
Liver cryosections of ApoE−/− mice after 6 weeks on a WTD with or without aleglitazar treatment were stained with oil red O and the ratio of the lipid droplet area compared to the total area was calculated from a minimum of 10 sections per animal as a measure of hepatic steatosis. All images were captured using a Nikon E600 epifluorescence microscope (Düsseldorf, Germany) and NIS 3.0 BR software.
Right femoral artery ligation (FAL) and hindlimb perfusion assessment
Collateral artery growth (arteriogenesis) is a natural escape mechanism to overcome the consequences of arterial obstruction or occlusion (Schirmer et al., 2009). A murine hindlimb model was used to investigate the effects of aleglitazar treatment before femoral artery occlusion (FAL) on perfusion restoration in WT and ApoE−/− mice. WT mice were treated with vehicle or aleglitazar (10 mg·kg−1·day−1) for 14 days before and for 7 more days after FAL. ApoE−/− mice on WTD were treated for 5 weeks (aleglitazar 10 mg·kg−1·day−1 or vehicle) before and for one more week after FAL.
Following anaesthesia with ketamine and xylazine, C57Bl/6 and ApoE−/− mice were subjected to FAL as described previously (Schirmer et al., 2012). Briefly, after a 5 mm incision below the inguinal ligament, the femoral artery was dissected and ligated twice just distal to the branch of the deep femoral artery, thereby insuring sufficient collateral blood flow at rest to prevent tissue necrosis in the lower leg.
Laser Doppler perfusion imaging (MoorLDI-VR, Moor Instruments, Axminster, UK) was used to assess the limb perfusion recovery as described previously (van der Laan et al., 2012). Animals were placed on a 37°C heating pad to reduce heat loss during measurements. Directly before and after, and 3 and 7 days after ligation, laser Doppler intensities of the foot area, where adequate penetration depth of the laser is ensured, were recorded and expressed as R/L ratio (ligated to non-ligated hindlimb).
Mice underwent collateral-dependent perfusion measurements of the lower extremities under conditions of maximal vasodilatation 7 days after FAL using fluorescent microspheres (Invitrogen) as described previously (Schirmer et al., 2008; 2012). Briefly, the abdominal aorta was cannulated above the bifurcation with a polyethylene catheter and different coloured fluorescent microspheres were injected into the catheter at different pressure levels under conditions of maximal vasodilatation (adenosine 1 mg·kg−1 body weight min−1). After death, hindlimb tissue was harvested, digested and microspheres were counted in a flow cytometer. Hindlimb perfusion is expressed as a ratio of ligated versus non-ligated hindlimb and thus represents perfusion restoration within 1 week after femoral ligation.
Telomerase activity
Telomerase activity was measured in CAC protein extracts (1 μg) in 1× CHAPS lysis buffer (Trapeze, Merck Millipore, Darmstadt, Germany) using the Telomerase Repeat Amplification Protocol as described previously (Werner et al., 2008; 2011). Each 20 μL PCR reaction contained 0.1 μg of Primer TS (template), 0.05 μg Primer ACX, 1.5 mmol·L−1 MgCl2 and 2 μL Lightcycler Fast Start SYBR Green PCR Master Mix (Roche). The samples were incubated at 30°C for 30 min to allow template elongation by telomerase activity. Telomerase activity was terminated and the PCR reaction initiated by incubation at 95°C for 10 min in a Lightcycler instrument (Roche) followed by 40 amplification cycles (20 s at 95°C, 30 s at 60°C and 50 s at 72°C). Protein extracts from HEK cells (HEK293, Gibco, Darmstadt, Germany) were included as positive controls in each assay.
Western blot analysis
Cultivated CAC were homogenized with 50 μL lysis buffer (100 mmol·L−1 Tris pH 6.8, 4% SDS, 20% glycerol) containing the protease inhibitors PMSF 0.1 mmol·L−1, leupeptin 0.5 μL and aprotinin 0.5 μL (all from Sigma-Aldrich). Proteins were separated on 8–12% SDS–PAGE and transferred to nitrocellulose membranes (Biorad), blocked with 5% dry milk for 30 min and exposed to primary antibodies. The antibodies used were mouse monoclonal IgG anti-GAPDH (6C5: sc-32233, Santa Cruz Biotechnology, Heidelberg, Germany; dilution 1:4000), rabbit polyclonal IgG anti-eNOS (Acris Antibodies, Herford, Germany; dilution 1:500), mouse monoclonal IgG anti-p-eNOS S1177 (BDI250 Acris Antibodies, Germany; dilution 1:500), rabbit polyclonal IgG anti-total Akt (Cell Signaling Technology, Danvers, MA, USA; #9272, dilution 1:1000), rabbit polyclonal IgG anti-p-Akt (Ser473) (CellSignaling Technology, #9271, dilution 1:1000), rabbit polyclonal IgG anti-P53 (FL-393 SantaCruz, sc-6243, dilution 1:500) and rabbit polyclonal IgG anti-Bcl-2 (SantaCruz, sc-492, dilution 1:500). Immunodetection was accomplished using appropriate secondary antibodies (1:5000 dilution; Sigma-Adrich) and an enhanced chemiluminescence kit (ECL, GE Healthcare, München, Germany). Western blot band intensities were analysed by densitometry and all data were normalized to GAPDH.
Semi-quantitative and quantitative PCR
To detect PPARα and PPARγ in human CAC, 2 × 107 human MNC were seeded on fibronectin-coated dishes in EBM with supplements after Ficoll isolation. After 4 days in culture, CAC were harvested and total RNA was isolated (peqGOLD RNAPure, Peqlab, Erlangen, Germany). To detect PPARα and PPARγ in MNC from untreated C57Bl/6 WT mice, Ficoll isolation of spleen homogenates, bone marrow and blood (four animals were pooled) was performed and the pellets were directly used for RNA isolation. HUVEC (passage 2) were used as a positive control as these cells are known to express PPARα and γ (Inoue et al., 1998; Kato et al., 1999). The High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Darmstadt, Germany) was used for reverse transcription of 2 μg total RNA and semi-quantitative PCRs were performed (60°C annealing temperature, 40 cycles) using the following primers (Hoekstra et al., 2003): PPARα forward: 5′-TGAACAAAGACGGGATG-3′; PPARα reverse: 5′-TCAAACTTGGGTTCCATGAT-3′; PPARγ forward: 5′-CATGCTTGTGAAGGATGCAAG-3′; PPARγ reverse: 5′-TTCTGAAACCGACAGTACTGACAT-3′. Reaction products were visualized on ethidium bromide-stained agarose gels.
Mouse liver and aortic tissue from ApoE−/− mice treated with and without aleglitazar that had been snap-frozen and stored at −80°C were homogenized and total RNA was isolated. Quantitative RT-PCR reactions were carried out on a StepOnePlus Real-Time PCR System (Applied Biosystems) to detect the expression of PPAR response genes in the liver and inflammatory regulators in the vessel wall. The thermal cycling profile consisted of a 95°C activation step, followed by 40 cycles of 95°C, 15 s and 60°C, 60 s. The PCR data are expressed using the comparative Ct method (2−ΔΔCt) to calculate relative differences in the mRNA expression between the aleglitazar-treated animals and controls, using 18 s as housekeeping mRNA. The following primer pairs were used in hepatic tissue (adapted from (Lee et al., 2006): Acyl-CoA oxidase (ACO) forward: 5′-TGTTAAGAAGAGTGCCACCAT-3′; reverse: 5′-ATAAGTGCCCGTGATCTCCA-3′; fatty acid-binding protein 3 (FABP3) forward: 5′-CCCCTCAGCTCAGCACCAT-3′; reverse: 5′-CAGAAAAATCCCAACCCAAGAAT-3′; carnitine palmitoyl transferase 1 (CPT1) forward: 5′-AACCCAGTGCCTTAACGATG-3′; reverse: 5′-GAACTGGTGGCCAATGAGAT-3′.
Monocyte-chemoattractant protein-1 (MCP1) and TNF-α were amplified in aortic tissue using the following primer sequences: CCL2- forward: 5′-TGGCTCAGCCAGATCCAGT-3′; reverse: 5′-TTGGGATCATCTTGCTGGTG-3′; TNF-α forward: 5′-GATTATGGCTCAGGGTCCAA-3′; reverse: 5′-CTCCCTTTGCAGAACTCAGG-3′. Furthermore, real-time PCRs were performed to detect and quantify PPARα and PPARγ mRNA expression in MNC from murine spleen, bone marrow and blood of untreated WT mice as compared with HUVEC using the above-listed primer sequences.
Statistical analysis
Unless otherwise stated, all data are presented as mean ± SEM. For statistical analysis, Student's two-tailed, unpaired t-test and anova for multiple comparisons were employed where applicable. Post hoc comparisons were performed with the Neuman–Keuls test. Results of serial glucose measurements during the glucose tolerance test, aortic endothelial relaxation in response to different dosages of carbachol and nitroglycerin as well as time course of laser Doppler perfusion measurements before and after right FAL were compared using a general linear model for repeated measurements using Wilks' Lambda for group comparisons. P-values of <0.05 indicate statistical significance. Statistical analyses were performed with GraphPrism software version 5.0 and with SPSS software version 19.0 (SPSS Inc., Chicago, IL, USA).
Results
The dual PPARα/γ agonist increases adiponectin, reduces hepatic fat accumulation and normalizes glucose tolerance
During the treatment period, heart rate and BP measured by the tail-cuff method remained unchanged in all groups (Table 1). In WT mice, body weight remained constant during the 3 week treatment period, but ApoE−/− mice showed approximately 20% body weight gain during 6 weeks study duration, which was similar in both the aleglitazar and vehicle treatment arms (Table 1).
Table 1.
No effects of aleglitazar on body weight, heart rate and BP
| C57Bl/6 + vehicle | C57Bl/6 + aleglitazar 10 mg·Kg−1·day−1 | |||||
|---|---|---|---|---|---|---|
| Week 1 | Week 2 | Week 3 | Week 1 | Week 2 | Week 3 | |
| Body weight (g) | 21.6 ± 1.5 | 21.1 ± 1.0 | 21.7 ± 1.0 | 21.8 ± 1.9 | 22.0 ± 0.3 | 22.1 ± 0.6 |
| Heart rate (min−1) | 576 ± 29 | 651 ± 14 | 635 ± 36 | 641 ± 34 | 663 ± 13 | 703 ± 21 |
| Systolic BP (mmHg) | 113 ± 6 | 128 ± 1 | 116 ± 3 | 118 ± 5 | 118 ± 6 | 120 ± 1 |
| Diastolic BP (mmHg) | 77 ± 6 | 90 ± 5 | 77 ± 2 | 75 ± 5 | 83 ± 5 | 81 ± 2 |
| ApoE−/− + vehicle | ||||||
| Week 1 | Week 2 | Week 3 | Week 4 | Week 5 | Week 6 | |
| Body weight (g) | 21.7 ± 2.1 | 22.7 ± 2.0 | 23.5 ± 2.0 | 24.5 ± 2.0 | 25.3 ± 2.1 | 26.0 ± 2.1 |
| Heart rate (min−1) | 584 ± 17 | 641 ± 7 | 599 ± 19 | 619 ± 30 | 582 ± 22 | 581 ± 23 |
| Systolic BP (mmHg) | 129 ± 6 | 140 ± 3 | 111 ± 3 | 123 ± 4 | 118 ± 5 | 120 ± 8 |
| Diastolic BP (mmHg) | 88 ± 4 | 96 ± 4 | 76 ± 3 | 81 ± 1 | 85 ± 6 | 80 ± 6 |
| ApoE−/− + Aleglitazar 10 mgKg−1day−1 | ||||||
| Week 1 | Week 2 | Week 3 | Week 4 | Week 5 | Week 6 | |
| Body weight (g) | 18.9 ± 0.8 | 20.1 ± 0.7 | 20.7 ± 0.8 | 21.1 ± 0.8 | 22.1 ± 0.7 | 22.9 ± 0.7 |
| Heart rate (min−1) | 516 ± 12 | 527 ± 20 | 575 ± 15 | 629 ± 22 | 608 ± 10 | 604 ± 17 |
| Systolic BP (mmHg) | 113 ± 7 | 116 ± 5 | 122 ± 3 | 124 ± 8 | 122 ± 4 | 119 ± 6 |
| Diastolic BP (mmHg) | 81 ± 4 | 79 ± 3 | 73 ± 3 | 77 ± 4 | 82 ± 2 | 76 ± 2 |
ApoE−/− denotes apolipoprotein E knockout mice; values are mean ± SEM.
Adiponectin, a vascular protective adipokine, is up-regulated by PPARγ agonists such as pioglitazone and was studied as a positive control (Werner et al., 2007). Aleglitazar treatment 10 mg·kg·day−1 i.p. for 3 weeks in C57Bl/6 WT mice and for 6 weeks in hypercholesterolaemic ApoE−/− mice on WTD significantly increased serum adiponectin concentrations in both strains (Figure 1A). In WT mice, adiponectin levels rose by 29 ± 1.8 ng·mL−1 versus baseline (compared with 5 ± 0.7 ng·mL−1 in vehicle-treated controls, P < 0.001) and in ApoE−/− mice, adiponectin increased by 30 ± 7.5ng·mL−1 versus baseline (compared with 9 ± 4.6 ng·mL−1 in control animals, P < 0.001). The effects of aleglitazar on the PPARα target genes ACO, FABP3 and CPT1 (Lee et al., 2006; Rakhshandehroo et al., 2010) were evaluated by real-time PCR in ApoE−/− mice on WTD. Aleglitazar treatment for 6 weeks led to an up-regulation of ACO (319 ± 43%), FABP3 (633 ± 60%) and CPT1 (133 ± 6%) mRNA expression in the liver (Figure 1B). Liver dry weight was decreased from 583 ± 53 mg to 475 ± 30 mg in aleglitazar-treated ApoE−/− mice (P = 0.04). Oil red O staining of liver cryosections revealed that ApoE−/− mice on the WTD were characterized by hepatic steatosis, which was potently reduced by aleglitazar treatment for 6 weeks (Figure 1C). C57Bl/6 mice on standard chow had no relevant liver steatosis (data not shown).
Figure 1.
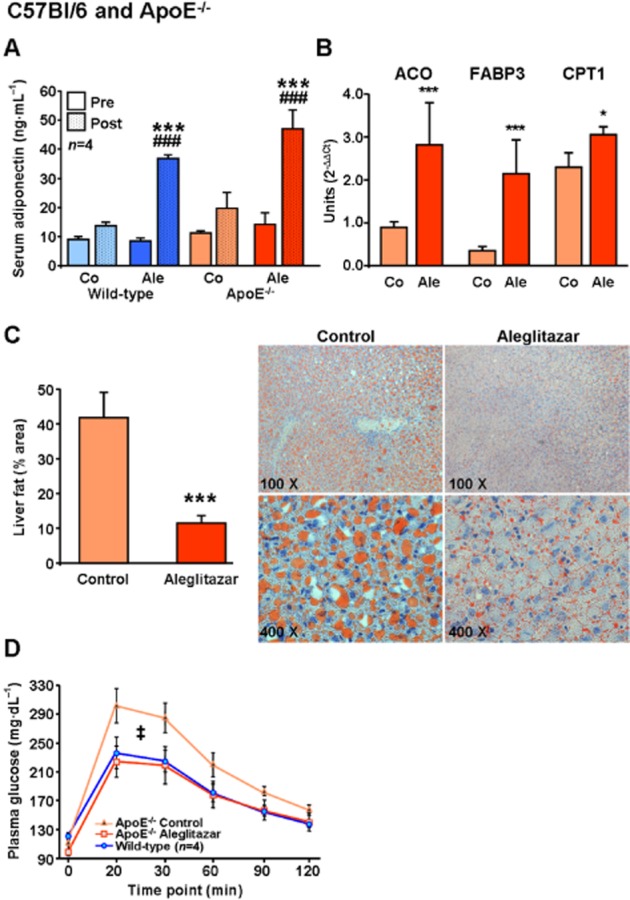
The dual PPARα/γ agonist aleglitazar upregulates adiponectin and PPARα downstream targets, reduces liver fat and improves glucose tolerance in ApoE−/− mice. (A) Serum adiponectin concentrations were measured by elisa before and after treatment of C57Bl/6 WT mice with aleglitazar 10 mg·kg−1 i.p. or vehicle daily for 3 weeks, and in ApoE−/− mice on WTD treated for 6 weeks. (B) Real-time quantitative PCR was used to assess mRNA expression of PPARα target genes ACO, FABP3 and CPT1 in liver tissue of ApoE−/− mice on WTD treated for 6 weeks with aleglitazar or vehicle. The 18 s mRNA was used as loading control and data were analysed using the comparative Ct method. (C) Hepatic steatosis was measured by oil red O staining of liver sections and estimated by calculating the area of the lipid droplets compared with the total area. (D) The time course for serum glucose concentrations after i.p. glucose injection (1.5 mg·kg−1) in untreated C57Bl/6 WT and ApoE−/− mice on WTD treated with aleglitazar 10 mg·kg−1 i.p. or vehicle daily for 6 weeks. Significant differences were calculated for the AUC. n = 6 unless otherwise indicated, *P < 0.05 and ***P < 0.001 versus vehicle-injected control mice, ###P < 0.001 versus aleglitazar-treated mice. ‡P < 0.001 versus C57Bl/6 WT mice and aleglitazar-treated ApoE−/− mice.
PeritonealGTTs revealed that ApoE−/− mice on WTD for 6 weeks had developed impaired glucose tolerance, which was normalized by aleglitazar treatment (Figure 1D). In contrast to ApoE−/−, WT mice showed no impairment of glucose tolerance and aleglitazar had no effect on fasting serum glucose in WT mice.
Aleglitazar increases the number of CAC in spleen, blood and bone marrow
In mice, the spleen serves as a haematopoietic organ. We measured the number of CAC in the spleen, blood and bone marrow of C57Bl/6 WT mice treated with aleglitazar for 3 weeks. CAC were generated by differentiation of spleen-derived MNC in culture for 4 days. Expression of PPARα and γ in murine MNC derived from spleen, bone marrow and venous blood was confirmed by semi-quantitative and real-time PCR assays (Supporting Information Figure S1). Aleglitazar treatment increased the numbers of diLDL+/lectin+ cells (182 ± 8% vs. control mice, P < 0.05, Figure 2A,B). Furthermore, FACS analyses using stem cell antigen-1 (Sca-1) and VEGFR2 as markers revealed that 3 weeks of aleglitazar treatment also increased CAC both in the blood (153 ± 10%, P < 0.05; absolute values: control 518 ± 110 vs. aleglitazar 794 ± 126 gated events per 100 000 events) and in the bone marrow (197 ± 22%, P < 0.05; absolute values: control 120 ± 15 vs. aleglitazar 237 ± 26 gated events per 100 000 events; Figure 2C,D).
Figure 2.

Enhancement of CAC number, functional capacity and neoangiogenesis by aleglitazar. (A) Quantification and (B) representative fluorescence microscopy (100 × magnification) of spleen-derived diLDL/lectin positive CAC after treatment of WT mice with aleglitazar 10 mg·kg−1 i.p. daily or vehicle (controls) for 3 weeks. (C) Effects of aleglitazar treatment in WT mice compared with controls on the number of Sca-1/VEGFR2 positive EPC in the blood and (D) the bone marrow as measured by FACS analyses. (E) Effect of aleglitazar 10 mg·kg−1 i.p. daily for 3 weeks in C57Bl/6 WT mice on CAC migration in modified Boyden chambers using SDF-1 (100 ng·mL−1) as a chemoattractant. (F) Quantification of disc neoangiogenesis 2 weeks after s.c. implantation of polyvinyl sponges and after perfusion with fluorescent microspheres (0.2 μm) in WT mice treated with aleglitazar or vehicle. Lower panels show representative microscopic images (20 × magnification) of the vascularized border zone of perfused discs for (G) a control and (H) an aleglitazar-treated animal. n = 6, *P < 0.05 versus control mice.
Aleglitazar improves CAC functional capacity and formation of new blood vessels in WT mice
CAC have the ability to migrate, extravasate and stimulate angiogenesis (Vasa et al., 2001). In C57Bl/6 WT mice, aleglitazar treatment for 3 weeks enhanced SDF-1-induced CAC migration through the pores of modified Boyden chambers (186 ± 6% vs. vehicle-treated mice, P < 0.01; Figure 2E). Neoangiogenesis was assessed in vivo by implanting disc-shaped polyvinyl alcohol sponges subcutaneously 2 weeks before sacrifice. The in-growth of new vessels was quantified by calculating the vascularized area of the discs after perfusion of the animals with space-filling fluorescent microspheres. Treatment with the dual PPARα/γ agonist aleglitazar increased neoangiogenesis by almost twofold (178 ± 18%; P < 0.05; Figure 2F–H).
Aleglitazar improves CAC functional capacity and partially restores impaired endothelial function in ApoE−/− mice
ApoE knockout mice (ApoE−/−) on a WTD is a well-characterized model of hypercholesterolaemia-induced vascular damage (Custodis et al., 2008). In ApoE−/− mice on WTD, aleglitazar treatment 10 mg·kg−1·day−1 for 6 weeks led to an increase in CAC number (diLDL+/lectin+ cells: 171 ± 6% vs. controls, P < 0.001; Figure 3A) and functional capacity (migration: 149 ± 4% vs. controls, P < 0.05; Figure 3B). Organ bath experiments demonstrated that endothelium-dependent vasorelaxation of aortic rings in response to carbachol was impaired in ApoE−/− mice fed with WTD for 6 weeks. Concomitant aleglitazar treatment partially restored endothelial function (Figure 3C). The endothelium-independent vasorelaxation of aortic rings induced by nitroglycerin was comparable in WT and ApoE−/− mice and remained unchanged by aleglitazar treatment (Figure 3D). After hindlimb ischaemia induced by right femoral artery ligation, Apo−/− mice treated with aleglitazar were characterized by an improvement of endothelial-dependent laser Doppler perfusion (right/left foot ratio 0.40 ± 0.03) after 7 days compared with vehicle-treated animals (right/left foot ratio 0.24 ± 0.01; Figure 3E). Collateral-dependent perfusion measurements of the lower extremities under conditions of maximal vasodilatation 7 days after FAL using fluorescent microspheres demonstrated that compared with WT mice, Apo−/− control mice showed an impairment of perfusion restoration (right/left leg ratio in WT 78 ± 13 vs. Apo−/− 56 ± 6), which was normalized by aleglitazar treatment (R/L ratio 79 ± 5; Figure 3F).
Figure 3.

Aleglitazar improves CAC, restores aortic endothelial function and improves perfusion recovery after hindlimb ischaemia in ApoE−/− mice. Effects of aleglitazar 10 mg·kg−1 i.p. daily for 6 weeks in ApoE−/− mice on WTD on (A) the number of diLDL/lectin positive CAC and (B) CAC migration in modified Boyden chambers; *P < 0.05 and ***P < 0.001 versus control mice. (C) Endothelium-dependent vasorelaxation of aortic rings in response to increasing concentrations of carbachol and (D) endothelium-independent vasorelaxation of aortic rings in response to increasing concentrations of nitroglycerin (shown as percentage of maximal phenylephrine-induced constriction) in untreated C57Bl/6 WT mice (n = 3), ApoE−/− control mice after 6 weeks WTD (n = 6), and ApoE−/− mice on WTD and concomitant aleglitazar treatment (10 mg·kg−1 i.p. daily) for 6 weeks (n = 6). †P < 0.001 WT versus ApoE−/− mice, #P < 0.05 for difference between ApoE−/− groups. (E) Laser Doppler measurement of endothelial-dependent hindlimb perfusion before, directly after, 3 days and 7 days after right FAL in ApoE−/− mice on WTD treated with vehicle (n = 10) or aleglitazar 10 mg·kg−1 i.p. (n = 12) daily for 5 weeks prior to ligation until the end of the study. Bars represent the mean Doppler perfusion ratio of the right ligated leg versus the left unligated leg. Lower panels show representative Doppler images. (F) Microsphere perfusion measurement of endothelial-independent hindlimb blood flow recovery 7 days after right FAL in C57Bl/6 mice on standard diet (treated with vehicle or aleglitazar 10 mg·kg−1 i.p. daily for 2 weeks before ligation until the end of the study) and ApoE−/− mice on WTD (treated with vehicle or aleglitazar 10 mg·kg−1 i.p. daily for 5 weeks before ligation until the end of the study). Bars represent the mean perfusion ratio of the right ligated leg versus the left unligated leg. ***P < 0.001 versus WT control mice, ###P < 0.001 versus ApoE−/− control mice; n = 6.
Aleglitazar prevents atherosclerosis in apolipoprotein E−/− mice
ApoE knockout mice (ApoE−/−) develop atherosclerosis on a normal diet that is accelerated on a WTD. Assessment of expression of two major inflammatory markers by real-time PCR showed that aleglitazar treatment almost completely repressed aortic MCP-1 mRNA expression to 6 ± 1% versus. control mice. TNF-α mRNA was also significantly down-regulated by aleglitazar (62 ± 12% vs. controls; Figure 4A). Oil red O staining of plaques in aortic root sections of ApoE−/− mice on WTD revealed that aleglitazar (10 mg·kg·day−1 i.p. for 6 weeks) potently prevented atherosclerotic plaque formation (plaque area/total lumen area 2.3 ± 0.8% vs. 10.1 ± 1.9% after 6 weeks treatment). A second study, performed in a separate cohort of animals at a later time point (8 weeks), confirmed that aleglitazar reduces plaque development (plaque area: aleglitazar, 22 ± 2.2% vs. vehicle, 36 ± 2.1%; Figure 4B–D).
Figure 4.

Aleglitazar retards development of atherosclerosis. (A) Real-time PCR assays of aortic MCP-1 and TNF-α mRNA expression in ApoE−/− mice on a WTD and treated with aleglitazar 10 mg·kg−1 i.p. or vehicle (n = 6 per group) for 8 weeks. Effects of aleglitazar for 6 or 8 weeks on atherosclerotic lesion formation in ApoE−/− mice on a WTD. (B) Histomorphometric quantification of atherosclerotic plaques in the aortic sinus after 6 weeks (n = 6) or 8 weeks (n = 7) aleglitazar 10 mg·kg−1 i.p. daily, shown as percentage of plaque area compared with total lumen area. *P < 0.05, **P < 0.01 and ***P < 0.001 versus vehicle-treated controls. Representative aortic root sections with oil red O staining of atherosclerotic plaques (40 × magnification) after (C) 6 weeks or (D) 8 weeks of aleglitazar treatment are shown.
The effects of aleglitazar on CAC number and function are absent in eNOS−/− mice
In knockout mice without functional eNOS (eNOS−/−), there was no effect of aleglitazar 10 mg·kg·day−1 for 3 weeks on diLDL+/lectin+ CAC numbers in culture (Figure 5A) and on CAC migration (Figure 5B). FACS measurements of Sca-1/VEGFR2 positive cells in the blood and bone marrow of WT and eNOS−/− mice revealed that CAC numbers in the blood were different in eNOS−/− compared with WT mice, but were not increased by aleglitazar treatment (Figure 5C). eNOS−/− mice were characterized by elevated Sca-1/VEGFR2 positive cells in the bone marrow and aleglitazar was not able to improve CAC mobilization from the bone marrow to other compartments (Figure 5D), indicating that eNOS is crucial for PPAR-mediated CAC mobilization from the bone marrow and augmentation of peripheral CAC numbers and function. Furthermore, measurement of BP with the tail-cuff system confirmed that eNOS knockout mice had higher systolic and diastolic BP compared with WT mice and showed that aleglitazar treatment had no haemodynamic effect in these mice (Figure 5E,F).
Figure 5.

eNOS is required for aleglitazar's effects on CAC. Effects of vehicle or aleglitazar 10 mg·kg−1 i.p. daily for 3 weeks in eNOS−/− mice on standard diet on (A) the number of diLDL/lectin positive CAC and (B) migration in modified Boyden chambers. Comparison of the effects of 3 weeks aleglitazar treatment in C57Bl/6 and eNOS−/− mice on Sca-1/VEGFR2 positive EPC in (C) the blood and (D) the bone marrow as measured by FACS analyses. *P < 0.05 and **P < 0.01 versus WT control. Repetitive measurements of (E) systolic and (F) diastolic BP at baseline and weekly during treatment with aleglitazar 10 mg kg−1 i.p. daily in eNOS−/− mice, compared with untreated C57Bl/6 mice.
Effects on human CAC: aleglitazar improves function and reduces apoptosis of human CAC in vitro
To bridge the translation of the effects of aleglitazar in mice to humans, we performed studies on cultivated human CAC isolated from the blood of healthy donors. All experiments were performed after 4 days of differentiation of CAC in endothelial basal medium. This was followed by treatment with aleglitazar and other PPAR ligands for 24 h at the concentrations as indicated in Figure 6. CAC migratory and colony-forming capacity were enhanced by low, nM concentrations of aleglitazar (Figure 6A,B). Within CAC colonies, aleglitazar enhanced the number of Ki67-positive nuclei (166 ± 10% vs. controls), suggesting that the dual PPARα/γ agonist stimulates proliferation of these cells (Figure 7A). Aleglitazar concentrations below 0.1 nmol·L−1 did not induce CAC colony-forming units (CFU; data not shown). Increased CAC numbers may be a result of both enhanced CAC proliferation and improved survival. Therefore, basal and oxidative stress-induced CAC apoptosis were measured by annexin V/propidium iodide FACS. Treatment of CAC with 10 nmol·L−1 aleglitazar had no effect on basal CAC apoptosis, but reduced H2O2-induced apoptosis (apoptosis rate: 47 ± 5% in H2O2-treated CAC vs. 33 ± 3% in aleglitazar/H2O2 co-treated CAC, P < 0.05; Figure 6C). The PPARγ agonist pioglitazone is known to induce CAC telomerase and survival proteins (Werner et al., 2011). Treatment of CAC with 10 μmol·L−1 pioglitazone induced CAC telomerase, eNOS and Akt phosphorylation, up-regulated Bcl2 expression and reduced expression of the pro-apoptotic regulator p53 (Figure 6D–H). CAC telomerase activity was also upregulated by the dual PPARα/γ agonist aleglitazar at very low concentrations (10 nmol·L−1: 155 ± 11%, P < 0.01; 100 nmol·L−1: 140 ± 7%, P < 0.05; Figure 6D). With respect to the protein expression and phosphorylation state of survival factors, aleglitazar induced similar or stronger effects than pioglitazone in cultivated CAC (aleglitazar 100 nmol·L−1: peNOS/eNOS ratio: 190 ± 16%, P < 0.01; pAkt/Akt ratio: 315 ± 49%, P < 0.01; p53: 44 ± 6%, P < 0.01; each vs. control CAC treated with vehicle; Figure 6E–H).
Figure 6.

Effects of aleglitazar on survival and apoptosis of human CAC. CAC were isolated from healthy donors by differentiation in cell culture for 4 days and treated as indicated. Effects of aleglitazar treatment for 24 h on (A) CAC migration in modified Boyden chambers and (B) CAC CFU. (C) CAC apoptosis as determined by FACS analysis shown as percentage of annexin V+/propidium iodide− cells. Basal and hydrogen peroxide-induced (H2O2, 500 μmol·L−1, 24 h) apoptosis were compared in control and aleglitazar-treated (10 nmol·L−1, 24 h) CAC. (D) Effects of pioglitazone (10 μmol·L−1, 24 h) and aleglitazar (10 nmol·L−1 and 100 nmol·L, 24 h) on CAC telomerase activity as measured by TRAP assays (n = 3). Representative Western blots and quantification of aleglitazar effects compared with pioglitazone (10 μmol·L−1) on CAC protein expression of (E) the ratio of phospho-eNOS/total eNOS and (F) phospho-Akt/total Akt, (G) the apoptosis regulator p53, and (H) the anti-apoptotic protein Bcl-2 (all Western blots normalized to GAPDH). n = 4 unless otherwise indicated, *P < 0.05, **P < 0.01, ***P < 0.001 versus controls (CAC treated for 24 h with 1% DMSO).
Figure 7.

Aleglitazar effects on CAC function in cell culture involve both PPARα and PPARγ and require Akt (PKB). (A) Effects of CAC treatment with 10 nmol·L−1 aleglitazar on the expression of the proliferation marker Ki67 in CAC colonies derived from healthy donors (n = 4). Right panels show an example of Ki67 + CAC-CFU (the asterisk marks a mitotic fission). Effects of treatment for 24 h with aleglitazar (10 nmol·L−1), pioglitazone (10 μmol·L−1), fenofibric acid (150 μmol·L−1) and the PPAR-γ antagonist GW9662 (1 μmol/L) on (B) CAC migration in modified Boyden chambers (n = 6) and (C) CAC proliferative capacity as shown by the number of CFU (n = 4). CAC from healthy donors (n = 4) were treated with 10 nmol·L−1 aleglitazar for 24 h and/or with the NOS inhibitor L-NAME (1 mmol·L−1), the phosphoinositide 3-kinase inhibitor LY294002 (10 μmol·L−1) or the direct Akt inhibitor 1L-6-hydroxymethyl-chiro-inositol2-[(R)-2-O-methyl-3-ooctadecylcarbonate] (10 μmol·L−1). Effects of these treatments on (D) CAC migration in modified Boyden chambers and (E) CFU were determined; *P < 0.05, **P < 0.001 and ***P < 0.001 versus controls.
Aleglitazar effects on human CAC function require both PPARα and γ signalling and are mediated by the PK Akt
As in murine MNC, both PPARα and γ mRNA were present in human CAC as detected by semiquantitative PCR (not shown). The mechanisms of aleglitazar-mediated improvement of CAC function were studied by co-incubation of CAC for 24 h with aleglitazar (10 nmol·L−1) in comparison to pioglitazone (10 μmol·L−1) or fenofibric acid (150 μmol·L−1), in the presence or absence of the PPARγ-selective antagonist GW9662 (1 μmol·L−1).
The PPARγ-selective agonist pioglitazone increased CAC migration (200 ± 11% vs. vehicle-treated control cells) and the number of CFU (166 ± 35%) to a similar extent as the PPARα-selective agonist fenofibric acid (186 ± 10% and 176 ± 8% respectively). The dual PPARα/γ agonist mediated a greater increase in CAC migration (279 ± 14%) and CAC CFU formation (251 ± 20%) compared with pioglitazone or fenofibric acid alone. Treatment of CAC with pioglitazone and fenofibric acid together led to a similar up-regulation of CAC migration and CFU formation (264 ± 28% and 261 ± 85%, respectively) as with aleglitazar. The effects of pioglitazone were completely abolished by co-incubation with the PPARγ antagonist GW9662, while the effects of aleglitazar were only partially reduced by GW9662 to the levels observed with stimulation of the PPARα pathway alone by fenofibric acid (165 ± 12% and 165 ± 19% respectively; Figure 7B,C). Together, these data indicate that the combined activation of PPARα and PPARγ by aleglitazar contributes to its greater effects on CAC function compared with the effects of agents that activate only a single receptor subtype.
To further unravel the signal transduction involved in aleglitazar-mediated improvement of CAC proliferation and migration, CAC were treated with vehicle or aleglitazar 10 nmol·L−1 and/or inhibitors targeting the NOS or the PI3K-Akt pathway. These experiments showed that under culture conditions, PPAR-mediated increase in CAC migration and CFU could not be inhibited by the NOS-inhibitor L-NAME (1 mmol·L−1) but was inhibited by both an upstream inhibitor of the Akt pathway (LY294002, 10 μmol·L−1) and a direct Akt inhibitor (10 μmol·L−1; Figure 7D,E).
Aleglitazar reverses CAC dysfunction in patients with CAD
Patients with CAD, especially such individuals with diabetes mellitus (DM), are characterized by a functional impairment of EPC (Adams et al., 2004; Ii et al., 2006; Sorrentino et al., 2007). Therefore, it is a relevant question whether aleglitazar improves the function of CAC from patients with CAD and DM. In a pilot study, MNC were isolated from healthy donors and CAD patients with or without DM (n = 3 per group; baseline characteristics in Table 2). Assessment of CAC migration and CFU confirmed that CAD patients had a marked decline in CAC function compared with healthy individuals. Importantly, aleglitazar treatment (10 nmol·L−1) in cell culture partially reversed CAC dysfunction in CAD patients, both in subjects with and without DM (migration: CAD without DM: control 24 ± 4 vs. ale 90 ± 15 migrated cells per 105 seeded cells; CAD with DM: control 72 ± 7 vs. ale 158 ± 7 migrated cells per CFU: CAD without DM: control 69 ± 6 vs. ale 119 ± 12 colonies per 24-well; CAD with DM: control 43 ± 7 vs. ale 127 ± 9 colonies per 24-well; Figure 8A,B).
Table 2.
Baseline characteristics of CAD patients in CAC culture study
| All patients (n = 6) | ||
|---|---|---|
| Medication (%) | ||
| Platelet inhibitors | 100 | |
| ACE inhibitors/ARBs | 100 | |
| β-blockers | 100 | |
| Statins | 100 | |
| Clinical characteristics | ||
| Age (years) | 66 ± 5 | |
| Male gender (%) | 83 | |
| Active smoking (%) | 17 | |
| Positive family history (%) | 17 | |
| Hyperlipidaemia (%) | 83 | |
| Arterial hypertension (%) | 100 | |
| Systolic BP (mmHg) | 183 ± 16 | |
| Diastolic BP (mmHg) | 88 ± 12 | |
| LV ejection fraction (%) | 64 ± 7 | |
| Affected coronaries (n) | 2.5 ± 0.2 | |
| Body mass index (kg·m−2) | 32 ± 3.3 | |
| Type II diabetes | ||
| Clinical chemistry | No (n = 3) | Yes (n = 3) |
| Fasting glucose (mmol L−1) | 5.9 ± 0.3 | 11.2 ± 2.9 |
| Total cholesterol (mmol L−1) | 5.4 ± 0.5 | 4.6 ± 0.5 |
| HDL cholesterol (mmol L−1) | 1.0 ± 0.1 | 1.0 ± 0.1 |
| LDL cholesterol (mmol L−1) | 3.8 ± 0.6 | 2.8 ± 0.4 |
| Triglycerides (mmol L−1) | 1.9 ± 0.8 | 2.0 ± 0.3 |
| Creatinin (mg·L−1) | 13.6 ± 3 | 12.1 ± 4 |
| Haemoglobin (mg·L−1) | 133 ± 6 | 131 ± 15 |
| Leukocytes (nL−1) | 7.4 ± 1.0 | 7.4 ± 1.8 |
| C-reactive protein (mg·L−1) | 6.4 ± 2.1 | 9.1 ± 6.1 |
Figure 8.

Aleglitazar improves CAC function in patients with CAD. Cultured CAC from healthy donors and from patients with stable CAD with or without DM were treated with vehicle or 10 nmol·L−1 aleglitazar for 24 h and as read-outs of CAC function (A) migration in modified Boyden chambers and (B) the number of CFU were measured; n = 3 per group; *P < 0.05, **P < 0.001 and ***P < 0.001 versus control CAC of healthy donors; ##P < 0.01 and ###P < 0.001 versus CAD without DM; †††P < 0.001 versus CAD with DM.
Discussion and conclusions
The main finding of the present study is that treatment with the dual PPARα/γ agonist aleglitazar increases both the number and the function of EPC in healthy WT mice and in atherosclerotic ApoE−/− mice, but not in eNOS−/− mice. The up-regulation of CAC is associated with important read-outs of CAC function, namely increased neoangiogenesis, improved endothelium-dependent vasodilatation and enhanced arteriogenesis. In addition, the PPARα/γ agonist potently inhibited the development of atherosclerotic plaques. In human CAC from healthy volunteers and from patients with CAD and DM, aleglitazar at nanomolar concentrations improved migration, proliferation and survival. Inhibitor experiments showed that both PPARα and PPARγ signalling contributed to these effects.
PPARα and PPARγ are nuclear receptors that regulate the transcription of genes important for metabolism and inflammation, especially in patients at risk of atherosclerotic vascular diseases (Brown and Plutzky, 2007). Dual-selective agonists of PPARα and PPARγ are of interest since they may combine the glucose-lowering and lipid-modifying properties of both agents. In a Phase II study (SYNCHRONY) in patients with type 2 diabetes, aleglitazar dose-dependently improved the lipid profile and decreased glycated haemoglobin and fasting plasma glucose, as well as other markers of cardiovascular risk, such as plasminogen activator inhibitor-1, fibrinogen and C-reactive protein (Henry et al., 2011). Similarly, aleglitazar improved insulin sensitivity and plasma lipids in diabetic rodents and monkeys (Benardeau et al., 2009; Hansen et al., 2011). However, the vascular effects of aleglitazar have not been studied. In the present study, we provide data regarding the effects of PPARα/γ agonism on vascular biology, including effects on EPC numbers and function, neoangiogenesis, endothelial function, arteriogenesis and atherogenesis.
Several markers are used to identify EPC (Fadini et al., 2012). In this study, Sca-1/VEGFR2 positive EPC were quantified in mice because their human equivalents (CD34/KDR positive EPC) independently predict cardiovascular outcomes in patients with CAD (Werner et al., 2005). According to the current definition proposed by Fadini et al., we investigated the CAC subpopulation of EPC (Fadini et al., 2012). Complementary independent methods were employed to characterize the effects of aleglitazar on CAC beyond evaluating the numbers of Sca-1/VEGFR2 positive cells. Culturing MNC and selection in endothelial growth medium, adhesion to fibronectin, the ability to uptake LDL and bind to lectin, as well as quantification of CAC CFU confirmed up-regulation by aleglitazar (Asahara et al., 1997; Fadini et al., 2012). The effect was observed in different mouse strains. In addition to the number of CAC, several functions of CAC are important in cardiovascular disease (Hill et al., 2003; Werner et al., 2005; Fadini et al., 2012) and have been shown to be impaired by cardiovascular risk factors independently of CAC numbers (Adams et al., 2004; Ii et al., 2006), particularly in patients with diabetes mellitus who are characterized by a specific impairment of CAC function (Ii et al., 2006; Sorrentino et al., 2007). In our study, aleglitazar improved CAC function in mice. In a translational approach, human isolated CAC were exposed to the PPARα/γ agonist. These experiments confirmed the improvement of CAC function observed in the mice models, as aleglitazar concentration-dependently enhanced migration and clonogenic potential of cultured human CAC. Further analyses showed that reactive oxygen species-induced apoptotic cell death of cultured human CAC was reduced, phosphorylation of eNOS and the survival kinase Akt were increased and expression of the apoptosis regulator p53 was suppressed by aleglitazar treatment. Furthermore, CAC telomerase activity was augmented by aleglitazar in vitro, which may cause aleglitazar's effects on the survival and replicative potential of CAC.
Importantly, the improvement of CAC number and function observed in vivo and in vitro was associated with an improvement of endothelial-dependent vasodilatation, with increased neoangiogenesis, improved arteriogenesis after hindlimb ischaemia and with reduced atherosclerotic lesion formation in mice treated with aleglitazar.
Expression analysis confirmed the expression of PPARα and PPARγ in murine and human CAC. The presented in vivo experiments in mice cannot precisely identify whether both PPAR subtypes are equally responsible for aleglitazar's effects in mice, but the in vitro experiments, with human CAC from healthy donors show that both PPARα and PPARγ are involved. In agreement with previous studies, treatment of CAC with the PPARγ agonist pioglitazone improved CAC migration and proliferation (Werner et al., 2007; 2011). The PPARγ antagonist GW9662 fully inhibited the effect of pioglitazone (in μM concentrations) but only partially abolished the effects of aleglitazar (in nM concentrations) on CAC function, confirming that the effects of aleglitazar are not mediated by PPARγ alone. The effect of PPARα agonists on CAC has not previously been studied. We showed that fenofibric acid increased migration and CFU in human CAC, indicating that PPARα is also a regulator of CAC function. Indeed, in this model of human cultured CAC, aleglitazar exhibited more potent effects on CAC function than pioglitazone or fenofibric acid alone.
An interesting finding of the present study is the marked reduction of hepatic fat content in the ApoE−/− mice on high-fat/high-cholesterol diet. Non-alcoholic fatty liver disease is present in the majority of patients with diabetes or metabolic syndrome and is associated with increased cardiovascular risk. Patients with non-alcoholic fatty liver disease are characterized by activation of pro-inflammatory pathways (Targher et al., 2010). In the present mechanistic study, the reduction of hepatic fat content in the treated mice was associated with a quantitatively very relevant decrease in inflammation as exemplified by a decrease in TNF-α and CCL2 (MCP-1) expression in the vessel wall. Non-alcoholic fatty liver disease has been shown to trigger systemic inflammation, which impairs CAC numbers and function and promotes atherogenesis providing a potential additional mechanistic link between PPARα/γ agonism and the observed prevention of atherogenesis in the ApoE−/− mice.
Aleglitazar enhanced the hepatic expression of PPARα-regulated genes such as acyl-CoA oxidase, fatty acid-binding protein 1 and carnitine palmitoyl transferase 1 (Rakhshandehroo et al., 2010). Serum adiponectin, one of the best-characterized PPARγ target genes, was markedly up-regulated in the serum showing potent PPARγ agonism. ApoE−/− mice have normal fasting serum glucose levels but show impaired glucose tolerance (Li et al., 2011). Aleglitazar normalized glucose tolerance in ApoE−/− mice. These results are in agreement with previous data in rodents, monkeys and humans and confirm effective dual PPARα/γ agonism by aleglitazar in our experimental set-up. In WT mice, both fasting and stimulated glucose levels were normal and were not lowered by aleglitazar. In the CAC cell culture studies, the observed effects of aleglitazar occurred with stable glucose and lipid concentrations in the culture medium. These data suggest that the effects of aleglitazar on CAC may be independent of its glucose- and lipid-lowering effects.
Limitations of the study
The presented findings obtained using animal studies and experiments conducted in human CAC are hypothesis-generating and provide mechanistic insight into the importance of PPAR signalling for the regulation CAC numbers and function. However, it is important to stress that they cannot be extrapolated to patients. Since glucose and cholesterol lowering per se improve CAC biology, it may be of interest to test the effects of aleglitazar on CAC in patients with normal glucose metabolism and lipid levels in order to investigate whether our findings can be extended to humans.
Bone marrow-derived progenitor cells significantly contribute to vascular homeostasis and repair and have emerged as a cellular read-out for vascular health (Asahara et al., 1997). However, their number and, more importantly, their function, are impaired by cardiovascular risk factors. This study shows that dual PPARα/γ agonism improves CAC numbers and function and provides evidence for a protective regenerative mechanism mediated by aleglitazar. To assess the clinical effect of aleglitazar on patients, especially in relation to the known unwanted effects of this class of drugs, clinical trials are needed.
Acknowledgments
The study was funded by a research grant from F. Hoffmann-La Roche to the University of the Saarland and by the European Stroke Network. We thank Ellen Becker, Simone Jäger, Jennifer Kieffer, Catrin Pittke and Julia Marhofer for their excellent technical assistance.
Glossary
- ApoE
apolipoprotein E
- CAC
circulating angiogenic cells
- eNOS
endothelial NOS
- EPC
endothelial progenitor cell(s)
- FAL
femoral artery ligation
- GTT
glucose tolerance test
- CCL2 (MCP-1)
monocyte-chemoattractant protein 1;
- MNC
mononuclear cells
- WT
wild-type
- WTD
Western-type diet
Conflict of interest
M W is an employee of F. Hoffmann-La Roche, Basel, Switzerland.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12608
PPARα and PPARγ are expressed in murine mononuclear cells. PPARα and PPARγ expression was measured in MNC isolated from C57Bl/6 mice. (A) Real-time PCR standard curves for the housekeeping mRNA 18 s (left panel), for PPARα (middle panel) and PPARγ (right panel) were created using spleen-derived MNC. (B) Live image of the amplification plot of these standard curves. (C) DNA gel electrophoresis of semi-quantitative PCR products (40 amplification cycles) showing KB+ (marker), WC (water controls), 1 (MNC isolated from spleen), 2 (MNC isolated from bone marrow) and 3 (MNC isolated from venous blood). (D) Real-time PCRbased quantification of PPARα and PPARγ mRNA in murine MNC isolated from spleen, bone marrow (BM) and blood, compared with HUVECs.
References
- Adams V, Lenk K, Linke A, Lenz D, Erbs S, Sandri M, et al. Increase of circulating endothelial progenitor cells in patients with coronary artery disease after exercise-induced ischemia. Arterioscler Thromb Vasc Biol. 2004;24:684–690. doi: 10.1161/01.ATV.0000124104.23702.a0. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear Hormone Receptors. Br J Pharmacol. 2013;170:1652–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Benameur T, Tual-Chalot S, Andriantsitohaina R, Martinez MC. PPARalpha is essential for microparticle-induced differentiation of mouse bone marrow-derived endothelial progenitor cells and angiogenesis. PLoS ONE. 2010;5:e12392. doi: 10.1371/journal.pone.0012392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benardeau A, Benz J, Binggeli A, Blum D, Boehringer M, Grether U, et al. Aleglitazar, a new, potent, and balanced dual PPAR alpha/gamma agonist for the treatment of type II diabetes. Bioorg Med Chem Lett. 2009;19:2468–2473. doi: 10.1016/j.bmcl.2009.03.036. [DOI] [PubMed] [Google Scholar]
- Brown JD, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation. 2007;115:518–533. doi: 10.1161/CIRCULATIONAHA.104.475673. [DOI] [PubMed] [Google Scholar]
- Cavender MA, Lincoff AM. Therapeutic potential of aleglitazar, a new dual PPAR-alpha/gamma agonist: implications for cardiovascular disease in patients with diabetes mellitus. Am J Cardiovasc Drugs. 2010;10:209–216. doi: 10.2165/11539500-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Charbonnel B. PPAR-alpha and PPAR-gamma agonists for type 2 diabetes. Lancet. 2009;374:96–98. doi: 10.1016/S0140-6736(09)61040-0. [DOI] [PubMed] [Google Scholar]
- Custodis F, Baumhäkel M, Schlimmer N, List F, Gensch C, Böhm M, et al. Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2008;117:2377–2387. doi: 10.1161/CIRCULATIONAHA.107.746537. [DOI] [PubMed] [Google Scholar]
- Dietz M, Mohr P, Kuhn B, Maerki HP, Hartman P, Ruf A, et al. Comparative molecular profiling of the PPARalpha/gamma activator aleglitazar: PPAR selectivity, activity and interaction with cofactors. Chem Med Chem. 2012;7:1101–1111. doi: 10.1002/cmdc.201100598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S. Regulation of bone marrow-derived vascular progenitor cell mobilization and maintenance. Arterioscler Thromb Vasc Biol. 2010;30:1088–1093. doi: 10.1161/ATVBAHA.109.191668. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–397. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadini GP, Losordo D, Dimmeler S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ Res. 2012;110:624–637. doi: 10.1161/CIRCRESAHA.111.243386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foteinos G, Hu Y, Xiao Q, Metzler B, Xu Q. Rapid endothelial turnover in atherosclerosis-prone areas coincides with stem cell repair in apolipoprotein E-deficient mice. Circulation. 2008;117:1856–1863. doi: 10.1161/CIRCULATIONAHA.107.746008. [DOI] [PubMed] [Google Scholar]
- Gensch C, Clever YP, Werner C, Hanhoun M, Böhm M, Laufs U. The PPAR-gamma agonist pioglitazone increases neoangiogenesis and prevents apoptosis of endothelial progenitor cells. Atherosclerosis. 2007;192:67–74. doi: 10.1016/j.atherosclerosis.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Gertz K, Kronenberg G, Kalin RE, Baldinger T, Werner C, Balkaya M, et al. Essential role of interleukin-6 in post-stroke angiogenesis. Brain. 2012;135(Pt 6):1964–1980. doi: 10.1093/brain/aws075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamed S, Brenner B, Roguin A. Nitric oxide: a key factor behind the dysfunctionality of endothelial progenitor cells in diabetes mellitus type-2. Cardiovasc Res. 2011;91:9–15. doi: 10.1093/cvr/cvq412. [DOI] [PubMed] [Google Scholar]
- Hansen BC, Tigno XT, Benardeau A, Meyer M, Sebokova E, Mizrahi J. Effects of aleglitazar, a balanced dual peroxisome proliferator-activated receptor alpha/gamma agonist on glycemic and lipid parameters in a primate model of the metabolic syndrome. Cardiovasc Diabetol. 10:7. doi: 10.1186/1475-2840-10-7. doi: 10.1186/1475-2840-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry RR, Lincoff AM, Mudaliar S, Rabbia M, Chognot C, Herz M. Effect of the dual peroxisome proliferator-activated receptor-alpha/gamma agonist aleglitazar on risk of cardiovascular disease in patients with type 2 diabetes (SYNCHRONY): a phase II, randomised, dose-ranging study. Lancet. 2011;374:126–135. doi: 10.1016/S0140-6736(09)60870-9. 2009. [DOI] [PubMed] [Google Scholar]
- Herz M, Gaspari F, Perico N, Viberti G, Urbanowska T, Rabbia M, et al. Effects of high dose aleglitazar on renal function in patients with type 2 diabetes. Int J Cardiol. 2011;151:136–142. doi: 10.1016/j.ijcard.2010.08.037. [DOI] [PubMed] [Google Scholar]
- Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- Hoekstra M, Kruijt JK, Van Eck M, Van Berkel TJ. Specific gene expression of ATP-binding cassette transporters and nuclear hormone receptors in rat liver parenchymal, endothelial, and Kupffer cells. J Biol Chem. 2003;278:25448–25453. doi: 10.1074/jbc.M301189200. [DOI] [PubMed] [Google Scholar]
- Ii M, Takenaka H, Asai J, Ibusuki K, Mizukami Y, Maruyama K, et al. Endothelial progenitor thrombospondin-1 mediates diabetes-induced delay in reendothelialization following arterial injury. Circ Res. 2006;98:697–704. doi: 10.1161/01.RES.0000209948.50943.ea. [DOI] [PubMed] [Google Scholar]
- Inoue I, Shino K, Noji S, Awata T, Katayama S. Expression of peroxisome proliferator-activated receptor alpha (PPAR alpha) in primary cultures of human vascular endothelial cells. Biochem Biophys Res Commun. 1998;246:370–374. doi: 10.1006/bbrc.1998.8622. [DOI] [PubMed] [Google Scholar]
- Jarajapu YP, Grant MB. The promise of cell-based therapies for diabetic complications: challenges and solutions. Circ Res. 2010;106:854–869. doi: 10.1161/CIRCRESAHA.109.213140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Satoh H, Endo Y, Yamada D, Midorikawa S, Sato W, et al. Thiazolidinediones down-regulate plasminogen activator inhibitor type 1 expression in human vascular endothelial cells: a possible role for PPARgamma in endothelial function. Biochem Biophys Res Commun. 1999;258:431–435. doi: 10.1006/bbrc.1999.0648. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan AM, Schirmer SH, de Vries MR, Koning JJ, Volger OL, Fledderus JO, et al. Galectin-2 expression is dependent on the rs7291467 polymorphism and acts as an inhibitor of arteriogenesis. Eur Heart J. 2012;33:1076–1084. doi: 10.1093/eurheartj/ehr220. [DOI] [PubMed] [Google Scholar]
- Laufs U, Werner N, Link A, Endres M, Wassmann S, Jurgens K, et al. Physical training increases endothelial progenitor cells, inhibits neointima formation, and enhances angiogenesis. Circulation. 2004;109:220–226. doi: 10.1161/01.CIR.0000109141.48980.37. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Kim M, Park HS, Kim HS, Jeon MJ, Oh KS, et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem Biophys Res Commun. 2006;340:291–295. doi: 10.1016/j.bbrc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Lenski M, Kazakov A, Marx N, Böhm M, Laufs U. Effects of DPP-4 inhibition on cardiac metabolism and function in mice. J Mol Cell Cardiol. 2011;51:906–918. doi: 10.1016/j.yjmcc.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Li J, Wang Q, Chai W, Chen MH, Liu Z, Shi W. Hyperglycemia in apolipoprotein E-deficient mouse strains with different atherosclerosis susceptibility. Cardiovasc Diabetol. 2011;10:117. doi: 10.1186/1475-2840-10-117. doi: 10.1186/1475-2840-10-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr M, Niederseer D, Niebauer J. From bench to bedside: what physicians need to know about endothelial progenitor cells. Am J Med. 2011;124:489–497. doi: 10.1016/j.amjmed.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Pistrosch F, Herbrig K, Oelschlaegel U, Richter S, Passauer J, Fischer S, et al. PPARgamma-agonist rosiglitazone increases number and migratory activity of cultured endothelial progenitor cells. Atherosclerosis. 2005;183:163–167. doi: 10.1016/j.atherosclerosis.2005.03.039. [DOI] [PubMed] [Google Scholar]
- Pöss J, Werner C, Lorenz D, Gensch C, Böhm M, Laufs U. The renin inhibitor aliskiren upregulates pro-angiogenic cells and reduces atherogenesis in mice. Basic Res Cardiol. 2010;105:725–735. doi: 10.1007/s00395-010-0120-5. [DOI] [PubMed] [Google Scholar]
- Rakhshandehroo M, Knoch B, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010;2010 doi: 10.1155/2010/612089. doi: 10.1155/2010/612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanwald-Ducray P, Liogier D'ardhuy X, Jamois C, Banken L. Pharmacokinetics, pharmacodynamics, and tolerability of aleglitazar in patients with type 2 diabetes: results from a randomized, placebo-controlled clinical study. Clin Pharmacol Ther. 2010;88:197–203. doi: 10.1038/clpt.2009.259. [DOI] [PubMed] [Google Scholar]
- Schirmer SH, Fledderus JO, Bot PT, Moerland PD, Hoefer IE, Baan J, Jr, et al. Interferon-beta signaling is enhanced in patients with insufficient coronary collateral artery development and inhibits arteriogenesis in mice. Circ Res. 2008;102:1286–1294. doi: 10.1161/CIRCRESAHA.108.171827. [DOI] [PubMed] [Google Scholar]
- Schirmer SH, van Nooijen FC, Piek JJ, van Royen N. Stimulation of collateral artery growth: travelling further down the road to clinical application. Heart. 2009;95:191–197. doi: 10.1136/hrt.2007.136119. [DOI] [PubMed] [Google Scholar]
- Schirmer SH, Degen A, Baumhakel M, Custodis F, Schuh L, Kohlhaas M, et al. Heart-rate reduction by If-channel inhibition with ivabradine restores collateral artery growth in hypercholesterolemic atherosclerosis. Eur Heart J. 2012;33:1223–1231. doi: 10.1093/eurheartj/ehr255. [DOI] [PubMed] [Google Scholar]
- Sorrentino SA, Bahlmann FH, Besler C, Muller M, Schulz S, Kirchhoff N, et al. Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus: restoration by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2007;116:163–173. doi: 10.1161/CIRCULATIONAHA.106.684381. [DOI] [PubMed] [Google Scholar]
- Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. 2010;363:1341–1350. doi: 10.1056/NEJMra0912063. [DOI] [PubMed] [Google Scholar]
- Urbich C, Heeschen C, Aicher A, Sasaki K, Bruhl T, Farhadi MR, et al. Cathepsin L is required for endothelial progenitor cell-induced neovascularization. Nat Med. 2005;11:206–213. doi: 10.1038/nm1182. [DOI] [PubMed] [Google Scholar]
- Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1–E7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- Walter DH, Rittig K, Bahlmann FH, Kirchmair R, Silver M, Murayama T, et al. Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105:3017–3024. doi: 10.1161/01.cir.0000018166.84319.55. [DOI] [PubMed] [Google Scholar]
- Werner C, Kamani CH, Gensch C, Böhm M, Laufs U. The peroxisome proliferator-activated receptor-gamma agonist pioglitazone increases number and function of endothelial progenitor cells in patients with coronary artery disease and normal glucose tolerance. Diabetes. 2007;56:2609–2615. doi: 10.2337/db07-0069. [DOI] [PubMed] [Google Scholar]
- Werner C, Hanhoun M, Widmann T, Kazakov A, Semenov A, Pöss J, et al. Effects of physical exercise on myocardial telomere-regulating proteins, survival pathways, and apoptosis. J Am Coll Cardiol. 2008;52:470–482. doi: 10.1016/j.jacc.2008.04.034. [DOI] [PubMed] [Google Scholar]
- Werner C, Fürster T, Widmann T, Pöss J, Roggia C, Hanhoun M, et al. Physical exercise prevents cellular senescence in circulating leukocytes and in the vessel wall. Circulation. 2009;120:2438–2447. doi: 10.1161/CIRCULATIONAHA.109.861005. [DOI] [PubMed] [Google Scholar]
- Werner C, Gensch C, Pöss J, Haendeler J, Böhm M, Laufs U. Pioglitazone activates aortic telomerase and prevents stress-induced endothelial apoptosis. Atherosclerosis. 2011;216:23–34. doi: 10.1016/j.atherosclerosis.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Werner N, Junk S, Laufs U, Link A, Walenta K, Böhm M, et al. Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ Res. 2003;93:e17–e24. doi: 10.1161/01.RES.0000083812.30141.74. [DOI] [PubMed] [Google Scholar]
- Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PPARα and PPARγ are expressed in murine mononuclear cells. PPARα and PPARγ expression was measured in MNC isolated from C57Bl/6 mice. (A) Real-time PCR standard curves for the housekeeping mRNA 18 s (left panel), for PPARα (middle panel) and PPARγ (right panel) were created using spleen-derived MNC. (B) Live image of the amplification plot of these standard curves. (C) DNA gel electrophoresis of semi-quantitative PCR products (40 amplification cycles) showing KB+ (marker), WC (water controls), 1 (MNC isolated from spleen), 2 (MNC isolated from bone marrow) and 3 (MNC isolated from venous blood). (D) Real-time PCRbased quantification of PPARα and PPARγ mRNA in murine MNC isolated from spleen, bone marrow (BM) and blood, compared with HUVECs.