

Abstract
A generic two-step lyssavirus real-time reverse transcriptase polymerase chain reaction (qRT-PCR), based on a nested PCR strategy, was validated for the detection of different lyssavirus species. Primers with 17 to 30% of degenerate bases were used in both consecutive steps. The assay could accurately detect RABV, LBV, MOKV, DUVV, EBLV-1, EBLV-2, and ABLV. In silico sequence alignment showed a functional match with the remaining lyssavirus species. The diagnostic specificity was 100% and the sensitivity proved to be superior to that of the fluorescent antigen test. The limit of detection was ≤1 50% tissue culture infectious dose. The related vesicular stomatitis virus was not recognized, confirming the selectivity for lyssaviruses. The assay was applied to follow the evolution of rabies virus infection in the brain of mice from 0 to 10 days after intranasal inoculation. The obtained RNA curve corresponded well with the curves obtained by a one-step monospecific RABV-qRT-PCR, the fluorescent antigen test, and virus titration. Despite the presence of degenerate bases, the assay proved to be highly sensitive, specific, and reproducible.
1. Introduction
Rabies is a fatal viral encephalitis that results from infection with negative strand RNA-viruses belonging to the genus Lyssavirus, family Rhabdoviridae, order Mononegavirales. So far, 12 species have been classified in the genus Lyssavirus. Traditionally, these include Rabies virus (RABV), Lagos bat virus (LBV), Mokola virus (MOKV), Duvenhage virus (DUVV), European bat lyssaviruses-1 and -2 (EBLV-1 and EBLV-2), and Australian bat lyssavirus (ABLV). More recently, Aravan virus (ARAV), Khujand virus (KHUV) [1], Irkut virus (IRKV) [1], Shimoni bat virus (SHIBV), and West Caucasian bat virus (WCBV) [1] were also added. Ikoma virus (IKOV) [2] and Bokeloh bat lyssavirus (BBLV) [3, 4] await classification in the genus. The genus is subdivided into phylogroups 1 and 2 [5, 6]. Phylogroup 1 includes RABV, DUVV, EBLV-1, EBLV-2, ABLV, ARAV, KHUV, and IRKV. Phylogroup 2 includes LBV, MOKV, and SHIBV. WCBV and IKOV do not cross-react serologically with any of the two phylogroups.
The classic rabies virus (RABV) has a worldwide distribution and uses carnivores and bats as main reservoir. The other lyssavirus species are mainly maintained in bats and have a more restricted distribution: DUVV, LBV, MOKV, SHIBV, and IKOV have been detected exclusively in Africa, EBLV-1, -2 and BBLV in Europe, ABLV in Australia, and ARAV, KHUV, IRKV, and WCBV in Asia. It is assumed that most lyssaviruses can cause the rabies syndrome in humans and other mammals [6–8]. In Western Europe, most cases of rabies in humans or pets are imported and may be caused by any species within the Genus Lyssavirus [9, 10]. For example, in 2007, in The Netherlands, a patient died from infection with the rare DUVV upon return from Kenya [11]. Moreover, locally acquired infections in humans and cats with EBLV-1 or EBLV-2 are also possible on the European territory [12]. A diagnostic assay that can rapidly detect all species is therefore highly recommended.
Currently, the gold standard methods for the diagnosis of rabies recommended by the World Health Organisation (WHO) are the fluorescent antibody test (FAT), the rabies tissue culture infection test (RTCIT), and the mouse inoculation test (MIT) [13–18]. The FAT is convenient for postmortem examination and detects the presence of viral nucleocapsid antigens in the brain or spinal cord tissue by staining with specific fluorescent antibodies [14]. For antemortem diagnosis of rabies, the presence of viral antigen can be detected with the FAT in tissue sections of skin biopsies, typically in the nerve endings surrounding the hair follicles. The viral antigens are however often only detectable at the end of the disease or cannot be detected at all by this method [17]. Repeated sampling is necessary to improve the diagnostic sensitivity. This is not practical for skin biopsies [17] but easier for body fluids, such as saliva, urine, or cerebrospinal fluid. The sensitivity of the FAT method is considered high for RABV but may be lower for other lyssavirus species [19–21].
RTCIT and MIT are based on the isolation and propagation of virus, respectively, in cell culture or in mice [22]. Isolation of the virus from body fluids requires the presence of infectious virus in the sample and the absence of viral inhibitors or antibodies and is time consuming. Antirabies virus antibodies acquired either by natural seroconversion, by treatment with immunoglobulins, or after a postexposure vaccination can interfere with the virus isolation from clinical samples, possibly yielding false negative results in the RTCIT and MIT. In our experience, MIT and RTCIT are very specific methods but are restricted to samples containing live and uninhibited virus. Furthermore, neither the FAT, RTCIT, or MIT can directly distinguish between different lyssavirus species. Seroconversion during the course of disease is highly indicative for rabies, but patients often receive treatment with antirabies immunoglobulins and vaccine, compromising the interpretation of serology. Molecular techniques have recently been developed for rabies virus diagnosis. Viral RNA can be extracted from several matrices, such as saliva, urine, cerebrospinal fluid (CSF), or skin tissue, and do not require the presence of live virus. RT-PCR can therefore be used under a broad range of conditions. RT-PCR has been shown to detect RNA in decomposed samples [23] or after long-term storage [24], giving a better chance of successful diagnosis than RTCIT [25]. Also, the qRT-PCR method can allow to distinguish different lyssavirus species.
Here, we aimed to develop and validate a nested two-step generic lyssavirus real-time RT-PCR (qRT-PCR) protocol, combining the use of degenerate primers with real-time PCR detection. A two-step approach was chosen to maximize the sensitivity of the assay. Degenerate bases were included in the primers at key positions to account for the variability in the sequence of the different lyssavirus species. During the first amplification round (PCR1), the primers amplified a 343 bp fragment of the nucleoprotein N gene, whereas in the following real-time PCR a 158 bp fragment was amplified and detected using SYBR Green. The overall sensitivity, specificity, selectivity, and reproducibility of the assay were assessed by comparison with FAT. More specifically for RABV detection, the performance of the generic lyssavirus qRT-PCR was compared with a RABV-specific qRT-PCR protocol, using specific primers without degenerate bases. A large set of samples obtained from humans, naturally and experimentally infected wild and domestic animals were included to validate the assay.
2. Materials and Methods
2.1. Samples
2.1.1. Negative Samples
Brain, serum, and cerebrospinal fluid (CSF) samples from different species (bat, red fox, dog, cat, mouse, and human) were used as negative controls (Table 1). All samples tested negative for rabies in the FAT and/or RTCIT gold standard methods. To further test for specificity, a virus suspension of vesicular stomatitis virus (VSV) (genus Vesiculovirus) was also tested to demonstrate the selectivity of the primers. VSV belongs to another genus within the family Rhabdoviridae and shares biological and genetic features with the lyssaviruses.
Table 1.
FAT and RTCIT-negative samples from various species used for the evaluation of the specificity of the lyssavirus qRT-PCR.
| Species | Matrix | Provider | n | FAT/RTCIT | Generic lyssavirus qRT-PCR |
|---|---|---|---|---|---|
| Bats (Pipistrellus, Myotis, and Eptesicus serotinus)1 | Brain tissue | Rabies NRC, WIV-ISP, Belgium | 100 | Negative | Negative |
| Red Fox (Vulpes vulpes)1 | Brain tissue | Rabies NRC, WIV-ISP, Belgium | 10 | Negative | Negative |
| Dog (Canis Lupus familiaris)1 | Brain tissue | Rabies NRC, WIV-ISP, Belgium | 10 | Negative | Negative |
| Cat (Felix cati)1 | Brain tissue | Rabies NRC, WIV-ISP, Belgium | 10 | Negative | Negative |
| Mouse (Mus Musculus)2 | Brain tissue | Rabies NRC, WIV-ISP, Belgium | 20 | Negative | Negative |
| Human (Homo Sapiens)3 | Cerebrospinal fluid | Rabies NRC, WIV-ISP, Belgium | 10 | Negative | Negative |
| Human (Homo Sapiens)3 | Saliva | Rabies NRC, WIV-ISP, Belgium | 5 | Negative | Negative |
| Human (Homo Sapiens)3 | Skin tissue | Rabies NRC, WIV-ISP, Belgium | 1 | Negative | Negative |
1Samples collected on the Belgian territory between 2007 and 2012 in the frame of the national surveillance system to guarantee the rabies-free status of Belgium.
2Specific-pathogen-free female Swiss outbred laboratory mice obtained from Harlan (Boxmeer, The Netherlands).
3Patients with encephalitis symptoms sent to the national reference centre for rabies virus, Belgium.
2.1.2. Positive Samples
To test the performance of the generic lyssavirus qRT-PCR, samples spiked with different lyssaviruses were used (Table 2). Challenge Virus Standard-11 (CVS-11), a virulent neurotropic lyssavirus, was obtained from the American Type Culture Collection (ATCC: reference VR959). CVS-11 was grown in baby hamster kidney (BHK)-21 cells, as described previously [29].
Table 2.
Rabies virus-positive samples used to assess the diagnostic sensitivity of the lyssavirus qRT-PCR.
| Positive samples | Provider | Strain reference | Matrix | n |
|---|---|---|---|---|
| Classical rabies virus RABV |
ANSES, France1 | Ariana, Tunisia | Dog brain homogenate | 3 |
| ANSES, France1 | CVS-27 | Mouse brain homogenate | 3 | |
| ANSES, France1 | GS7, France | Fox brain homogenate | 7 | |
| ANSES, France1 | Raccoon, Poland | Raccoon dog brain homogenate | 1 | |
| ANSES, France1 | 201020958, Spain | Mouse brain homogenate | 2 | |
| ANSES, France1 | Cn Viv Estonie 10–12, Estonia | Mouse brain homogenate | 1 | |
| Rabies NRC, ISP-WIV | Strain fox, Belgium (1995) | Red fox brain homogenate | 1 | |
| Rabies NRC, ISP-WIV | Strain fox, Luxembourg (2000) | Pony brain homogenate | 1 | |
| Rabies NRC, ISP-WIV | CB-1, Maroc (2007) | Dog brain homogenate | 1 | |
| ATCC, USA | CVS-11 (VR959) | Mouse brain homogenate (experimentally infected mice at Rabies NRC, WIV-ISP, Belgium) | 20 | |
|
| ||||
| Lagos bat virus LBV |
Centre des Ressources Biologiques, Pasteur Paris Institute, France | CRBIP8.14 | Cell culture lysate | 1 |
|
| ||||
| Mokola virus MOKV |
Centre des Ressources Biologiques, Pasteur Paris Institute, France | CRBIP8.27 | Cell culture lysate | 1 |
|
| ||||
| Duvenhage virus DUVV |
ANSES, France1 | 96132, SA (fixed strain) | Mouse brain homogenate | 1 |
| Centre des Ressources Biologiques, Pasteur Paris Institute, France | CRBIP8.28 | Cell culture lysate | 1 | |
|
| ||||
| European bat lyssavirus-1 EBLV-1 |
ANSES, France1 | EBLV-1a, France | Mouse brain homogenate | 3 |
| ANSES, France1 | EBLV-1b, France | Mouse brain homogenate | 4 | |
| WIV-ISP, Belgium2 | AF-2010, Spain | Bat brain homogenate (Naturally infected Eptesicus serotinus bat) | 1 | |
| ANSES, France1 | R75, Spain | Mouse brain homogenate | 1 | |
| Pasteur Paris Institute, France3 | 8919FRA, France (isolated from Eptesicus serotinus bat) | Mouse brain homogenate (experimentally infected mice at Rabies NRC, WIV-ISP, Belgium) | 5 | |
|
| ||||
| European bat lyssavirus-2 EBLV-2 |
ANSES, France1 | EBLV-2 VLA P3, United Kingdom | Mouse brain homogenate | 1 |
| ANSES, France1 | EBL2 RV1787, United Kingdom | Mouse brain homogenate | 1 | |
| ANSES, France1 | EBLV-2, United Kingdom | Mouse brain homogenate | 5 | |
|
| ||||
| Australian bat lyssavirus ABLV |
ANSES, France1 | ABLV, Australia | Mouse brain homogenate | 4 |
1Samples obtained through participation to consecutive an interlaboratory proficiency tests organised by the European Union reference laboratory of rabies (ANSES, Nancy, France) between 2009 and 2013. Samples were reconstituted in 1 mL sterile, nuclease-free, and distilled water (Robardet et al., 2011 [26]).
2Van Gucht et al., 2013 [27].
3Bourhy et al., 1992 [28].
Experimentally infected brain samples were prepared as described next. Female Swiss outbred mice (Harlan, The Netherlands) were inoculated intranasally at the age of 6 to 8 weeks, according to Rosseels et al. [29]. The experimental procedure was approved by the Local Ethical Committee of the WIV-ISP (advice nr. 060217-03). Sixty mice were each inoculated with 3 × 102 50% TCID50 of CVS-11 and euthanized by cervical dislocation from 0 to 10 days after inoculation (DPI). Five control mice received only phosphate-buffered saline (PBS). The evolution of the load of infectious virus, viral antigen, and viral RNA was followed for 10 days. The brain was cut in half and the inner part of the left half was pressed on a glass slide for FAT analysis. Both brain halves were then homogenized in 1 mL of PBS with a Potter homogenator. The homogenate underwent 3 consecutive freeze-thaw cycles at −80°C and was centrifuged at 20000 g for 20 min at 4°C. The supernatant was collected for virus titration (300 μL), RABV-specific qRT-PCR, and generic lyssavirus qRT-PCR (2 × 85 μL). Virus titration was performed according to the instructions of the Manual of Diagnostic Tests and Vaccines for Terrestrial Animals (Office International des Epizooties, 2013). Serial five-fold dilutions were added in triplicate to BHK-21 cells. After a 2-day incubation period, the cells were stained with fluorescein isothiocyanate (FITC)-coupled antinucleocapsid rabbit antibodies (Bio-Rad Laboratories, Hercules, USA) and the number of infected foci was counted. The titre of infectious virus was expressed in TCID50/mL.
2.2. Fluorescent Antibody Test (FAT)
The FAT was performed according to the instructions of the Manual of Diagnostic Tests and Vaccines for Terrestrial Animals (Office International des Epizooties, 2012). Briefly, the tissue slides were fixed in 75% acetone for 10 min at −20°C and stained with FITC-labelled antinucleocapsid rabbit antibodies (Bio-Rad Laboratories, Hercules, USA) for 30 min at 37°C. The slides were examined with a Nikon Diaphot 200 fluorescence microscope connected to a Moticam 2500 camera (Hong-Kong, China) at a magnification of 100x.
2.3. Generic Lyssavirus qRT-PCR
2.3.1. RNA Extraction
Total RNA from the various samples was extracted using the Qiagen RNeasy Mini kit (for brain and cell culture medium) or the Qiagen QIAamp viral RNA Mini kit (for serum and CSF) (Qiagen, Hilden, Germany). Brain samples (400 mg) were first homogenised in 1 mL of PBS with a Potter homogenator. Eighty-five microliter of the brain homogenate was mixed with 265 µL of lysis buffer (RLT) and used for RNA extraction. From here on, the instructions from the RNeasy Mini kit were followed. For RNA extractions from infected cell culture supernatants, a volume of 150 µL was homogenised in 200 µL of RLT buffer, as starting material for the RNA extraction. Starting from serum or CSF, a volume of 140 µL was used for RNA extraction with the Qiagen QIAamp viral RNA Mini kit following the manufacturer's instructions.
2.3.2. Reverse Transcription
The RNA extracts were quantified with a NanoVue spectrophotometer (GE healthcare, Bucks, United Kingdom). The reverse transcription reaction was performed using the qScript cDNA SuperMix (Quanta BioSciences, Gaithersburg, USA). Briefly, 18 µL containing 100 ng of RNA template in RNAse-free water and 2 µL of the qScript supermix were mixed and incubated for 5 min at 25°C followed by 30 min at 42°C and 5 min at 85°C.
2.3.3. Design of the Oligonucleotide Primers
The nucleoprotein N gene was used as target for the qRT-PCR. The primers were synthesized by Eurogentec (Seraing, Belgium). The lyssavirus primers were chosen based on multiple alignments of the N gene sequence from RABV, LBV, MOKV, DUVV, EBLV-1, EBLV-2, ABLV, WCBV, KHUV, and IRKV. Complete sequences of the nucleoprotein gene of each lyssavirus species were obtained from genomic databases and aligned by using the MEGA5 and the CLC Sequence Viewer software. The accession numbers of these viruses are shown in Table 3(a). External and nested primer sequences were chosen from regions conserved in all lyssavirus species. The RAB PCR1 F, RAB PCR1 R, and RAB qPCR F primers were designed in-house (Table 3(b)). The GRAB2R primer was described earlier by Vázquez-Morón et al. [30]. Degenerate bases were included in the primers to account for the variability in the sequences of different lyssavirus species. A first amplification round (PCR1) was performed using the RAB PCR1 F and RAB PCR1 R primers, which amplify a fragment of 343 bp of the N gene. 5 µL of the PCR product was diluted 10x and then used in a real-time PCR with the RAB qRT-PCR F and GRAB2R primers, amplifying a 158 bp fragment within the first PCR amplicon. The RABV monospecific primers (RAB CVS-11 F and RAB CVS-11 R) were designed to specifically target the nucleoprotein N gene of CVS-11 (accession number GU992321). They contain no degenerate bases (Table 3(b)). The primers VETINHF2 and VETINHR1 were designed in-house to amplify a conserved sequence of the r18S ribosomal RNA gene and were usedto check for PCR inhibition and RNA quality [31, 32]. They can be used for samples from multiple animal species (Table 3(b)).
Table 3.
(a) Accession numbers of analysed lyssavirus species. (b) Nucleotide sequence of the primers (with the IUPAC codes for the degenerate bases) that were used in this study.
(a)
| Rabies virus | Identification | GenBank accession number |
|---|---|---|
| RABV | CVS-11 | GQ918139 |
| RABV | 9174GSFRA | U22474 |
| RABV | 9107MAR | U22852 |
| LBV | LBVSA2006 | EF547452 |
| LBV | U22842 | |
| MOKV | NC006429 | |
| MOKV | U22843 | |
| DUVV | U22848 | |
| DUVV | EU623438 | |
| EBLV-1 | 891FRA | U22845 |
| EBLV-1 | 8615POL | U22844 |
| EBLV-2 | 9007FIN | U22846 |
| EBLV-2 | 9018HOL | U22847 |
| ABLV | AF006497 | |
| ARAV | EF614259 | |
| ARAV | AB094438 | |
| KHUV | EF614261 | |
| IRKV | EF614260 | |
| IRKV | THChina12 | JX442979 |
| WCBV | EF614258 | |
| SHV | GU170201 | |
| BOKV | 21961 | JF311903 |
| IKOV | Isolate RV2508 | JX193798 |
| IKOV | NC018629 |
(b)
| Name | Tm | Sequence 5′-3′ | Position (for RABV-CVS11) | Use of primers |
|---|---|---|---|---|
| RAB PCR1 F | 60,2°C | AYAARATGTGYGCIAAYTGGAGYA | 572–595 | Generic lyssavirus PCR1 |
| RAB PCR1 R | 61,8°C | ACIGCRTTSGANGARTAAGGAGA | 892–914 | Generic lyssavirus PCR1 |
| RAB qPCR F | 62,1°C | GTIGGVACDGTIGTIACHGCHTA | 676–698 | Generic lyssavirus qRT-PCR |
| GRAB2R | 61°C | TCYTGHCCIGGCTCRAACAT | 814–833 | Generic lyssavirus qRT-PCR |
| RAB CVS11 F | 68,1°C | GTGGGCACAGTCGTCACCGCTTA | 676–698 | RABV-specific qRT-PCR |
| RAB CVS11 R | 60,85°C | TCTTGCCCTGGCTCGAACAT | 814–833 | RABV-specific qRT-PCR |
| VETINHF2 | 60,8°C | GTTGATTAAGTCCCTGCCCTTT | / | r18S qPCR |
| VETINHR1 | 60,8°C | GATAGTCAAGTTCGACCGTCTT | / | r18S qPCR |
The r18S ribosomal RNA gene primers were designed to amplify RNA from multiple species.
2.3.4. Nested qPCR
The protocol consisted of a nested PCR strategy with two amplification steps. In the first amplification round (PCR1), 5 μL of cDNA was mixed with a PCR master mix containing 10 μL of 5x reaction buffer, 1.5 mM of MgCl2, 0.2 U Taq polymerase (Promega, Madison, USA), 0.5 μM of the primers RAB PCR1 F and RAB PCR1 R, 0.2 mM of dNTP mix (Roche, Basel, Switzerland),and nuclease-free water to obtain a final volume of 50 µL. The amplification was performed on an iCycler (Biorad, Hemel Hempstead, United Kingdom) according to the following program: 1 min at 95°C for initial denaturation, followed by 25 cycles of 20 s at 95°C (denaturation) and 40 s at 60°C (annealing and extension), and a final step of 10 min at 72°C. The second amplification consisted of a real-time PCR performed on a CFX96 real-time PCR system (Biorad, Hemel Hempstead, United Kingdom). The reaction mix consisted of 12.5 µL of 2x SyberGreen Master Mix (Quanta BioSciences, Gaithersburg, USA), 5 µL of 10x diluted PCR1 product and 0.8 µM of each primer (RAB qRT-PCR F and GRAB2R), and nuclease-free water to obtain a final volume of 25 µL. All samples were analyzed in duplicate. The program consisted of 2 min at 95°C for Taq activation and initial denaturation, followed by 45 cycles of 20 s at 95°C and 30 s at 61°C. To check for the presence of primer dimers and nonspecific amplicons, a melting curve analysis was performed after each run (Bio-Rad CFX manager 2.1 software).
A total of 45 cycles were ran in the qPCR. The interpretation of the qPCR results was done as follows: a sample with a Cq value ≤40 was considered positive and a Cq value >40 or an undetectable Cq was considered negative. Samples with a Cq value between 40 and 45 were always retested. The melting curve was analysed to check the specificity of the amplification.
2.3.5. Sequencing Species Identification
The obtained amplicons were purified using the GFX PCR DNA kit (GE Healthcare, PQ, USA) and quantified with the NanoVue spectrophotometer (GE Healthcare, PQ, USA). The sequencing reactions were performed using the RAB qRT-PCR F primer and the big dye terminator v.3.1 cycle sequencing kit (Applied Biosystem, CA, USA), according to the manufacturer's instructions. As starting material, 1–3 ng of the purified PCR fragment was used. The cycle PCR reactions were performed on an iCycler from Biorad, programmed as follows: a first denaturation step of 1 min at 96°C and 25 cycles consisting of 10 s at 96°C and 4 min at 60°C. Final products were purified by precipitation by adding 5 µL of 125 mM EDTA/sodium acetate 3 M solution and 60 µL of ethanol 100%. The sequencing reactions were analysed on an ABI Prism 3130 Genetic Analyzer (Applied Biosystem, California, USA). The obtained sequences were matched with the sequences stored in the National Center for Biotechnology Information (NCBI) database, using the Basic Local Alignment Search Tool (BLAST).
2.4. RABV-Specific qRT-PCR
The RABV-specific qRT-PCR consisted of a single amplification step and was performed on a CFX96 real-time system from Biorad (Hemel Hempstead, United Kingdom). Each 25 μL reaction mixture consisted of 12.5 µL of 2x Sybr Green Master Mix (Quanta BioSciences, Gaithersburg, USA), 5 μL of diluted cDNA, and 0.4 μM of each primer. All samples were analyzed in duplicate. The amplification program consisted of 2 min initial denaturation at 95°C, followed by 45 cycles of 20 s at 95°C and 30 s of annealing and extension at 62°C. A melting curve analysis was performed in order to verify the absence of primer dimers and the specificity of the obtained amplicons after each run (Bio-Rad CFX manager 2.1 software). The correlation between Cq values of both qRT-PCR methods was calculated with the Pearson's correlation test (GraphPad Prism 6 software).
3. Results
3.1. Diagnostic Sensitivity
3.1.1. Spectrum of Lyssavirus Detection
All positive samples (n = 69) that contained either one of 7 different lyssavirus species (Table 2) were tested positive with the lyssavirus qRT-PCR, yielding a sensitivity of 100% for the assay. Some of the rare species (WCBV, IRKV, KHUV, IKOV, SHIBV, and BBLV) were not tested, but in silico alignment of the targeted region in the nucleoprotein gene of these species demonstrated a sufficient match with the degenerate primers to allow amplification (Figure 1). No primer dimers were detected. The obtained amplicons were also checked by agarose gel analysis and had the expected size of 158 bp (data not shown).
Figure 1.

Sequence alignment of the degenerate primers with the targeted region of the nucleoprotein gene of 24 isolates of 14 different lyssavirus species.
To demonstrate the superior sensitivity of the two-step approach, the lyssavirus qRT-PCR was compared with a previously published one-step lyssavirus qRT-PCR [33]. Serial dilutions of 2 virus-positive samples from the 2013 annual international proficiency test from ANSES (Nancy, France) were compared. The samples contained a RABV isolate from a dog (Canis lupus familiaris), or an EBLV-1 isolate from a bat (Eptesicus serotinus). The virus-positive samples were diluted ten-fold and tested by both protocols. The obtained results are presented in Table 4. The two-step approach was 10 times more sensitive than the one-step approach [33].
Table 4.
Comparison of the sensitivity between the two-step lyssavirus qRT-PCR and the one-step qRT-PCR published by Hayman et al. in 2011 [33].
| Dilutions | RABV strain | EBLV-1 strain | ||
|---|---|---|---|---|
| Two-step developed assay | One-step published assay | Two-step developed assay | One-step published assay | |
| Cq value | Cq value | Cq value | Cq value | |
| 1.00E + 00 | 9.52 | 17.41 | 6.77 | 19.51 |
| 1.00E − 02 | 12.58 | 23.80 | 11.40 | 26.92 |
| 1.00E − 03 | 17.59 | 27.31 | 17.07 | 30.70 |
| 1.00E − 04 | 21.97 | 30.95 | 21.37 | 33.70 |
| 1.00E − 05 | 26.16 | 34.29 | 24.98 | 37.36 |
| 1.00E − 06 | 30.94 | 38.10 | 28.35 | ND |
| 1.00E − 07 | 34.30 | ND | ND | ND |
RNA was extracted from infected cell culture supernatants, serially diluted and used to generate cDNA. The cDNA samples were tested by both qRT-PCR protocols. ND: no signal detected.
3.1.2. Comparison of the Course of an Experimental RABV Virus Infection in Mice Determined by Generic Lyssavirus qRT-PCR, RABV Monospecific qRT-PCR, and FAT
Mice were inoculated intranasally with rabies virus and brain tissue was sampled at different DPI for analysis. qRT-PCR results were expressed in delta Cq values, which were calculated as follows: 45 − Cqsample (45 = the total number of cycles run in the qPCR). Viral RNA was first detected at 3 DPI (mean delta Cq of 4.2), which was followed by an increase of the viral RNA load at 5 DPI (mean delta Cq of 23.2 at 5 DPI) (Figure 2(a)). The viral load reached a plateau from 7 DPI onwards (mean delta Cq of 26). Starting from 5 DPI, viral antigens could also be detected with the FAT and small amounts of infectious virus could be isolated from the brain (Figure 2(b)). At 5 DPI, the virus titer ranged from 102 to 104 TCID50/mL. The titer increased during the following days to reach a plateau, ranging between 104 and 106 TCID50/mL at 8 DPI (Figure 2(c)). The Cq values obtained with the one-step monospecific qRT-PCR correlated well with the values obtained with the two-step lyssavirus qRT-PCR (Pearson value r = 0.9773; P < 0.001, GraphPad Prism 6) (Figure 2(d)). The generic lyssavirus qRT-PCR proved to be more sensitive than the FAT. Eleven mice that tested positive with the generic lyssavirus qRT-PCR at 3 and 4 DPI, tested negative by the FAT. Uninfected control mice tested negative with both qRT-PCR methods and the FAT.
Figure 2.

Kinetic profile of RABV infection in mice determined by generic lyssavirus qRT-PCR, FAT and virus titration. Mice were infected with 3 × 102 TCID50 of RABV (CVS-11) by intranasal inoculation and sacrificed 0 to 10 days later. The brain was collected for analysis. The course of (a) viral RNA by generic lyssavirus qRT-PCR, (b) load of viral antigen (FAT), (c) infectious virus (RTCIT), and (d) correlation of Cq values between the generic lyssavirus and RABV-specific qRT-PCR are presented. The correlation between the Cq values obtained by both qRT-PCR methods was excellent (Pearson's correlation coefficient r = 0.9773, P < 0.0001).
3.2. Selectivity
For all samples, the sequence of the obtained amplicon could be assigned unequivocally to the correct species by comparing the nucleotide homology in the NCBI database (BLAST, highly similar sequences). Homology was in the range of 95–100% for RABV, MOKV, EBLV-1, EBLV-2, and ABLV and in the range of 85–87% for LBV and DUVV. Moreover, melting temperature profiles of the respective amplicons differed for the different species and no primer dimers were observed in any PCR run (Figure 3). The vesicular stomatitis virus (VSV) tested negative (undetectable Cq) with the generic lyssavirus qRT-PCR.
Figure 3.

Melting peaks obtained for 7 lyssavirus species. The melting temperatures for RABV, MOKV, LBV, DUVV, EBLV-1, -2, and ABLV were, respectively, 77°C, 77.5°C, 76°C, 76.5°C, 78.5°C, 78°C, and 76.5°C. No primer dimers were observed.
3.3. Limit of Detection
The limit of detection was determined by analysing 10-fold serial dilutions of a suspension of RABV (CVS-11) and EBLV-1. RABV and EBLV-1 suspensions were produced in BHK-21 and neuroblastoma N2a cells, respectively. The titer of infectious virus was determined in cell culture. The virus was diluted from 106 to 10−2 TCID50 for RABV and from 104 to 10−2 TCID50 for EBLV-1. For RABV, the viral RNA load was determined by both the generic and the RABV-specific qRT-PCR, while for EBLV-1, only the generic lyssavirus qRT-PCR was used. The obtained Cq values correlated well with the logarithm of the corresponding TCID50 values using a linear fit model (GraphPad Prism 6) (Figure 4(a)).
Figure 4.
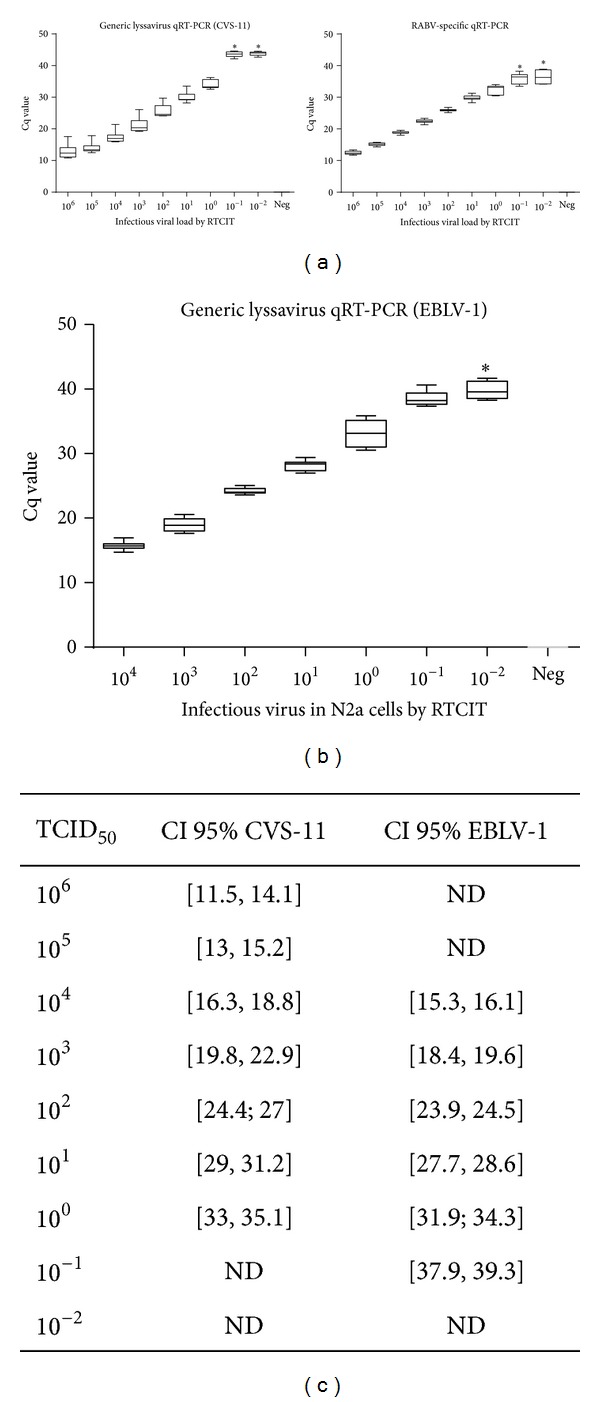
Analytical sensitivity of the monospecific RABV and the generic lyssavirus qRT-PCR for RABV (CVS-11) (a) and EBLV-1 (b). Six independent runs with each time two repeats were performed per virus dilution (10-fold serial dilution). There was an excellent linear regression between the load of infectious virus, determined by virus titration, and the Cq value for RABV and EBLV-1 (regression coefficient of 0.965 and 0.989, resp.). For both qRT-PCR methods, the limit of detection of RABV and EBLV-1 was ≤ 100 TCID50. The Cq remained undetectable in the negative control samples.*Mean and standard deviation are calculated based on the runs/repeats with a positive signal (Cq ≤ 40). ND = not determined.
The limit of detection for RABV was 100 TCID50 for both qRT-PCR methods, with 100% positive replicates (6 different runs in duplicate). At the 10−1 TCID50 and 10−2 TCID50 dilutions, all replicates were negative (Cq value > 40 or undetectable) with the generic lyssavirus qRT-PCR. For the RABV monospecific qRT-PCR, 8 replicates were positive and 4 replicates were negative at the 10−1 TCID50 dilution. At the 10−2 TCID50 dilution, 4 replicates were positive and 8 replicates were negative with the RABV-specific qRT-PCR (Figure 4(a)). Negative control samples (viral RNA substituted by water) gave no signal with both qRT-PCR methods. The detection limit of EBLV-1 was determined at 10−1 TCID50 for the generic lyssavirus qRT-PCR. At 10−1 TCID50, all replicates yielded positive results. At 10−2 TCID50, 5 replicates yielded positive and 7 replicates yielded negative results (Cq value > 40 or undetectable) (Figure 4(b)).
The two-step protocol was also compared with a one-step qPCR round based on the same PCR1 or qPCR primers, using samples from the 2011 and 2013 annual ring test from ANSES (Nancy, France). A sample containing an ABLV isolate from an Australian bat (Pteropus Alecto) was diluted 1/10, 1/100, 1/500, and 1/1000 and a sample containing a RABV isolate from a European red fox (Vulpes vulpes) was diluted 1/8, 1/100, and 1/500. All dilutions tested positive with the two-step protocol. Depending on the dilution, Cq values ranged from 16 to 24. In contrast, the one-step protocol gave either no signal or high Cq values, ranging between 31 and 44 (data not shown). This underlines the need for the two amplification rounds to obtain acceptable sensitivity.
3.4. Diagnostic Specificity
The diagnostic specificity was 100%. A total of 150 brains of rabies virus-free mammals, belonging to 5 different species, tested negative by FAT and generic lyssavirus qRT-PCR (Table 1). Ten human cerebrospinal fluid samples, 6 saliva samples, and 1 skin biopsy obtained from rabies-free patients with neurological symptoms tested negative by qRT-PCR, yielding a diagnostic specificity of 100%. The qRT-PCR always yielded negative results upon substitution of the RNA by water in the different steps of the protocol (RNA extraction, reverse transcription, and two-step qPCR).
3.5. Repeatability (Intra-Assay Variation)
To assess the intra-assay variation, 3 samples (CVS-11, EBLV1, and water as a negative control) were tested in 10 replicates. The variation coefficient (Pearson r-GraphPad Prism 6) was 1.7% for CVS-11 (at a mean Cq of 21.5) and 0.89% for EBLV-1 (at a mean Cq of 24.3). The negative control remained undetectable in all replicates.
3.6. Reproducibility (Inter-Assay Variation)
The inter-assay precision was assessed by testing 3 samples (CVS-11, EBLV-1, and water as a negative control) in 6 independent runs (Figure 4). The virus suspensions were diluted from 106 to 10−2 TCID50 for CVS-11 and from 104 to 10−2 TCID50 for EBLV-1. The Cq values increased with the viral load. The 95% confidence interval was calculated for all CVS-11 and EBLV-1 tested dilutions (Figure 4(c)).
3.7. Matrix Effect
Brain tissue is the preferred specimen for postmortem diagnosis in both humans and animals, but this sample is not feasible for antemortem diagnosis. In the latter case, diagnosis is based on detection of virus or viral RNA in saliva, neck skin biopsy, or an impression of the cornea. To validate the generic lyssavirus qPCR on other matrices than brain tissue, CSF, saliva, and urine were spiked with different virus doses, ranging from 105 to 10−1 TCID50 for RABV (CVS-11) and from 104 to 10−2 TCID50 for EBLV-1. Each sample was analysed in 3 different runs (RNA extraction followed by qPCR) and results were expressed in mean Cq values (Table 5). PCR inhibition and RNA quality were checked by using r18S ribosomal RNA gene amplification. Virus could be detected in all tested matrices (saliva, CSF, and urine). The limit of detection for EBLV-1 was 100 TCID50 in CSF, saliva, and urine. The limit of detection for RABV was also 100 TCID50 in urine but appeared higher in CSF (101 TCID50) and in saliva (102 TCID50).
Table 5.
Effect of the matrix on qRT-PCR performance.
(a)
| RABV (TCID50) | CSF | Saliva | Urine |
|---|---|---|---|
| 1.00E + 05 | 19.65 | 18.80 | 18.53 |
| 1.00E + 04 | 24.50 | 23.39 | 23.13 |
| 1.00E + 03 | 28.12 | 27.17 | 26.85 |
| 1.00E + 02 | 33.15 | 32.75 | 30.56 |
| 1.00E + 01 | 40.53 | ND | 39.67 |
| 1.00E + 00 | 43.54 | ND | 39.78 |
| 1.00E − 01 | ND | ND | ND |
| 1.00E − 02 | ND | ND | ND |
(b)
| EBLV-1 (TCID50) | CSF | Saliva | Urine |
|
| |||
| 1.00E + 04 | 14.56 | 15.31 | 14.50 |
| 1.00E + 03 | 18.11 | 18.34 | 18.84 |
| 1.00E + 02 | 22.72 | 23.79 | 22.17 |
| 1.00E + 01 | 27.74 | 28.90 | 28.06 |
| 1.00E + 00 | 33.10 | 28.17 | 28.67 |
| 1.00E − 01 | ND | ND | ND |
| 1.00E − 02 | ND | ND | ND |
Saliva, CSF, and urine samples were spiked with decreasing doses of RABV or EBLV-1. Three independent runs with each time two repeats were performed per virus dose (10-fold serial dilution). The limit of detection for RABV was 100 TCID50 in urine, 101 TCID50 in CSF, and 102 TCID50 in saliva. For EBLV-1, the limit of detection was 100 TCID50 in CSF, saliva, and urine. ND: signal not detected.
4. Discussion
Real-time PCR provides significant methodological benefits for virus detection [34]. RT-PCR offers many advantages for rabies diagnosis, due to its high sensitivity, rapidity, no interference by inhibitors of virus infectivity or antibodies, practicability for samples which may contain only minute amounts of virus, such as cerebrospinal fluid or bat brain, and the possibility to quickly determine the species and molecular phylogeny of the isolate. Several RT-PCR protocols for the detection of rabies virus have been published during the past decade. All protocols that recognise multiple lyssavirus species use degenerate primers [30, 33, 35–39] and most of them use the JW12 primer published by Heaton et al. 1997 [35]. A cocktail of nondegenerate/degenerate primers is only used in the protocols published by Heaton et al. in 1997 [35] and Black et al. in 2003 [36]. Three protocols are designed as a two-step PCR and all of them use a gel-based DNA system detection [30, 35, 39] while others are real-time one-step protocols [33, 36–38]. Only two protocols were validated for 7 species [30, 33]. The RT-PCR of Vázquez-Morón et al. [30] uses a nested classical PCR system with degenerate primers and a gel-based DNA detection system. Real-time RT-PCR is however a more rapid and potentially more sensitive technique [34, 40]. Recently, Hayman et al. [33] validated the use of a real-time RT-PCR protocol for the detection of 7 lyssavirus species. They used a set of mildly degenerate primers that contained one or two degenerate bases per primer and were originally designed for the recognition of RABV, EBLV-1, and -2.
In this study, a two-step generic lyssavirus qRT-PCR, capable of detecting 7 lyssavirus species (RABV, LBV, MOKV, DUVV, EBLV-1, EBLV-2, and ABLV), was developed and validated. The two-step approach allows maximum sensitivity in order to detect virus in typically small-size samples, such as bat brains or samples which contain only minute amounts of virus, such as CSF. A good in silico match of the primers with the N gene of the remaining 6 species (WCBV, IRKV, KHUV, IKOV, SHIBV, and BBLV) was also demonstrated. A cocktail of primers with 17 to 30% of degenerate bases, taking into account the variability of the different lyssavirus species, was designed for the assay. The primers target well-conserved regions of the N gene. Indeed, ample sequence data are available for the N gene and most of the published RT-PCR protocols use primers for the N gene [30, 33, 36–39, 41–43]. A large set of samples obtained from wild and domestic animals (bat, red fox, dog, and cat), human patients with nonrabies encephalitis, interlaboratory ring trials, and naturally and experimentally infected animals (bat and mice) was tested. To check for PCR inhibition and RNA quality, the cellular r18S ribosomal RNA gene was used as a target. The r18S ribosomal RNA has been shown to be more reliable than β-actin [42, 44]. The diagnostic specificity was 100% and sensitivity proved superior to the fluorescent antigen test. The limit of detection was ≤1 TCID50. Sequence analysis of the amplicon unequivocally assigned the correct species. The vesicular stomatitis virus (belonging to the related genus Vesiculovirus) was not recognized, confirming the selectivity of the degenerate primers.
This qRT-PCR protocol is now routinely used for rabies surveillance in Belgium. Since 2001, Belgium has been officially declared free of the classic rabies virus. However, bats are an important reservoir of lyssaviruses in Europe [45] and are still collected and tested in the frame of the Belgian rabies surveillance system [46].
In experimentally infected mice, viral RNA was first detected at 3 DPI in the brain, whereas first symptoms appeared much later at 8 DPI. Symptoms involved depression, loss of body weight, ruffled fur, and paralysis of the hind limbs. In general, results of the generic lyssavirus qRT-PCR, RABV-specific qRT-PCR, virus titration, and FAT correlated well and provided similar kinetic profiles throughout the infection. The qRT-PCR proved however to be more sensitive than virus titration and FAT. Indeed, 11 out of 55 mice tested negative by FAT and positive by qRT-PCR. Also, 11 out 55 qRT-PCR-positive mice tested negative by RTCIT. Cycle thresholds obtained by the generic and the RABV-specific qRT-PCR were highly correlated.
The virus species in the positive samples could be identified by sequencing. For some species, we observed different melting temperatures and curves, providing an early indication of the species prior to sequencing. The melting temperature, determined with the CFX96 system from Biorad, was different for RABV, LBV, MOKV, EBLV-1, and EBLV-2, whereas DUVV and ABLV had the same melting temperature (Figure 3). The number of strains tested per species was however insufficient to accurately define the melting curve differentiation. It is also possible to obtain a more precise calculation by high resolution melt analysis [47], which may theoretically allow to discriminate all species prior to sequencing. This was not done for this study.
To further assess the exactitude of our qPCR method, we participated to the consecutive international ring trials organised by ANSES (Nancy, France) from 2009 to 2013. Up to now, we reported very good compliant results in each trial. Moreover, we participated in the Epizone ring trial organised by the Friedrich-Loeffler-Institute (FLI, Germany) in 2011. The panel consisted of 30 samples prepared by the FLI and included RNA of 26 RABV isolated in different countries and years, an EBLV-1 and an EBLV-2 RNA sample, and a log10 serial dilution of RABV. We correctly reported all negative samples and 28 of the 30 positive samples, representing a sensitivity of 93.3%. Two samples with a Cq value >40 were considered as negative samples. These two false negatives could have been due to the fact that the samples were extracted and prepared in RSB50, a buffer different from the one used in our protocol. The RSB50 buffer contains the carrier poly A, which forced us to use a gene-specific reverse transcription kit (qScript Flex cDNA Synthesis kit, Quanta BioSciences, Gaithersburg, USA). Only 3 of the 16 laboratories submitted 100% concordant results for RABV diagnosis [48].
Despite the presence of 17 to 30% of degenerate bases in some of the primers, the lyssavirus qRT-PCR proved to be highly sensitive, specific, and reproducible. In our national reference laboratory, this technique is now used as the method of choice for antemortem rabies diagnosis and analysis of small-size samples, such as bat brain.
Acknowledgments
The “Agence Nationale de la Sécurité Sanitaire” (ANSES) (Nancy, France) is sincerely acknowledged for allowing the use of the samples sent in the frame of an interlaboratory proficiency test for rabies diagnosis. The authors thank Dr. X. Saelens (Molecular Virology Unit, Flemish Institute of Biotechnology (VIB), Department for Molecular Biomedical Research UGent, Gent, Belgium) for kindly providing the vesicular stomatitis virus. The authors also thank Dr. Sigrid De Keersmaecker and M. Xavier Libert (Platform biotechnology and molecular biology, Scientific Public Health Institute, Brussels, Belgium) for the alignment sequences. S. Lamoral was supported by the Research Foundation-Flanders (FWO-Vlaanderen) convention n°G.0657.08. F. Nazé received a fellowship from “Le fond pour la formation à la Recherche dans l'Industrie et dans l'Agriculture” (FRIA-Wallonie). The National Reference Centre of Rabies is partially supported by the Belgian Ministry of Social Affairs through a fund from the Health Insurance System.
Conflict of Interests
The authors declare no conflict of interests.
References
- 1.Kuzmin IV, Hughes GJ, Botvinkin AD, Orciari LA, Rupprecht CE. Phylogenetic relationships of Irkut and West Caucasian bat viruses within the Lyssavirus genus and suggested quantitative criteria based on the N gene sequence for Lyssavirus genotype definition. Virus Research. 2005;111(1):28–43. doi: 10.1016/j.virusres.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Marston DA, Horton DL, Ngeleja C, et al. Ikoma Lyssavirus, highly divergent novel Lyssavirus in an African civet. Emerging Infectious Diseases. 2012;18(4):664–667. doi: 10.3201/eid1804.111553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freuling CM, Beer M, Conraths FJ, et al. Novel Lyssavirus in Natterer’s bat, Germany. Emerging Infectious Diseases. 2011;17(8):1519–1522. doi: 10.3201/eid1708.110201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freuling CM, Abendroth B, Beer M, et al. Molecular diagnostics for the detection of Bokeloh bat Lyssavirus in a bat from Bavaria, Germany. Virus Research. 2013;177(2):201–204. doi: 10.1016/j.virusres.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 5.Johnson N, Vos A, Freuling C, Tordo N, Fooks AR, Müller T. Human rabies due to Lyssavirus infection of bat origin. Veterinary Microbiology. 2010;142(3-4):151–159. doi: 10.1016/j.vetmic.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Nel LH, Markotter W. Lyssaviruses. Critical Reviews in Microbiology. 2007;33(4):301–324. doi: 10.1080/10408410701647602. [DOI] [PubMed] [Google Scholar]
- 7.Banyard AC, Hayman D, Johnson N, McElhinney L, Fooks AR. Bats and Lyssaviruses. Advances in Virus Research. 2011;79:239–289. doi: 10.1016/B978-0-12-387040-7.00012-3. [DOI] [PubMed] [Google Scholar]
- 8.Calisher CH, Ellison JA. The other rabies viruses: the emergence and importance of Lyssaviruses from bats and other vertebrates. Travel Medicine and Infectious Disease. 2012;10(2):69–79. doi: 10.1016/j.tmaid.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson N, Freuling C, Horton D, Müller T, Fooks AR. Imported rabies, European Union and Switzerland, 2001–2010. Emerging Infectious Diseases. 2011;17(4):753–754. doi: 10.3201/eid1706.101154. [DOI] [PubMed] [Google Scholar]
- 10.Carrara P, Parola P, Brouqui P, Gautret P. Imported human rabies cases worldwide, 1990–2012. PLOS Neglected Tropical Diseases. 2013;7(5) doi: 10.1371/journal.pntd.0002209.e2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Thiel PP, van den Hoek JA, Eftimov F, et al. Fatal case of human rabies (Duvenhage virus) from a bat in Kenya: The Netherlands, December 2007. Euro Surveillance. 2008;13(2) [PubMed] [Google Scholar]
- 12.Fooks AR, Brookes SM, Johnson N, McElhinney LM, Hutson AM. European bat Lyssaviruses: an emerging zoonosis. Epidemiology and Infection. 2003;131(3):1029–1039. doi: 10.1017/s0950268803001481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.OIE. Rabies OIE Manual of Diagnostic Tests and Vaccine for Terrestrial Animals. 6th edition 2013. [Google Scholar]
- 14.Goldwasser RA, Kissling RE. Fluorescent antibody staining of street and fixed rabies virus antigens. Proceedings of the Society for Experimental Biology and Medicine. 1958;98(2):219–223. doi: 10.3181/00379727-98-23996. [DOI] [PubMed] [Google Scholar]
- 15.Harkess G, Fooks AR. Lyssaviruses: special emphasis on rabies virus and other members of the Lyssavirus genus. Methods in Molecular Biology. 2011;665:279–307. doi: 10.1007/978-1-60761-817-1_16. [DOI] [PubMed] [Google Scholar]
- 16.Lackay SN, Kuang Y, Fu ZF. Rabies in small animals. Veterinary Clinics of North America: Small Animal Practice. 2008;38(4):851–861. doi: 10.1016/j.cvsm.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woldehiwet Z. Clinical laboratory advances in the detection of rabies virus. Clinica Chimica Acta. 2005;351(1-2):49–63. doi: 10.1016/j.cccn.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 18.WHO. WHO Expert Consultation on Rabies: Second Report. 2013. [PubMed] [Google Scholar]
- 19.Serra-Cobo J, Amengual B, Carlos Abellán B, Bourhy H. European bat Lyssavirus infection in Spanish bat populations. Emerging Infectious Diseases. 2002;8(4):413–420. doi: 10.3201/eid0804.010263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dacheux L, Larrous F, Mailles A, et al. European bat Lyssavirus transmission among cats, Europe. Emerging Infectious Diseases. 2009;15(2):280–284. doi: 10.3201/eid1502.080637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rønsholt L, Sørensen KJ, Bruschke CJM, et al. Clinically silent rabies infection in (zoo) bats. Veterinary Record. 1998;142(19):519–520. doi: 10.1136/vr.142.19.519. [DOI] [PubMed] [Google Scholar]
- 22.Webster LT. Diagnostic and immunological tests of rabies in mice. American Journal of Public Health and the Nations Health. 1936;26(12):1207–1210. doi: 10.2105/ajph.26.12.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.David D, Yakobson B, Rotenberg D, Dveres N, Davidson I, Stram Y. Rabies virus detection by RT-PCR in decomposed naturally infected brains. Veterinary Microbiology. 2002;87(2):111–118. doi: 10.1016/s0378-1135(02)00041-x. [DOI] [PubMed] [Google Scholar]
- 24.Lopes MC, Venditti LLR, Queiroz LH. Comparison between RT-PCR and the mouse inoculation test for detection of rabies virus in samples kept for long periods under different conditions. Journal of Virological Methods. 2010;164(1-2):19–23. doi: 10.1016/j.jviromet.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 25.Dacheux L, Reynes J-M, Buchy P, et al. A reliable diagnosis of human rabies based on analysis of skin biopsy specimens. Clinical Infectious Diseases. 2008;47(11):1410–1417. doi: 10.1086/592969. [DOI] [PubMed] [Google Scholar]
- 26.Robardet E, Picard-Meyer E, Andrieu S, Servat A, Cliquet F. International interlaboratory trials on rabies diagnosis: An overview of results and variation in reference diagnosis techniques (fluorescent antibody test, rabies tissue culture infection test, mouse inoculation test) and molecular biology techniques. Journal of Virological Methods. 2011;177(1):15–25. doi: 10.1016/j.jviromet.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 27.Van Gucht S, Verlinde R, Colyn J, et al. Favourable outcome in a patient bitten by a rabid bat infected with the European bat Lyssavirus-1. Acta Clinica Belgica. 2013;68:54–58. doi: 10.2143/ACB.68.1.2062721. [DOI] [PubMed] [Google Scholar]
- 28.Bourhy H, Kissi B, Lafon M, Sacramento D, Tordo N. Antigenic and molecular characterization of bat rabies virus in Europe. Journal of Clinical Microbiology. 1992;30(9):2419–2426. doi: 10.1128/jcm.30.9.2419-2426.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosseels V, Nazé F, De Craeye S, Francart A, Kalai M, Van Gucht S. A non-invasive intranasal inoculation technique using isoflurane anesthesia to infect the brain of mice with rabies virus. Journal of Virological Methods. 2011;173(1):127–136. doi: 10.1016/j.jviromet.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 30.Vázquez-Morón S, Avellón A, Echevarría JE. RT-PCR for detection of all seven genotypes of Lyssavirus genus. Journal of Virological Methods. 2006;135(2):281–287. doi: 10.1016/j.jviromet.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Meerburg BG, De Craeye S, Dierick K, Kijlstra A. Neospora caninum and Toxoplasma gondii in brain tissue of feral rodents and insectivores caught on farms in the Netherlands. Veterinary Parasitology. 2012;184(2-4):317–320. doi: 10.1016/j.vetpar.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 32.De Craeye S, Speybroeck N, Ajzenberg D, et al. Toxoplasma gondii and Neospora caninum in wildlife: common parasites in Belgian foxes and Cervidae? Veterinary Parasitology. 2011;178(1-2):64–69. doi: 10.1016/j.vetpar.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 33.Hayman DTS, Banyard AC, Wakeley PR, et al. A universal real-time assay for the detection of Lyssaviruses. Journal of Virological Methods. 2011;177(1):87–93. doi: 10.1016/j.jviromet.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mackay IM, Arden KE, Nitsche A. Real-time PCR in virology. Nucleic Acids Research. 2002;30(6):1292–1305. doi: 10.1093/nar/30.6.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heaton PR, Johnstone P, McElhinney LM, Cowley R, O’Sullivan E, Whitby JE. Heminested PCR assay for detection of six genotypes of rabies and rabies-related viruses. Journal of Clinical Microbiology. 1997;35(11):2762–2766. doi: 10.1128/jcm.35.11.2762-2766.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Black EM, Lowings JP, Smith J, Heaton PR, McElhinney LM. A rapid RT-PCR method to differentiate six established genotypes of rabies and rabies-related viruses using TaqMan™ technology. Journal of Virological Methods. 2003;105(1):25–35. doi: 10.1016/S0166-0934(02)00062-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coertse J, Weyer J, Nel LH, Markotter W. Improved PCR methods for detection of african rabies and rabies-related Lyssaviruses. Journal of Clinical Microbiology. 2010;48(11):3949–3955. doi: 10.1128/JCM.01256-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wakeley PR, Johnson N, McElhinney LM, Marston D, Sawyer J, Fooks AR. Development of a real-time, differential RT-PCR TaqMan® assay for Lyssavirus genotypes 1, 5 and 6. Developments in Biologicals. 2006;126:227–236. [PubMed] [Google Scholar]
- 39.Picard-Meyer E, Bruyère V, Barrat J, Tissot E, Barrat MJ, Cliquet F. Development of a hemi-nested RT-PCR method for the specific determination of European Bat Lyssavirus 1: comparison with other rabies diagnostic methods. Vaccine. 2004;22(15-16):1921–1929. doi: 10.1016/j.vaccine.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 40.Mentel R, Wegner U, Bruns R, Gürtler L. Real-time PCR to improve the diagnosis of respiratory syncytial virus infection. Journal of Medical Microbiology. 2003;52(10):893–896. doi: 10.1099/jmm.0.05290-0. [DOI] [PubMed] [Google Scholar]
- 41.De Benedictis P, De Battisti C, Dacheux L, et al. Lyssavirus detection and typing using pyrosequencing. Journal of Clinical Microbiology. 2011;49(5):1932–1938. doi: 10.1128/JCM.02015-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nadin-Davis SA, Sheen M, Wandeler AI. Development of real-time reverse transcriptase polymerase chain reaction methods for human rabies diagnosis. Journal of Medical Virology. 2009;81(8):1484–1497. doi: 10.1002/jmv.21547. [DOI] [PubMed] [Google Scholar]
- 43.Wacharapluesadee S, Sutipanya J, Damrongwatanapokin S, et al. Development of a TaqMan real-time RT-PCR assay for the detection of rabies virus. Journal of Virological Methods. 2008;151(2):317–320. doi: 10.1016/j.jviromet.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 44.Smith J, McElhinney LM, Heaton PR, Black EM, Lowings JP. Assessment of template quality by the incorporation of an internal control into a RT-PCR for the detection of rabies and rabies-related viruses. Journal of Virological Methods. 2000;84(2):107–115. doi: 10.1016/s0166-0934(99)00124-x. [DOI] [PubMed] [Google Scholar]
- 45.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: Important reservoir hosts of emerging viruses. Clinical Microbiology Reviews. 2006;19(3):531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Gucht S, Le Roux I. Rabies control in Belgium: From eradication in foxes to import of a contaminated dog. Vlaams Diergeneeskundig Tijdschrift. 2008;77(6):376–384. [Google Scholar]
- 47.Grando D, Said MM, Mayall BC, Gurtler V. High resolution melt analysis to track infections due to ribotype 027 Clostridium difficile. Journal of Microbiological Methods. 2012;89(2):87–94. doi: 10.1016/j.mimet.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 48.Fischer M, Wernike K, Freuling CM, et al. A step forward in molecular diagnostics of Lyssaviruses—results of a ring trial among European laboratories. PLoS ONE. 2013;8(3) doi: 10.1371/journal.pone.0058372.e58372 [DOI] [PMC free article] [PubMed] [Google Scholar]