

Abstract
Epithelial—mesenchymal transition (EMT), a critical step in the acquisition of metastatic state, is an attractive target for therapeutic interventions directed against tumor metastasis. Honokiol (HNK) is a natural phenolic compound isolated from an extract of seed cones from Magnolia grandiflora. Recent studies from our lab show that HNK impedes breast carcinogenesis. Here, we provide molecular evidence that HNK inhibits EMT in breast cancer cells resulting in significant downregulation of mesenchymal marker proteins and concurrent upregulation of epithelial markers. Experimental EMT induced by exposure to TGFβ and TNFα in spontaneously immortalized nontumorigenic human mammary epithelial cells is also completely reversed by HNK as evidenced by morphological as well as molecular changes. Investigating the downstream mediator(s) that may direct EMT inhibition by HNK, we found functional interactions between HNK, Stat3, and EMT‐signaling components. In vitro and in vivo analyses show that HNK inhibits Stat3 activation in breast cancer cells and tumors. Constitutive activation of Stat3 abrogates HNK‐mediated activation of epithelial markers whereas inhibition of Stat3 using small molecule inhibitor, Stattic, potentiates HNK‐mediated inhibition of EMT markers, invasion and migration of breast cancer cells. Mechanistically, HNK inhibits recruitment of Stat3 on mesenchymal transcription factor Zeb1 promoter resulting in decreased Zeb1 expression and nuclear translocation. We also discover that HNK increases E‐cadherin expression via Stat3‐mediated release of Zeb1 from E‐cadherin promoter. Collectively, this study reports that HNK effectively inhibits EMT in breast cancer cells and provide evidence for a previously unrecognized cross‐talk between HNK and Stat3/Zeb1/E‐cadherin axis.
Keywords: Honokiol, EMT, Zeb1, E‐cadherin, Stat3, Breast cancer
Highlights
Shows that honokiol inhibits EMT in breast cancer cells.
Reveals that Stat3 inhibition is integral for honokiol‐mediated EMT inhibition.
Mechanistic insights show the involvement of Stat3/Zeb1/E‐cadherin axis.
Proposes that honokiol could potentially be a rational therapeutic strategy for breast cancer.
1. Introduction
Breast cancer is the most frequently diagnosed cancer in women worldwide, and according to the data from the World Health Organization, it comprises 16% of all female cancers. Owing to the advances in early diagnosis, improved surgical techniques, adjuvant therapies and the advent of various targeted therapeutic approaches, mortality from breast cancer has been progressively declining over the past two decades. A major challenge currently facing the scientific community is that despite these advances, a large number of breast cancer patients present with metastatic cancer or relapse and metastasize after initial response to standard‐of‐care therapy. An emerging hypothesis is that epithelial–mesenchymal transition (EMT) bestows metastatic potential to epithelial cancer cells enabling them to invade, migrate and subsequently disseminate to form distant metastases resulting in non‐curable disease. EMT facilitates an increase in the subpopulation of CSC (cancer stem cells) and is associated with a chemoresistant phenotype in breast cancer (Creighton et al., 2010). EMT is a multistep process involving gradual loss of cell–cell adhesions and tight junctions leading to cytoskeleton reorganization and loss of apical polarity which eventually enables a cancer cell of epithelial origin to attain spindeloid morphology (Creighton et al., 2010; Dave et al., 2012). Induction of EMT in transformed mammary epithelial cells and mouse models of breast cancer induces cells with CSC properties (Mani et al., 2008; Morel et al., 2008; Radisky and LaBarge, 2008; Santisteban et al., 2009). Breast cancer stem cells (BCSCs) isolated from breast tumors and metastatic pleural effusion show gene expression and miRNA expression profiles similar to the cells undergoing EMT (Blick et al., 2010; Shimono et al., 2009). Importantly, modulation of EMT‐related select miRNAs and genes to revert EMT results in suppression of BCSCs (Creighton et al., 2010, 2009). Pharmacological inhibition of EMT can potentially lead to reversion of aggressive breast cancer cells to a more‐differentiated epithelial phenotype by inducing mesenchymal–epithelial transition (MET) to prevent metastasis and diminish distant recurrence.
In recent years, bioactive components from plants used in traditional Asian medicine have shown efficacy as potential cancer preventive as well as therapeutic agents. Cones, bark, and leaves from Magnolia plant species have been successfully used in the traditional Chinese and Japanese medicine for their anti‐thrombocytic, anti‐inflammatory, anxiolytic, antidepressant, antioxidant, antispasmodic and antibacterial effects (Choi et al., 2012; Lin et al., 2005; Lo et al., 1994; Oh et al., 2009; Xu et al., 2008). Medicinal benefits of Magnolia species have been attributed to a natural phenolic compound, honokiol (HNK), isolated from an extract of seed cones from Magnolia grandiflora (Fujita et al., 1973). Previous studies from our lab have shown that HNK inhibits breast carcinogenesis in vitro and in vivo (Nagalingam et al., 2012). Anticancer activities of HNK such as suppression of angiogenesis (Nagase et al., 2001; Shigemura et al., 2007), migration, invasion (Hirano et al., 1994; Singh and Katiyar, 2013) and proliferation (Battle et al., 2005; Hibasami et al., 1998; Ishitsuka et al., 2005; Wang et al., 2004; Yang et al., 2002) have been reported in multiple cancer cell lines and tumor models (Arora et al., 2012). Cellular studies have provided novel insights into the mechanisms underlying anticancer effects of HNK (Battle et al., 2005; Hibasami et al., 1998; Ishitsuka et al., 2005; Singh et al., 2013; Wang et al., 2004; Yang et al., 2002). For example, our own work has revealed that HNK activates tumor suppressor and upstream kinase, LKB1 leading to AMPK activation in breast cancer cells (Nagalingam et al., 2012). Downstream effectors of HNK‐mediated apoptosis involve BAX and BAD upregulation, activation of caspase 8, 9 and 3, cleavage of Mcl‐1 and downregulation of XIAP (Battle et al., 2005; Ishitsuka et al., 2005). Biological actions of HNK also involve inhibition of phosphorylation of ERK, Akt and c‐Src (Fried and Arbiser, 2009) and inhibition of NF‐κB signaling in multiple cancers (Ahn et al., 2006; Arora et al., 2011; Lee et al., 2005; Sheu et al., 2008; Tse et al., 2005). Collectively, these studies have established HNK as a promising bioactive compound possessing anticancer effects.
Given that EMT plays an integral role in sustaining the CSCs as well as metastatic progression of breast tumors, developing more‐effective, non‐endocrine, non‐toxic therapeutic strategies to target EMT is highly desirable. In the present study, we specifically investigated the potential of HNK to inhibit EMT, an early stage in cancer metastasis and examine the underlying molecular mechanisms. We provide strong evidence that HNK inhibits EMT in breast cancer cells modulating the mesenchymal and epithelial marker profiles. Our in vitro and in vivo analyses show that HNK inhibits Stat3 activation in breast cancer cells and tumors which plays a key role in modulating Zeb1 expression and its recruitment on E‐cadherin promoter. Our studies provide evidence for a previously unrecognized cross‐talk between HNK and Stat3/Zeb1/E‐cadherin axis.
2. Materials and methods
2.1. Cell culture and reagents
MCF7, MDA‐MB‐231, and MCF10A were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured according to supplier's instructions. Cell line authentication was done by analysis of known genetic markers or response (e.g., expression of estrogen receptor and p53 and estrogen responsiveness) (Kim et al., 2011). MDA‐MB‐231 cell line is a highly invasive “basal B” type and estrogen‐independent fibroblastic human breast cancer cell line with stellate morphology. MCF7 cell line is a well‐accepted representative of estrogen‐receptor‐positive “luminal” type breast cancer and exhibits epithelial phenotype. MCF10A is nontumorigenic and widely used as a representative normal mammary epithelial cell line. MCF10A was isolated from fibrocystic breast disease and spontaneously immortalized. For treatment, cells were seeded at a density of 1 × 106/100‐mm tissue culture dish. We extract Honokiol (HNK) from seed cone of M. grandiflora according to the previously published study (Bai et al., 2003). In treatments of cell cultures, honokiol was dissolved in 100% ethanol as a vehicle and in 20% Intralipid (Baxter Healthcare) for animal treatments. TGFβ was purchased from Calbiochem (Billerica, MA) and TNFα was obtained from Sigma–Aldrich (St. Louis, MO). Stat3 inhibitor, Stattic was purchased from Sigma–Aldrich. Antibodies for E‐cadherin, occludin, vimentin, β‐catenin, cyclinD1, phosphorylated Stat3, Stat3 and Actin were purchased from Cell Signaling Technology (Danvers, MA), Sigma–Aldrich (St. Louis, MO) and Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
2.2. Spheroid‐migration assay
MDA‐MB‐231 and MCF7 cells (1.5 × 104) were seeded in 0.5% agar‐coated plates and cultured on an orbital shaker (100 rpm) for 48 h in a humidified atmosphere containing 5% CO2 at 37 °C. Intact tumor spheroids were selected and transferred to six‐well plates. The spheroids were treated with honokiol as indicated. After 24‐72h h of incubation, spheroids were fixed with 10% buffered formalin in PBS and stained with crystal violet. The migration of cells from spheroids was observed under light microscope.
2.3. Invasion assay
For an in vitro model system for metastasis, a matrigel‐invasion assay (Saxena et al., 2007a) was performed using a matrigel‐invasion chamber from BD Biocoat Cellware (San Jose, CA). The slides were coded to prevent counting bias, and the number of invaded cells on representative sections of each membrane were counted under light microscope. The number of invaded cells for each experimental sample represents the average of triplicate wells.
2.4. RNA isolation, RT‐PCR
Total cellular RNA was extracted using the TRIZOL Reagent kit (Life Technologies, Inc., Rockville, MD). RT‐PCR was performed using specific sense and antisense PCR primers. Primer details are provided in supplemental section.
2.5. Western blotting
Whole cell lysates (Saxena et al., 2007b) was prepared by scraping MCF7 and MDA‐MB‐231 cells in 250 μl of ice cold modified RIPA buffer. Equal amount of lysate protein was resolved on sodium‐dodecyl sulfate polyacrylamide gel, transferred to nitrocellulose membrane, and western blot analysis was performed. Immunodetection was performed using enhanced chemiluminescence (ECL system, Amersham Pharmacia Biotech Inc., Arlington Heights, IL) according to manufacturer's instructions.
2.6. Immunofluorescence and confocal imaging
Breast cancer cells (5 × 105 cells/well) were plated in 4‐well chamber slides (Nunc, Rochester, NY) followed by treatment with HNK as indicated and subjected to immunofluorescence analysis (Taliaferro‐Smith et al., 2009). Fixed and immunofluorescently stained cells were imaged (20× magnification) using a Zeiss LSM510 Meta (Zeiss) laser scanning confocal system configured to a Zeiss Axioplan 2 upright microscope with a 63XO (NA 1.4) plan‐apochromat objective. All experiments were performed multiple times using independent biological replicates.
2.7. Breast tumorigenesis assay
MDA‐MB‐231 (5 × 106) cells in 0.1 ml of HBSS were injected subcutaneously into the left and right gluteal region of 4–6‐week‐old female athymic nude mice, procured from Harlan Laboratories Inc (Indianapolis, IN). Two weeks after initial implantation, animals were placed into two experimental groups. Mice were treated with intraperitoneal injections of 1) control (saline and Intralipid); 2) HNK, at 3 mg/mouse/day in 20% Intralipid (Baxter Healthcare), three times per week for the duration of the experiment. The dose and route of HNK administration was selected from our previous studies documenting in vivo efficacy (Nagalingam et al., 2012). Tumors volume were measured using vernier calipers, and the formula (V = a/2 × b2), where V is the tumor volume in mm3, a and b are the largest and smallest diameters in mm, respectively. All animals were sacrificed after 5 weeks of treatment, tumors were collected and weighed. One part of tumor was fixed in 10% neutral‐buffered formalin for further analysis by Immunohistochemistry (IHC), other parts were utilized for RNA and protein isolation for analysis using RT‐PCR or western blotting. All animal studies were in accordance with the guidelines of Johns Hopkins University IACUC.
2.8. Immunohistochemical analysis
We used tumor sections to determine the effect of HNK on the expression of mesenchymal and epithelial markers as well as expression of key signaling molecules by immunohistochemistry. Immunohistochemistry was performed essentially as described by us previously for other proteins (Saxena et al., 2010; Sharma et al., 2010). At least four nonoverlapping representative images (20× magnification) from each tumor section from five mice of each group were captured using ImagePro software for quantitation of protein expression. Total cell lysates were isolated from tumor samples and subjected to immunoblot analysis.
2.9. Transfection
The plasmid expressing a constitutively active form of Stat3 (Stat3‐CA) and wild‐type Stat3 (Stat3‐WT) were kind gifts from Dr. James. E. Darnell (The Rockefeller University, New York, New York) (Bromberg et al., 1999). Cells were transfected with Stat3‐CA or Stat3‐WT using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to manufacturer's instructions and treated as indicated. Forty eight hours post‐transfection, the cells were harvested and cell lysates were subjected to RT‐PCR analysis, Immunoblot analysis or ChIP analysis.
2.10. Chromatin immunoprecipitation assay
ChIP analyses were performed using our published procedure (Sharma et al., 2006). Chromatin samples were sonicated on ice three times for 10 s each (average length of sheared genomic DNA ∼ 1–1.5 kb) followed by centrifugation for 10 min. The immunoprecipitated DNA was ethanol precipitated and resuspended in 25 μl of H2O. Total input samples were resuspended in 100 μl of H2O and diluted 1:100 before real‐time quantitative PCR analysis.
2.11. Statistical analysis
All experiments were performed thrice in triplicates. Statistical analysis was performed using Microsoft Excel software. Significant differences were analyzed using student's t test and two‐tailed distribution. Results were considered to be statistically significant if p < 0.05. Results were expressed as mean ± SE between triplicate experiments performed thrice.
3. Results
3.1. Honokiol inhibits invasion, migration potential and modulates the expression of mesenchymal and epithelial markers of EMT in breast cancer cells
Metastatic progression of tumor involves invasion of basement membrane by cancer cells and migration to distant sites to form metastatic lesions. Upon examining the effect of honokiol (HNK) on invasion potential of breast cancer cells using matrigel‐invasion assay, we found that treatment with 5 μM honokiol inhibited invasion of breast cancer cells through matrigel in comparison to untreated cells (Figure 1A). Spheroid‐migration assay was utilized to investigate if honokiol impacted migration capacity of breast cancer cells. Significant migration of MCF7 and MDA‐MB‐231 cells from the spheroids was observed in untreated conditions while honokiol treatment resulted in inhibition of migration (Figure 1B). Epithelial to mesenchymal transition (EMT) of cancer cells of epithelial origin is a crucial step that precedes the induction of motility and invasive potential during metastatic progression of malignant cells (Dave et al., 2012). Since HNK inhibited invasion and migration of MCF7 and MDA‐MB‐231 cells, we aimed to examine whether HNK treatment influenced EMT in breast cancer cells. Biochemical hallmarks of EMT reversal include gain of expression of epithelial marker proteins such as E‐cadherin, occludin and cytokeratin‐18 with a concomitant decrease in mesenchymal marker (e.g., vimentin, fibronectin, and N‐cadherin) expression (Dave et al., 2012). Interestingly, treatment of breast cancer cells with HNK resulted in inhibition of mesenchymal markers, fibronectin and vimentin expression (Figure 2A and B) and an accompanied increase in the expression of occludin and CK‐18 (Figure 2D and E). Immunocytochemical analysis of HNK‐treated breast cancer cells provided additional evidence to support HNK‐mediated EMT reversal showing loss of expression of vimentin (Figure 2C) and gain of occludin expression (Figure 2F).
Figure 1.
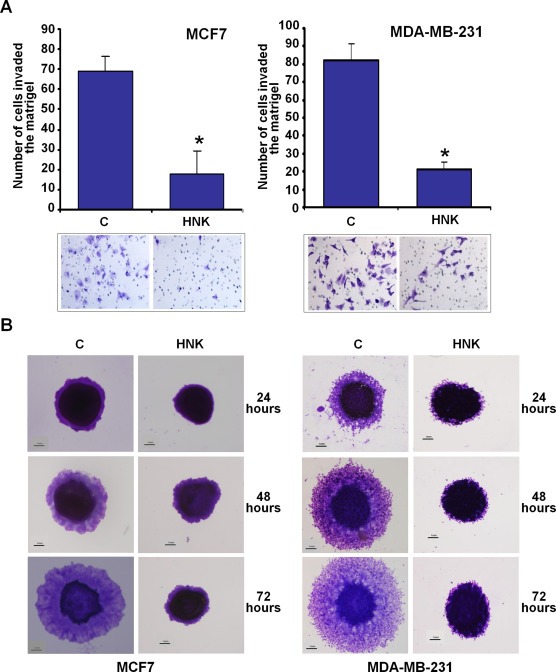
Honokiol inhibits invasion and migration of breast cancer cells. A, MCF7 and MDA‐MB‐231 cells were cultured in matrigel‐invasion chambers followed by treatment with 5 μM honokiol (HNK) for 24 h as indicated. C represents vehicle controls. The number of cells that invaded through the matrigel was counted in five different regions. *P < 0.005, compared with untreated controls. B, MCF7 and MDA‐MB‐231 cells were subjected to spheroid‐migration assay. Culture media were replaced with media containing honokiol (5 μM) or vehicle control media (C). The spheroids were photographed at 24 h, 48 h and 72 h‐post treatment. The results shown are representative of three independent experiments performed in triplicates.
Figure 2.
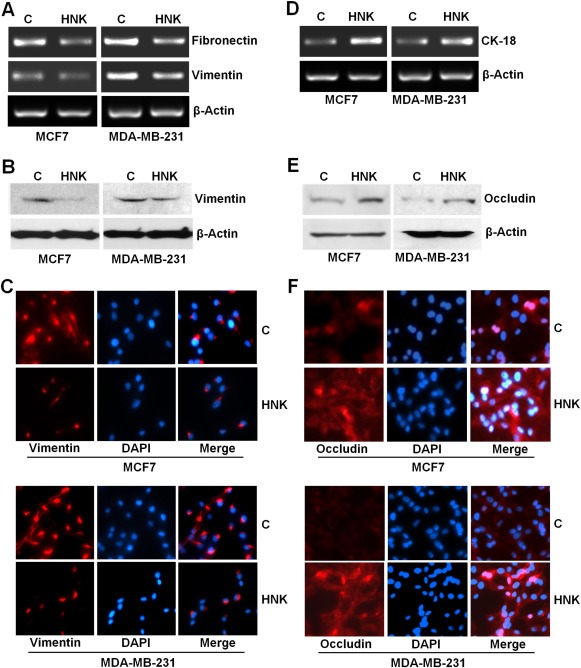
Honokiol inhibits the expression of mesenchymal genes and induces the expression of epithelial markers in breast cancer cells. A, D, MCF7 and MDA‐MB‐231 cells were treated with vehicle (C) or 5 μM honokiol (HNK). Total RNA was isolated and examined for the expression of fibronectin, vimentin and CK‐18 using specific primers. B, E, MCF7 and MDA‐MB‐231 cells were treated with vehicle (C) or 5 μM honokiol (HNK). Total lysates were immunoblotted for vimentin and occludin expression levels. C, F, MCF7 and MDA‐MB‐231 cells were treated with vehicle (C) or 5 μM honokiol (HNK) and subjected to immunofluorescence analysis of vimentin and occludin.
3.2. Inhibition of EMT in TGFβ/TNFα‐treated mammary epithelial cells by honokiol
Treatment with a combination of TGFβ and TNFα has been shown to induce EMT in some cell types (Bates and Mercurio, 2003; Sehrawat and Singh, 2011; Willis et al., 2005). When MCF10A normal mammary epithelial cells were treated with TGFβ/TNFα, morphologic changes from an epithelial‐like to mesenchymal‐like appearance were induced. Vehicle‐treated MCF10A cells showed round, well‐packed cobblestone appearance, a characteristic of epithelial cells while TGFβ/TNFα‐treated MCF10A cells exhibited mesenchymal phenotype acquiring spindle‐like appearance and increased intracellular separation signifying loss of intercellular adhesion (Figure 3A). We tested the possibility that HNK treatment is able to block TGFβ/TNFα‐induced EMT in MCF10A cells. As evident in Figure 3A, HNK prevented the morphologic transition from an epithelial‐like to mesenchymal‐like appearance caused by TGFβ/TNFα treatment. HNK alone did not affect the morphology of MCF10A (Figure 3A). TGFβ/TNFα‐treated MCF10A cells showed elevated levels of mesenchymal markers (vimentin and fibronectin) and reduced levels of epithelial markers (occludin and CK‐18). HNK blocked TGFβ/TNFα‐induced modulation of mesenchymal and epithelial markers leading to decreased expression of vimentin and fibronectin and elevated expression of occludin and CK‐18 (Figure 3B, C). Immunofluorescence analyses showed that HNK abrogated TGFβ/TNFα‐induced increased expression of vimentin. HNK treatment antagonized TGFβ/TNFα‐mediated inhibition of epithelial marker expression leading to increased E‐cadherin and occludin expression (Figure 3D). All these findings confirm the potential of HNK as a novel EMT inhibitor using an experimental system involving TGFβ/TNFα and MCF10A cells.
Figure 3.
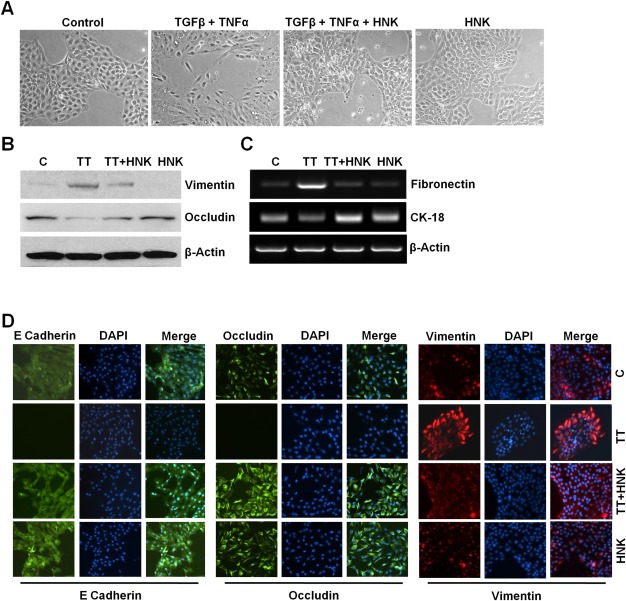
Honokiol abrogates TGFβ/TNFα‐induced epithelial–mesenchymal transition in mammary epithelial cells. A, MCF10A cells were treated with vehicle control (control), TGFβ + TNFα (10 ng/ml of each), 5 μM honokiol (HNK) or TGFβ + TNFα +HNK for 24 h. Morphological changes associated with EMT are shown in phase‐contrast images. The presence of spindle‐shaped cells, increased intercellular separation and pseudopodia were noted in TGFβ + TNFα‐treated cells but not in HNK‐treated or TGFβ + TNFα + HNK‐treated cells. B, MCF10A cells were treated as in A and total lysates were immunoblotted for vimentin and occludin expression levels. Actin was used as control. C, MCF10A cells were treated as in A, total RNA was isolated and expression of fibronectin and CK‐18 was analyzed. Actin was included as control. D, MCF10A cells were treated as in A, and subjected to immunofluorescence analysis of E‐Cadherin, occludin and vimentin.
3.3. Honokiol treatment inhibits breast tumorigenesis and modulates EMT markers in vivo
We further examined the physiological relevance of our in vitro findings by evaluating whether HNK inhibits breast carcinoma in vivo. Growth of MDA‐MB‐231 xenografts in female athymic nude mice was inhibited significantly by intraperitoneal HNK administration compared with the vehicle‐treated group (Figure 4A, B). In our in vitro analyses, we discovered that HNK modulates the expression of various epithelial and mesenchymal markers. Tumor samples from HNK and vehicle‐treated groups were used to determine the effect of HNK administration on expression of EMT markers. We observed that CK‐18 expression was very low in the tumors from control mice whereas HNK administration caused upregulation of CK‐18 expression in MDA‐MB‐231 xenografts (Figure 4C). Expression of mesenchymal gene markers, vimentin and fibronectin was markedly decreased in MDA‐MB‐231 tumors from HNK‐treated group compared with vehicle‐treated group (Figure 4C).
Figure 4.
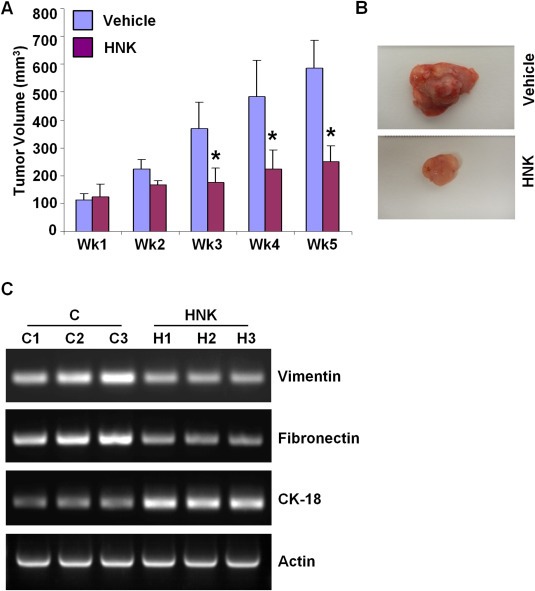
Honokiol treatment inhibits breast tumor growth in nude mice. MDA‐MB‐231 cells derived tumors were developed in nude mice and treated with vehicle or honokiol (HNK). At the end of five weeks of treatment, tumors were collected, measured, weighed and photographed. A, Tumor growth was monitored by measuring the tumor volume for 5 weeks (n = 8 mice per group), *P < 0.001, compared with vehicle‐treated controls. B, Representative tumor images are shown here. C, Total RNA was isolated from tumor samples and subjected to RT‐PCR analysis. Expression of epithelial and mesenchymal markers (vimentin, fibronectin and CK‐18) was analyzed.
3.4. Abrogation of Stat3 is integral to honokiol‐mediated inhibition of EMT, invasion and migration of breast cancer cells
Constitutive activation of signal transducer and activator of transcription 3 (Stat3) has been reported in many malignancies, including breast cancer (Haura et al., 2005). Stat3, a DNA‐binding transcription factor, regulates cell proliferation and survival, functions as a major player in driving the growth of breast cancer stem cells (Idowu et al., 2012; Marotta et al., 2011) and has been associated with epithelial–mesenchymal transition of cancer cells and malignant progression (Berclaz et al., 2001; Colomiere et al., 2009; Haura et al., 2005). We sought to determine whether HNK modulates Stat3 phosphorylation and activation. MCF7 and MDA‐MB‐231 cells treated with HNK exhibited decreased phosphorylation of Stat3 at Tyr‐705 whereas no change was observed in total Stat3 protein expression levels (Figure 5A). For in vivo evidence, we further examined tumor samples from HNK and vehicle‐treated mice. As evident in Figure 5B, HNK administration caused inhibition of Stat3 phosphorylation. Immunohistochemical analyses of HNK and vehicle‐treated tumors also showed reduced expression of phosphorylated Stat3 in HNK‐treated group (Figure 5C). Phosphorylation of Stat3 has been widely associated with its activation, dimerization and translocation to the nucleus. Once activated, Stat3 directly transactivates multiple genes involved in cell proliferation, and metastatic progression including cyclinD1 and β‐catenin (Haura et al., 2005). We examined the expression of cyclinD1 and β‐catenin to evaluate Stat3 activity in response to HNK treatment. Immunohistochemical analyses of tumor samples from HNK‐treated mice showed decreased expression of cyclinD1 and pStat3 (Figure 5C). Decreased expression of cyclinD1 and β‐catenin in tumor samples was observed in response to HNK treatment as compared with vehicle‐treated tumors (Figure 5D and E).
Figure 5.
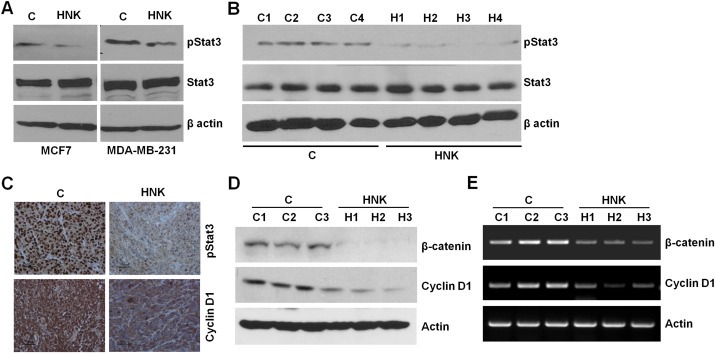
Evidence for honokiol‐mediated inhibition of signal transducer and activator of transcription 3. A, MCF7 and MDA‐MB‐231 cells were treated with vehicle (C) or 5 μM honokiol (HNK). Total lysates were immunoblotted for phosphorylated Stat3 (pStat3) and total Stat3 expression levels. B, MDA‐MB‐231 cells derived tumors were developed in nude mice, treated with vehicle or honokiol (HNK) for five weeks. Tumors were collected at the end of five weeks and subjected to western blot analysis for phosphorylated Stat3 (pStat3) and total Stat3 expression levels. C, MDA‐MB‐231 cells derived tumors, treated with vehicle or honokiol (HNK) were subjected to immunohistochemical analysis using phosphorylated Stat3 and cyclinD1 antibodies. D, Tumor lysates (from three different tumors from each set) were subjected to immunoblot analysis using β‐catenin and cyclinD1 antibodies. Actin antibody was used as control. E, Total RNA was isolated from three different tumors from each set were subjected to RT‐PCR analysis using β‐catenin and cyclinD1 primers. Actin primers were used as control.
To investigate whether Stat3 inhibition plays an important role in HNK‐mediated EMT inhibition in breast cancer cells, we used overexpression of constitutively active Stat3 (Stat3‐CA) and pharmacological inhibition of Stat3. Overexpression of Stat3‐CA competed with HNK and diminished HNK‐mediated activation of epithelial markers (CK‐18 and occludin) (Figure 6A). Pharmacological inhibition of Stat3 activation using a small molecule inhibitor, Stattic, potentiated HNK‐mediated inhibition of mesenchymal markers (Figure 6B). Given the association of Stat3 with invasive phenotype, we further aimed to evaluate the contribution of Stat3 in HNK‐mediated inhibition of invasion and migration potential. Breast cancer cells were treated with Stattic alone and in combination with HNK followed by matrigel‐invasion and spheroid‐migration assays. Results showed that co‐treatment with Stattic and HNK induced greater inhibition of invasion potential of both MCF7 and MDA‐MB‐231 cells as compared to HNK alone (Figure 6C). Stattic also potentiated HNK‐mediated inhibition of migration of breast cancer cells (Figure 6D). These findings suggest that indeed Stat3‐inhibition is integral for HNK‐mediated molecular changes in EMT markers and inhibition of invasion and migration of breast cancer cells.
Figure 6.
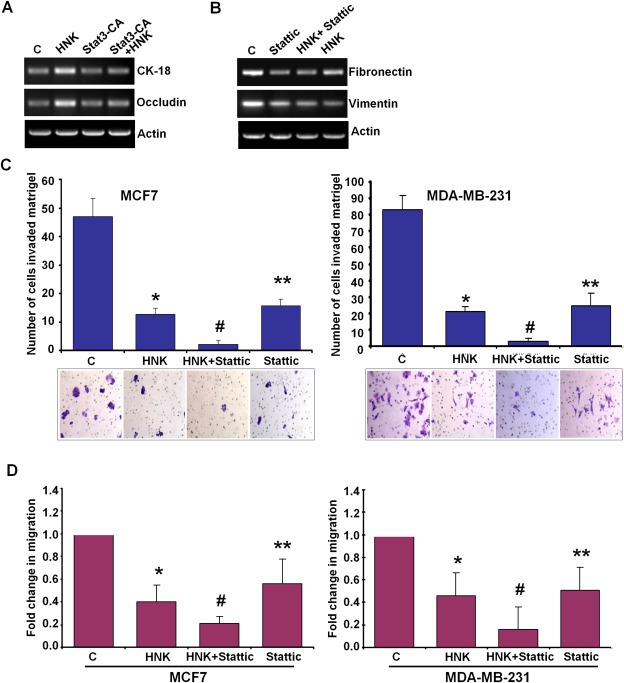
Stat3‐inhibition plays an important role in honokiol‐mediated modulation of EMT markers, and inhibition of invasion and migration of breast cancer cells. A, MCF7 cells were treated with vehicle (C) or 5 μM honokiol (HNK), transfected with constitutively active Stat3 (Stat3‐CA) and treated with Honokiol (Stat3‐CA + HNK). Total RNA was isolated and subjected to RT‐PCR analysis using CK‐18 and occludin primers. Actin was included as control. B, MDA‐MB‐231 cells were treated with vehicle (C), 10 μM Stattic, 5 μM honokiol (HNK) alone, or in combination (HNK + Stattic), total RNA was isolated and subjected to RT‐PCR analysis using fibronectin and vimentin primers. Actin was included as control. C, MCF7 and MDA‐MB‐231 cells were cultured in matrigel‐invasion chambers followed by treatment with 10 μM Stattic, 5 μM honokiol (HNK) alone, or in combination (HNK + Stattic) for 24 h as indicated. C represents vehicle controls. The number of cells that invaded through the matrigel was counted in five different regions. *P < 0.005, compared with vehicle‐treated controls; **P < 0.001, compared with vehicle‐treated controls; #P < 0.005, compared with HNK‐treated cells. D, MCF7 and MDA‐MB‐231 cells were subjected to spheroid‐migration assay. Culture media were replaced with media containing 10 μM Stattic, 5 μM honokiol (HNK) alone, in combination (HNK + Stattic) or vehicle control (C). The spheroids were photographed 48 h‐post treatment. The results are shown as fold‐change in migration of breast cancer cells in response to HNK and Stattic treatments. These are representative of three independent experiments performed in triplicates. *P < 0.01, compared with vehicle‐treated controls; **P < 0.05, compared with vehicle‐treated controls; #P < 0.05, compared with HNK‐treated cells.
3.5. Honokiol inhibits an important modifier of EMT, Zeb1 via de‐recruitment of Stat3 on Zeb1 promoter
Overexpression of zinc finger E‐box binding homeobox transcription factor 1 (Zeb1) has been linked with increased aggressiveness and higher metastatic potential in various primary human carcinomas, including breast cancer (Sanchez‐Tillo et al., 2011). A crucial mediator of EMT, Zeb1, exerts its effects on induction of EMT by regulating epithelial and mesenchymal gene expression balance (Schmalhofer et al., 2009). Interestingly, we found that HNK inhibited the expression of Zeb1 in breast cancer cells (Figure 7A). Immunofluorescence analyses showed that TGFβ/TNFα treatment induced the nuclear translocation of Zeb1 in MDA‐MB‐231 and MCF7 cells which was effectively abrogated by HNK (Figure 7B). Given our results showing a critical role of Stat3‐inhibition in mediating biological functions of HNK and its effect on molecular changes related to EMT markers, we reasoned that overexpression of constitutively active Stat3 might be able to mitigate HNK‐induced Zeb1 inhibition. Indeed, overexpression of constitutively active Stat3 alleviated HNK‐mediated inhibition of Zeb1 (Figure 7C). We utilized a small molecule inhibitor, Stattic, to inhibit Stat3 activation in MCF7 and MDA‐MB‐231 cells and assessed its impact on HNK‐mediated Zeb1 inhibition. Pharmacological inhibition of Stat3 inhibited Zeb1 expression and increased the effectiveness of HNK‐mediated inhibition of Zeb1 (Figure 7D). These data further indicate that Stat3 may directly participate in regulating Zeb1 expression in HNK‐treated breast cancer cells. Of particular interest, two putative Stat3‐binding sites have been reported on Zeb1 promoter (Xiong et al., 2012). ChIP analysis clearly showed that HNK treatment inhibited recruitment of Stat3 to Zeb1 gene promoter (Figure 7E). Cells overexpressing constitutively active Stat3 exhibited increased binding of pStat3 while Stattic treatment led to inhibition of Stat3 recruitment to Zeb1 gene promoter. We observed that overexpression of constitutively active Stat3 could compete with honokiol and provide protection against honokiol‐mediated release of Stat3 binding. On the other hand, Stattic treatment potentiated HNK‐mediated release of Stat3 from Zeb1 promoter (Figure 7E). Cells overexpressing wild‐type Stat3 exhibited increased binding of pStat3 on Zeb1 promoter which could be abrogated with HNK treatment (10 and 25 μM) (Figure 7E). These data strongly suggest a regulatory role of Stat3 in HNK‐mediated inhibition of Zeb1.
Figure 7.
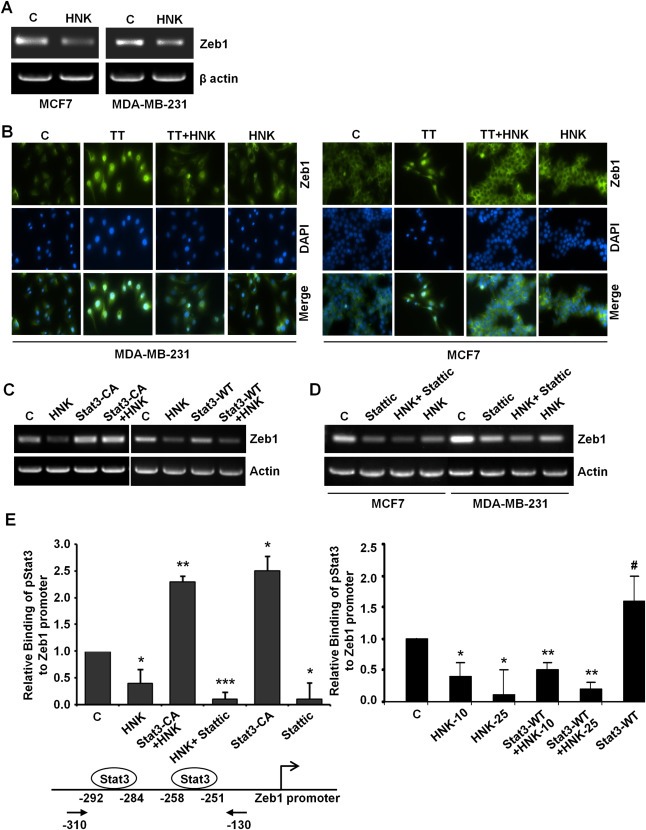
Honokiol inhibits Zeb1 expression, nuclear translocation and releases Stat3 from Zeb1 promoter. A, MCF7 and MDA‐MB‐231 cells were treated with vehicle (C) or 5 μM honokiol (HNK). Total RNA was isolated and examined for the expression of Zeb1 using specific primers. B, MCF7 and MDA‐MB‐231 cells were treated with vehicle control (C), TGFβ and TNFα (10 ng/ml of each) (TT), 5 μM honokiol (HNK) or TT + HNK and subjected to immunofluorescence analysis of Zeb1. TT induces nuclear translocation of Zeb1 which is abrogated by HNK treatment. C, MCF7 cells were treated with vehicle (C) or 5 μM honokiol (HNK), transfected with constitutively active Stat3 (Stat3‐CA) and treated with Honokiol (Stat3‐CA + HNK). In another set, MCF7 cells were treated with vehicle (C) or 5 μM honokiol (HNK), transfected with wild‐type Stat3 (Stat3‐WT) and treated with Honokiol (Stat3‐WT + HNK). Total RNA was isolated and subjected to RT‐PCR analysis using Zeb1 primers. Actin was included as control. D, MCF7 and MDA‐MB‐231 cells were treated with vehicle (C), 10 μM Stattic, 5 μM honokiol (HNK) alone, or in combination (HNK + Stattic), total RNA was isolated and subjected to RT‐PCR analysis using Zeb1 primers. Actin was included as control. E, Soluble chromatin was prepared from MCF7 cells treated with vehicle (C), 5 μM honokiol (HNK), transfected with constitutively active Stat3 (Stat3‐CA) alone or in combination with honokiol (Stat3‐CA + HNK), 10 μM Stattic alone or in combination (HNK + Stattic) and subjected to chromatin immunoprecipitation assay using pStat3 antibody. In another set, soluble chromatin was prepared from MCF7 cells treated with vehicle (C), 10 μM and 25 μM honokiol (HNK), transfected with wild‐type Stat3 (Stat3‐WT) alone or in combination with honokiol (Stat3‐WT + HNK‐10 and Stat3‐WT + HNK‐25) and subjected to chromatin immunoprecipitation assay using pStat3 antibody. The purified DNA was analyzed by real‐time quantitative PCR using primers spanning the Stat3‐binding sites at Zeb1 promoter.
3.6. Honokiol‐mediated Stat3 inhibition releases Zeb1 from E‐cadherin promoter
Modulation of E‐cadherin expression is a fundamental event during EMT and is accompanied with reorganization of intercellular complexes and synthesis of extracellular matrix components (fibronectin, collagen etc.) (Thiery et al., 2009). Western blot and RT‐PCR analyses of breast cancer cells treated with HNK showed that HNK treatment increased the expression of E‐cadherin (Figure 8A, B). Additional evidence was provided with immunofluorescence analysis showing elevated expression of E‐cadherin in HNK‐treated breast cancer cells (Figure 8C). Promoter region (nt‐520 to +70) of E‐cadherin has been reported to contain two putative Stat3‐binding sites and four putative Zeb1‐binding sites (Xiong et al., 2012). Zeb1 has been known to repress E‐cadherin and induce EMT (Sanchez‐Tillo et al., 2011). We questioned whether Stat3 and/or Zeb1 are involved in HNK‐mediated upregulation of E‐cadherin expression in breast cancer cells. ChIP analysis showed that Zeb1 got recruited to E‐cadherin promoter and this binding was diminished in the presence of HNK (Figure 8D). Overexpression of constitutively active Stat3 resulted in an increased binding of Zeb1 to E‐cadherin promoter and also alleviated HNK‐mediated decreased recruitment of Zeb1. Stattic treatment, on the other hand, inhibited Zeb1 recruitment on E‐cadherin promoter and potentiated HNK‐mediated release of Zeb1 from E‐cadherin promoter in breast cancer cells (Figure 8D). Cells overexpressing wild‐type Stat3 exhibited increased binding of Zeb1 on E‐cadherin promoter which could be abrogated with HNK treatment (10 and 25 μM) (Figure 8D). Interestingly, ChIP analysis of chromatin samples immunoprecipitated with anti‐Stat3 antibodies revealed no recruitment of Stat3 on E‐cadherin promoter (data not shown). These results indicate that Stat3 does not bind directly to E‐cadherin promoter but plays an important role in Zeb1 recruitment and release and hence E‐cadherin regulation.
Figure 8.
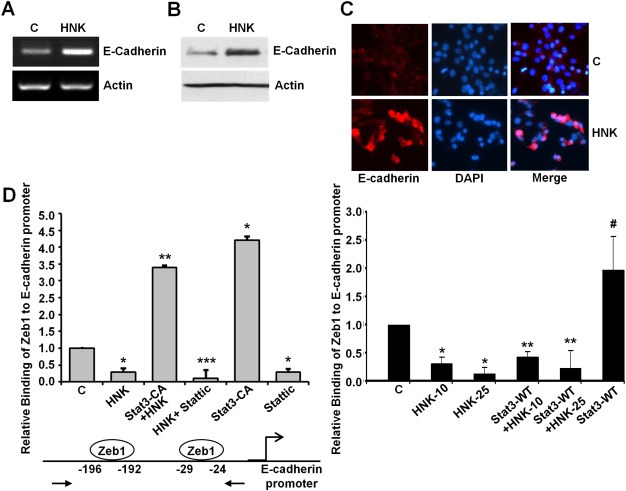
Honokiol increases E‐cadherin expression and inhibits the recruitment of Zeb1 on E‐cadherin promoter in a Stat3‐dependent manner. A, MCF7 cells were treated with vehicle (C) or 5 μM honokiol (HNK). Total RNA was isolated and examined for the expression of E‐cadherin using specific primers. B, MCF7 cells were treated as in A, total lysates were immunoblotted for E‐cadherin expression levels. C, MCF7 cells were treated as in A and subjected to immunofluorescence analysis of E‐cadherin. D, Soluble chromatin was prepared from MCF7 cells treated with vehicle (C), 5 μM honokiol (HNK), transfected with constitutively active Stat3 (Stat3‐CA) alone or in combination with honokiol (Stat3‐CA + HNK), 10 μM Stattic alone or in combination (HNK + Stattic) and subjected to chromatin immunoprecipitation assay using Zeb1 antibody. In another set, soluble chromatin was prepared from MCF7 cells treated with vehicle (C), 10 μM and 25 μM honokiol (HNK), transfected with wild‐type Stat3 (Stat3‐WT) alone or in combination with honokiol (Stat3‐WT + HNK‐10 and Stat3‐WT + HNK‐25) and subjected to chromatin immunoprecipitation assay using Zeb1 antibody. The purified DNA was analyzed by real‐time quantitative PCR using primers spanning the Zeb1‐binding sites at E‐cadherin promoter.
Based on our studies, we propose a model in which HNK inhibits Stat3 phosphorylation and recruitment of Stat3 on Zeb1 promoter resulting in decreased Zeb1 expression and nuclear translocation. HNK increases E‐cadherin expression via Stat3‐mediated release of Zeb1 from E‐cadherin promoter. Collectively, our study shows that HNK effectively inhibits EMT in breast cancer cells and provide evidence for the involvement of Stat3/Zeb1/E‐cadherin axis.
4. Discussion
A compelling body of evidence has put forth that EMT, a morphogenic process normally activated during embryonal development and wound healing, is an important event during metastatic progression of cancer. Indeed, EMT is cardinal manifestation of the molecular and phenotypic changes acquired by epithelium‐derived tumors switching to a mesenchymal phenotype capable of local invasion and distant metastasis. Many studies have examined the possible role of EMT in breast cancer. Overexpression of mesenchymal molecular markers of EMT in breast cancer biopsies is associated with tumor aggressiveness, adverse clinicopathological characteristics, increased recurrence, and shorter survival (Creighton et al., 2010; Tomaskovic‐Crook et al., 2009). Therefore, it is of great therapeutic interest to develop effective therapeutic strategies to target EMT in breast cancer cells to prevent malignant growth. In the current study, we investigated the potential of honokiol (HNK) in the inhibition of EMT in breast cancer cells and elucidated the underlying molecular mechanisms. Our findings present compelling evidence that HNK effectively inhibits EMT in breast cancer cells and reverses the acquisition of mesenchymal characteristics in TGFβ/TNFα‐stimulated mammary epithelial cells. The key mechanism that we elucidate to account for this important novel function of HNK is that it inhibits Stat3 phosphorylation and transactivation activity. Another important finding is that HNK inhibits the expression of Zeb1, a transcription factor that plays a pivotal role in EMT. We further show that HNK‐mediated inhibition of Zeb1 is achieved by blocking Stat3 recruitment on Zeb1 promoter. We also provide molecular evidence that HNK triggers release of Zeb1 from E‐cadherin promoter hence releasing the Zeb1‐mediated repressive effect in a Stat3‐dependent manner leading to increased E‐cadherin expression. The significance of our in vitro results is strengthened by the fact that key components of this HNK‐mediated pathway we describe are detected in breast tumors treated with HNK.
Given the general association between EMT and tumor aggressiveness and the observation that biopsies from breast tumors show gene expression profiles that are associated with EMT, a desirable approach is to revert EMT and inhibit metastatic potential. Silencing of mesenchymal genes and participating transcription factors promoting EMT has been shown to revert the EMT phenotype in several experimental systems (Zhuo et al., 2008, 2008). Another approach can be the targeted inhibition of pathways such as NF‐κB, which play an important role in EMT (Huber et al., 2004). Direct targeting of E‐cadherin with specific anti‐adhesive antibodies has also shown positive effects for EMT reversal (Green et al., 2004). Apart from the synthetic approaches, many recent studies have identified several bioactive molecules that demonstrate activity as EMT inhibitors, including genistein, garlic derivatives, green tea polyphenols and benzyl isothiocyanate (Belguise et al., 2007; Chu et al., 2006; Sehrawat and Singh, 2011; Zhang et al., 2008). Although lower toxicity associated with bioactive molecules is a much desired quality, their limited bioavailability hinders further development (Bar‐Sela et al., 2010). In contrast to various other natural bioactive products, HNK exhibits a much desirable spectrum of bioavailability, as significant systemic levels of HNK can be achieved in preclinical models and it can cross blood–brain barrier (Wang et al., 2011). These qualities of HNK make it a promising small‐molecular weight natural agent that can inhibit EMT in cancer cells. One of the major findings of this study is that inhibition of Stat3 phosphorylation plays an integral role in mediating the effect of HNK on EMT, migration and invasion of breast cancer cells.
Stat3 is constitutively active in many types of cancer, including breast, melanoma, prostate, head and neck squamous cell carcinoma, multiple myeloma, pancreatic, ovarian and brain tumors (Buettner et al., 2002; Sinibaldi et al., 2000; Turkson, 2004; Turkson and Jove, 2000; Yu and Jove, 2004; Yue and Turkson, 2009). Given the role of Stat3 in transformation, multiple aspects of tumor progression and metastasis including EMT, Stat3 represents an attractive target for cancer therapy. Over the past few years, various indirect and direct strategies have been used to block Stat3 activation including antisense methods, ectopic expression of dominant negative mutants, inhibition of upstream kinases and phosphotyrosyl peptides (Bowman et al., 2000). Tyrphostin AG490 has been shown to inhibit proliferation of human acute lymphocytic leukemia, human and mouse myeloma cells (Burdelya et al., 2002; Catlett‐Falcone et al., 1999; Meydan et al., 1996). Antisense Stat3 ODN (oligodeoxynucleotide) have been developed to specifically block the expression of Stat3 mRNA in human head and neck squamous carcinoma cell lines inhibiting proliferation (Grandis et al., 1998). A transcription factor decoy approach blocks Stat3 activation and results in decreased tumor growth (Xi et al., 2005). Screening of a virtual database of 429,000 compounds led to the identification of many potential candidate small molecule inhibitors for Stat3 function. Functional evaluation of these candidate Stat3 inhibitors showed that STA21, a small‐molecular inhibitor inhibits breast cancer cells expressing constitutively active STAT3 (Song et al., 2005). Despite these developments, there are relatively few reports in the literature on the translational use of Stat3 inhibitors, caveats being, limited efficacy, therapy‐related toxicity and off‐target effects. Our study shows that bioactive molecule, honokiol, effectively inhibits phosphorylation of Stat3 as well as transactivation function of Stat3 potentially blocking all downstream effectors molecules of Stat3‐network. This warrants further development of honokiol as an effective Stat3 antagonist and a therapeutic strategy ameliorating metastasis to secondary sites.
Sustained expression of E‐cadherin, the core molecule of adherens junctions, is important to retain an epithelial phenotype and inhibit invasion potential of tumor cells. E‐cadherin molecules connect neighboring epithelial cells using their extracellular domains and the cytoplasmic part of E‐cadherin interacts with the other components of adheren junctions (Thiery et al., 2009). The characteristic event of EMT is loss of E‐cadherin. E‐cadherin gene (CDH1) is commonly mutated in invasive lobular carcinoma and lack of E‐cadherin expression is a characteristic feature of invasive lobular carcinoma while other types of breast cancer show variable downregulation (Prasad et al., 2009). Infiltrating cells or the tumor cells themselves produce EMT inducers (such as TGFβ and TNFα) and trigger the expression of a variety of transcriptional repressors leading to inhibition of E‐cadherin. We show that MCF10A cells treated with TGFβ and TNFα exhibit onset of EMT and accompanied molecular alterations include inhibition of E‐cadherin expression. An important transcription factor involved in E‐cadherin repression is Zeb1, encoded by TCF8 gene (Creighton et al., 2010; Schmalhofer et al., 2009). Analysis of multiple breast cancer cell lines revealed an inverse correlation between TCF8 and CHD1 gene transcript levels and importantly, cell lines with higher Zeb1 expression exhibited more mesenchymal characteristics (Chua et al., 2007). We found that Zeb1 gets recruited to E‐cadherin promoter in breast cancer cells which was released upon HNK treatment. These results indicate that active removal of repressive effects of Zeb1 is involved in HNK‐mediated increased expression of E‐cadherin. Zeb1 interacts with the regulatory regions of responsive target genes and alters the expression of various genes involved in the development of hematopoietic cells and ECM components (Schmalhofer et al., 2009). Zeb1 expression itself is regulated by several upstream signaling pathways including TGFβ, TNFα, and IGF1 in various cancer cell lines. In addition, epidermal growth factor receptor (EGFR), estrogen and progesterone mediated activation of TCF8 gene has also been reported (reviewed in Schmalhofer et al., 2009) in cancer cell metastasis. A recent report demonstrated the involvement of Stat3 in regulating Zeb1 expression in colorectal carcinoma cells (Xiong et al., 2012). Since HNK treatment profoundly inhibits Stat3, we questioned if HNK‐mediated Zeb1 inhibition involves Stat3 in breast cancer cells. Our studies found that Stat3 gets recruited to Zeb1 promoter and it correlates with Zeb1 expression in breast cancer cells. HNK treatment releases Stat3 from Zeb1 promoter leading to inhibition of Zeb1 expression. Further research can show the interplay of specific coactivator molecules orchestrated by HNK.
4.1. Conclusions
Here, we report a novel function of HNK as an inhibitor of EMT in breast cancer cells providing in vitro as well as in vivo evidence of some novel and important players of the HNK network. Based on our findings, we also present a model for HNK‐mediated inhibition of EMT in breast cancer. We propose that HNK inhibits Stat3 phosphorylation and transactivation potential that causes decreased recruitment of Stat3 to Zeb1 promoter leading to reduced Zeb1 expression. In addition, Stat3 also regulates Zeb1 recruitment to E‐cadherin promoter and HNK‐mediated Stat3 inactivation triggers release of the repressive effects of Zeb1 on E‐cadherin, altering its expression and ultimately inhibiting mesenchymal transition of breast cancer cells. Furthermore, consistent with the critical role of Stat3 in HNK‐mediated EMT inhibition, alteration of Stat3 activity modulated HNK‐induced morphological as well as molecular changes. Collectively, this study gives insight into the specific pathways that are required for HNK‐mediated EMT inhibition and put forth HNK as a rational therapeutic strategy for breast carcinoma.
Conflict of interest
DBA, AN, MYB, NKS and DS declare no conflict of interest. JLA is listed as an inventor on patents filed by Emory University. Emory has licensed its honokiol technologies to Naturopathic Pharmacy. JLA has received stock in Naturopathic Pharmacy, which to the best of knowledge is not publically traded.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
This work was supported by NIDDK NIH, K01DK076742 and R03DK089130 (to NKS); NCI NIH R01AR47901 (to JLA); NCI NIH R01CA131294, Safeway Foundation, Avon Foundation, Breast Cancer Research Foundation (BCRF) 90047965 (to DS).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.01.004.
Avtanski Dimiter B., Nagalingam Arumugam, Bonner Michael Y., Arbiser Jack L., Saxena Neeraj K. and Sharma Dipali, (2014), Honokiol inhibits epithelial—mesenchymal transition in breast cancer cells by targeting signal transducer and activator of transcription 3/Zeb1/E‐cadherin axis, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.01.004.
Contributor Information
Neeraj K. Saxena, Email: nsaxena@medicine.umaryland.edu
Dipali Sharma, Email: dsharma7@jhmi.edu, Email: dsharma28@gmail.com.
References
- Ahn, K.S. , Sethi, G. , Shishodia, S. , Sung, B. , Arbiser, J.L. , Aggarwal, B.B. , 2006. Honokiol potentiates apoptosis, suppresses osteoclastogenesis, and inhibits invasion through modulation of nuclear factor-kappaB activation pathway. Mol. Cancer Res. MCR. 4, 621–633. [DOI] [PubMed] [Google Scholar]
- Arora, S. , Bhardwaj, A. , Srivastava, S.K. , Singh, S. , McClellan, S. , Wang, B. , Singh, A.P. , 2011. Honokiol arrests cell cycle, induces apoptosis, and potentiates the cytotoxic effect of gemcitabine in human pancreatic cancer cells. PLoS One. 6, e21573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora, S. , Singh, S. , Piazza, G.A. , Contreras, C.M. , Panyam, J. , Singh, A.P. , 2012. Honokiol: a novel natural agent for cancer prevention and therapy. Curr. Mol. Med.. 12, 1244–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, X. , Cerimele, F. , Ushio-Fukai, M. , Waqas, M. , Campbell, P.M. , Govindarajan, B. , Der, C.J. , Battle, T. , Frank, D.A. , Ye, K. , Murad, E. , Dubiel, W. , Soff, G. , Arbiser, J.L. , 2003. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J. Biol. Chem.. 278, 35501–35507. [DOI] [PubMed] [Google Scholar]
- Bar-Sela, G. , Epelbaum, R. , Schaffer, M. , 2010. Curcumin as an anti-cancer agent: review of the gap between basic and clinical applications. Curr. Med. Chem.. 17, 190–197. [DOI] [PubMed] [Google Scholar]
- Bates, R.C. , Mercurio, A.M. , 2003. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol. Biol. Cell. 14, 1790–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle, T.E. , Arbiser, J. , Frank, D.A. , 2005. The natural product honokiol induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia (B-CLL) cells. Blood. 106, 690–697. [DOI] [PubMed] [Google Scholar]
- Belguise, K. , Guo, S. , Yang, S. , Rogers, A.E. , Seldin, D.C. , Sherr, D.H. , Sonenshein, G.E. , 2007. Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, slug, and an invasive phenotype. Cancer Res.. 67, 11742–11750. [DOI] [PubMed] [Google Scholar]
- Berclaz, G. , Altermatt, H.J. , Rohrbach, V. , Siragusa, A. , Dreher, E. , Smith, P.D. , 2001. EGFR dependent expression of STAT3 (but not STAT1) in breast cancer. Int. J. Oncol.. 19, 1155–1160. [DOI] [PubMed] [Google Scholar]
- Blick, T. , Hugo, H. , Widodo, E. , Waltham, M. , Pinto, C. , Mani, S.A. , Weinberg, R.A. , Neve, R.M. , Lenburg, M.E. , Thompson, E.W. , 2010. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/−) stem cell phenotype in human breast cancer. J. Mammary Gland Biol. Neoplasia. 15, 235–252. [DOI] [PubMed] [Google Scholar]
- Bowman, T. , Garcia, R. , Turkson, J. , Jove, R. , 2000. STATs in oncogenesis. Oncogene. 19, 2474–2488. [DOI] [PubMed] [Google Scholar]
- Bromberg, J.F. , Wrzeszczynska, M.H. , Devgan, G. , Zhao, Y. , Pestell, R.G. , Albanese, C. , Darnell, J.E. , 1999. Stat3 as an oncogene. Cell. 98, 295–303. [DOI] [PubMed] [Google Scholar]
- Buettner, R. , Mora, L.B. , Jove, R. , 2002. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res.. 8, 945–954. [PubMed] [Google Scholar]
- Burdelya, L. , Catlett-Falcone, R. , Levitzki, A. , Cheng, F. , Mora, L.B. , Sotomayor, E. , Coppola, D. , Sun, J. , Sebti, S. , Dalton, W.S. , Jove, R. , Yu, H. , 2002. Combination therapy with AG-490 and interleukin 12 achieves greater antitumor effects than either agent alone. Mol. Cancer Ther.. 1, 893–899. [PubMed] [Google Scholar]
- Catlett-Falcone, R. , Landowski, T.H. , Oshiro, M.M. , Turkson, J. , Levitzki, A. , Savino, R. , Ciliberto, G. , Moscinski, L. , Fernandez-Luna, J.L. , Nunez, G. , Dalton, W.S. , Jove, R. , 1999. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 10, 105–115. [DOI] [PubMed] [Google Scholar]
- Choi, D.Y. , Lee, Y.J. , Hong, J.T. , Lee, H.J. , 2012. Antioxidant properties of natural polyphenols and their therapeutic potentials for Alzheimer's disease. Brain Res. Bull.. 87, 144–153. [DOI] [PubMed] [Google Scholar]
- Chu, Q. , Ling, M.T. , Feng, H. , Cheung, H.W. , Tsao, S.W. , Wang, X. , Wong, Y.C. , 2006. A novel anticancer effect of garlic derivatives: inhibition of cancer cell invasion through restoration of E-cadherin expression. Carcinogenesis. 27, 2180–2189. [DOI] [PubMed] [Google Scholar]
- Chua, H.L. , Bhat-Nakshatri, P. , Clare, S.E. , Morimiya, A. , Badve, S. , Nakshatri, H. , 2007. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 26, 711–724. [DOI] [PubMed] [Google Scholar]
- Colomiere, M. , Findlay, J. , Ackland, L. , Ahmed, N. , 2009. Epidermal growth factor-induced ovarian carcinoma cell migration is associated with JAK2/STAT3 signals and changes in the abundance and localization of alpha6beta1 integrin. Int. J. Biochem. Cell Biol.. 41, 1034–1045. [DOI] [PubMed] [Google Scholar]
- Creighton, C.J. , Chang, J.C. , Rosen, J.M. , 2010. Epithelial–mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J. Mammary Gland Biol. Neoplasia. 15, 253–260. [DOI] [PubMed] [Google Scholar]
- Creighton, C.J. , Li, X. , Landis, M. , Dixon, J.M. , Neumeister, V.M. , Sjolund, A. , Rimm, D.L. , Wong, H. , Rodriguez, A. , Herschkowitz, J.I. , Fan, C. , Zhang, X. , He, X. , Pavlick, A. , Gutierrez, M.C. , Renshaw, L. , Larionov, A.A. , Faratian, D. , Hilsenbeck, S.G. , Perou, C.M. , Lewis, M.T. , Rosen, J.M. , Chang, J.C. , 2009. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. U. S. A.. 106, 13820–13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave, B. , Mittal, V. , Tan, N.M. , Chang, J.C. , 2012. Epithelial–mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res.. 14, 202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried, L.E. , Arbiser, J.L. , 2009. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid. Redox Signal.. 11, 1139–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita, M. , Itokawa, H. , Sashida, Y. , 1973. Studies on the components of Magnolia obovata Thunb. 3. Occurrence of magnolol and honokiol in M. obovata and other allied plants. Yakugaku Zasshi. 93, 429–434. [DOI] [PubMed] [Google Scholar]
- Grandis, J.R. , Drenning, S.D. , Chakraborty, A. , Zhou, M.Y. , Zeng, Q. , Pitt, A.S. , Tweardy, D.J. , 1998. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J. Clin. Invest. 102, 1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, S.K. , Francia, G. , Isidoro, C. , Kerbel, R.S. , 2004. Antiadhesive antibodies targeting E-cadherin sensitize multicellular tumor spheroids to chemotherapy in vitro. Mol. Cancer Ther.. 3, 149–159. [PubMed] [Google Scholar]
- Haura, E.B. , Turkson, J. , Jove, R. , 2005. Mechanisms of disease: insights into the emerging role of signal transducers and activators of transcription in cancer. Nat. Clin. Pract. Oncol.. 2, 315–324. [DOI] [PubMed] [Google Scholar]
- Hibasami, H. , Achiwa, Y. , Katsuzaki, H. , Imai, K. , Yoshioka, K. , Nakanishi, K. , Ishii, Y. , Hasegawa, M. , Komiya, T. , 1998. Honokiol induces apoptosis in human lymphoid leukemia Molt 4B cells. Int. J. Mol. Med.. 2, 671–673. [DOI] [PubMed] [Google Scholar]
- Hirano, T. , Gotoh, M. , Oka, K. , 1994. Natural flavonoids and lignans are potent cytostatic agents against human leukemic HL-60 cells. Life Sci.. 55, 1061–1069. [DOI] [PubMed] [Google Scholar]
- Huber, M.A. , Azoitei, N. , Baumann, B. , Grunert, S. , Sommer, A. , Pehamberger, H. , Kraut, N. , Beug, H. , Wirth, T. , 2004. NF-kappaB is essential for epithelial–mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Invest.. 114, 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idowu, M.O. , Kmieciak, M. , Dumur, C. , Burton, R.S. , Grimes, M.M. , Powers, C.N. , Manjili, M.H. , 2012. CD44(+)/CD24(−/low) cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum. Pathol.. 43, 364–373. [DOI] [PubMed] [Google Scholar]
- Ishitsuka, K. , Hideshima, T. , Hamasaki, M. , Raje, N. , Kumar, S. , Hideshima, H. , Shiraishi, N. , Yasui, H. , Roccaro, A.M. , Richardson, P. , Podar, K. , Le Gouill, S. , Chauhan, D. , Tamura, K. , Arbiser, J. , Anderson, K.C. , 2005. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and -independent apoptosis. Blood. 106, 1794–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.H. , Nagalingam, A. , Saxena, N.K. , Singh, S.V. , Sharma, D. , 2011. Benzyl isothiocyanate inhibits oncogenic actions of leptin in human breast cancer cells by suppressing activation of signal transducer and activator of transcription 3. Carcinogenesis. 32, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. , Jung, E. , Park, J. , Jung, K. , Lee, S. , Hong, S. , Park, E. , Kim, J. , Park, S. , Park, D. , 2005. Anti-inflammatory effects of magnolol and honokiol are mediated through inhibition of the downstream pathway of MEKK-1 in NF-kappaB activation signaling. Planta Med.. 71, 338–343. [DOI] [PubMed] [Google Scholar]
- Lin, Y.R. , Chen, H.H. , Ko, C.H. , Chan, M.H. , 2005. Differential inhibitory effects of honokiol and magnolol on excitatory amino acid-evoked cation signals and NMDA-induced seizures. Neuropharmacology. 49, 542–550. [DOI] [PubMed] [Google Scholar]
- Lo, Y.C. , Teng, C.M. , Chen, C.F. , Chen, C.C. , Hong, C.Y. , 1994. Magnolol and honokiol isolated from Magnolia officinalis protect rat heart mitochondria against lipid peroxidation. Biochem. Pharmacol.. 47, 549–553. [DOI] [PubMed] [Google Scholar]
- Mani, S.A. , Guo, W. , Liao, M.J. , Eaton, E.N. , Ayyanan, A. , Zhou, A.Y. , Brooks, M. , Reinhard, F. , Zhang, C.C. , Shipitsin, M. , Campbell, L.L. , Polyak, K. , Brisken, C. , Yang, J. , Weinberg, R.A. , 2008. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell. 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta, L.L. , Almendro, V. , Marusyk, A. , Shipitsin, M. , Schemme, J. , Walker, S.R. , Bloushtain-Qimron, N. , Kim, J.J. , Choudhury, S.A. , Maruyama, R. , Wu, Z. , Gonen, M. , Mulvey, L.A. , Bessarabova, M.O. , Huh, S.J. , Silver, S.J. , Kim, S.Y. , Park, S.Y. , Lee, H.E. , Anderson, K.S. , Richardson, A.L. , Nikolskaya, T. , Nikolsky, Y. , Liu, X.S. , Root, D.E. , Hahn, W.C. , Frank, D.A. , Polyak, K. , 2011. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J. Clin. Invest.. 121, 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meydan, N. , Grunberger, T. , Dadi, H. , Shahar, M. , Arpaia, E. , Lapidot, Z. , Leeder, J.S. , Freedman, M. , Cohen, A. , Gazit, A. , Levitzki, A. , Roifman, C.M. , 1996. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 379, 645–648. [DOI] [PubMed] [Google Scholar]
- Morel, A.P. , Lievre, M. , Thomas, C. , Hinkal, G. , Ansieau, S. , Puisieux, A. , 2008. Generation of breast cancer stem cells through epithelial–mesenchymal transition. PLoS One. 3, e2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalingam, A. , Arbiser, J.L. , Bonner, M.Y. , Saxena, N.K. , Sharma, D. , 2012. Honokiol activates AMP-activated protein kinase in breast cancer cells via an LKB1-dependent pathway and inhibits breast carcinogenesis. Breast Cancer Res.. 14, R35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase, H. , Ikeda, K. , Sakai, Y. , 2001. Inhibitory effect of magnolol and honokiol from Magnolia obovata on human fibrosarcoma HT-1080. Invasiveness in vitro. Planta Med.. 67, 705–708. [DOI] [PubMed] [Google Scholar]
- Oh, J.H. , Kang, L.L. , Ban, J.O. , Kim, Y.H. , Kim, K.H. , Han, S.B. , Hong, J.T. , 2009. Anti-inflammatory effect of 4-O-methylhonokiol, compound isolated from Magnolia officinalis through inhibition of NF-kappaB [corrected]. Chem.-Biol. Interact.. 180, 506–514. [DOI] [PubMed] [Google Scholar]
- Prasad, C.P. , Rath, G. , Mathur, S. , Bhatnagar, D. , Parshad, R. , Ralhan, R. , 2009. Expression analysis of E-cadherin, Slug and GSK3beta in invasive ductal carcinoma of breast. BMC Cancer. 9, 325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky, D.C. , LaBarge, M.A. , 2008. Epithelial–mesenchymal transition and the stem cell phenotype. Cell Stem Cell. 2, 511–512. [DOI] [PubMed] [Google Scholar]
- Sanchez-Tillo, E. , Siles, L. , de Barrios, O. , Cuatrecasas, M. , Vaquero, E.C. , Castells, A. , Postigo, A. , 2011. Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am. J. Cancer Res.. 1, 897–912. [PMC free article] [PubMed] [Google Scholar]
- Santisteban, M. , Reiman, J.M. , Asiedu, M.K. , Behrens, M.D. , Nassar, A. , Kalli, K.R. , Haluska, P. , Ingle, J.N. , Hartmann, L.C. , Manjili, M.H. , Radisky, D.C. , Ferrone, S. , Knutson, K.L. , 2009. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res.. 69, 2887–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena, N.K. , Fu, P.P. , Nagalingam, A. , Wang, J. , Handy, J. , Cohen, C. , Tighiouart, M. , Sharma, D. , Anania, F.A. , 2010. Adiponectin modulates C-jun N-terminal kinase and mammalian target of rapamycin and inhibits hepatocellular carcinoma. Gastroenterology. 139, 1762–1773. 1773, 1761–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena, N.K. , Sharma, D. , Ding, X. , Lin, S. , Marra, F. , Merlin, D. , Anania, F.A. , 2007. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res.. 67, 2497–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena, N.K. , Vertino, P.M. , Anania, F.A. , Sharma, D. , 2007. Leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to CYCLIN D1 promoter via activation of Stat3. J. Biol. Chem.. 282, 13316–13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalhofer, O. , Brabletz, S. , Brabletz, T. , 2009. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev.. 28, 151–166. [DOI] [PubMed] [Google Scholar]
- Sehrawat, A. , Singh, S.V. , 2011. Benzyl isothiocyanate inhibits epithelial–mesenchymal transition in cultured and xenografted human breast cancer cells. Cancer Prev. Res. (Phila). 4, 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, D. , Saxena, N.K. , Davidson, N.E. , Vertino, P.M. , 2006. Restoration of tamoxifen sensitivity in estrogen receptor-negative breast cancer cells: tamoxifen-bound reactivated ER recruits distinctive corepressor complexes. Cancer Res.. 66, 6370–6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, D. , Wang, J. , Fu, P.P. , Sharma, S. , Nagalingam, A. , Mells, J. , Handy, J. , Page, A.J. , Cohen, C. , Anania, F.A. , Saxena, N.K. , 2010. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology. 52, 1713–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu, M.L. , Chiang, C.K. , Tsai, K.S. , Ho, F.M. , Weng, T.I. , Wu, H.Y. , Liu, S.H. , 2008. Inhibition of NADPH oxidase-related oxidative stress-triggered signaling by honokiol suppresses high glucose-induced human endothelial cell apoptosis. Free Radic. Biol. Med.. 44, 2043–2050. [DOI] [PubMed] [Google Scholar]
- Shigemura, K. , Arbiser, J.L. , Sun, S.Y. , Zayzafoon, M. , Johnstone, P.A. , Fujisawa, M. , Gotoh, A. , Weksler, B. , Zhau, H.E. , Chung, L.W. , 2007. Honokiol, a natural plant product, inhibits the bone metastatic growth of human prostate cancer cells. Cancer. 109, 1279–1289. [DOI] [PubMed] [Google Scholar]
- Shimono, Y. , Zabala, M. , Cho, R.W. , Lobo, N. , Dalerba, P. , Qian, D. , Diehn, M. , Liu, H. , Panula, S.P. , Chiao, E. , Dirbas, F.M. , Somlo, G. , Pera, R.A. , Lao, K. , Clarke, M.F. , 2009. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 138, 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, T. , Katiyar, S.K. , 2013. Honokiol inhibits non-small cell lung cancer cell migration by targeting PGE(2)-mediated activation of beta-catenin signaling. PLoS One. 8, e60749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, T. , Prasad, R. , Katiyar, S.K. , 2013. Inhibition of class I histone deacetylases in non-small cell lung cancer by honokiol leads to suppression of cancer cell growth and induction of cell death in vitro and in vivo. Epigenetics. 8, 54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinibaldi, D. , Wharton, W. , Turkson, J. , Bowman, T. , Pledger, W.J. , Jove, R. , 2000. Induction of p21WAF1/CIP1 and cyclin D1 expression by the Src oncoprotein in mouse fibroblasts: role of activated STAT3 signaling. Oncogene. 19, 5419–5427. [DOI] [PubMed] [Google Scholar]
- Song, H. , Wang, R. , Wang, S. , Lin, J. , 2005. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. U.S.A.. 102, 4700–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaferro-Smith, L. , Nagalingam, A. , Zhong, D. , Zhou, W. , Saxena, N.K. , Sharma, D. , 2009. LKB1 is required for adiponectin-mediated modulation of AMPK-S6K axis and inhibition of migration and invasion of breast cancer cells. Oncogene. 28, 2621–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery, J.P. , Acloque, H. , Huang, R.Y. , Nieto, M.A. , 2009. Epithelial–mesenchymal transitions in development and disease. Cell. 139, 871–890. [DOI] [PubMed] [Google Scholar]
- Tomaskovic-Crook, E. , Thompson, E.W. , Thiery, J.P. , 2009. Epithelial to mesenchymal transition and breast cancer. Breast Cancer Res.. 11, 213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse, A.K. , Wan, C.K. , Shen, X.L. , Yang, M. , Fong, W.F. , 2005. Honokiol inhibits TNF-alpha-stimulated NF-kappaB activation and NF-kappaB-regulated gene expression through suppression of IKK activation. Biochem. Pharmacol.. 70, 1443–1457. [DOI] [PubMed] [Google Scholar]
- Turkson, J. , 2004. STAT proteins as novel targets for cancer drug discovery. Expert Opin. Ther. Targets. 8, 409–422. [DOI] [PubMed] [Google Scholar]
- Turkson, J. , Jove, R. , 2000. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 19, 6613–6626. [DOI] [PubMed] [Google Scholar]
- Wang, T. , Chen, F. , Chen, Z. , Wu, Y.F. , Xu, X.L. , Zheng, S. , Hu, X. , 2004. Honokiol induces apoptosis through p53-independent pathway in human colorectal cell line RKO. World J. Gastroenterol. WJG. 10, 2205–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Duan, X. , Yang, G. , Zhang, X. , Deng, L. , Zheng, H. , Deng, C. , Wen, J. , Wang, N. , Peng, C. , Zhao, X. , Wei, Y. , Chen, L. , 2011. Honokiol crosses BBB and BCSFB, and inhibits brain tumor growth in rat 9L intracerebral gliosarcoma model and human U251 xenograft glioma model. PLoS One. 6, e18490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis, B.C. , Liebler, J.M. , Luby-Phelps, K. , Nicholson, A.G. , Crandall, E.D. , du Bois, R.M. , Borok, Z. , 2005. Induction of epithelial–mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am. J. Pathol.. 166, 1321–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi, S. , Gooding, W.E. , Grandis, J.R. , 2005. In vivo antitumor efficacy of STAT3 blockade using a transcription factor decoy approach: implications for cancer therapy. Oncogene. 24, 970–979. [DOI] [PubMed] [Google Scholar]
- Xiong, H. , Hong, J. , Du, W. , Lin, Y.W. , Ren, L.L. , Wang, Y.C. , Su, W.Y. , Wang, J.L. , Cui, Y. , Wang, Z.H. , Fang, J.Y. , 2012. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial–mesenchymal transition. J. Biol. Chem.. 287, 5819–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Q. , Yi, L.T. , Pan, Y. , Wang, X. , Li, Y.C. , Li, J.M. , Wang, C.P. , Kong, L.D. , 2008. Antidepressant-like effects of the mixture of honokiol and magnolol from the barks of Magnolia officinalis in stressed rodents. Prog. Neuropsychopharmacol. Biol. Psychiatry. 32, 715–725. [DOI] [PubMed] [Google Scholar]
- Yang, S.E. , Hsieh, M.T. , Tsai, T.H. , Hsu, S.L. , 2002. Down-modulation of Bcl-XL, release of cytochrome c and sequential activation of caspases during honokiol-induced apoptosis in human squamous lung cancer CH27 cells. Biochem. Pharmacol.. 63, 1641–1651. [DOI] [PubMed] [Google Scholar]
- Yu, H. , Jove, R. , 2004. The STATs of cancer – new molecular targets come of age. Nat. Rev. Cancer. 4, 97–105. [DOI] [PubMed] [Google Scholar]
- Yue, P. , Turkson, J. , 2009. Targeting STAT3 in cancer: how successful are we?. Expert Opin. Investig. Drugs. 18, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L.L. , Li, L. , Wu, D.P. , Fan, J.H. , Li, X. , Wu, K.J. , Wang, X.Y. , He, D.L. , 2008. A novel anti-cancer effect of genistein: reversal of epithelial mesenchymal transition in prostate cancer cells. Acta Pharmacol. Sin. 29, 1060–1068. [DOI] [PubMed] [Google Scholar]
- Zhuo, W. , Wang, Y. , Zhuo, X. , Zhang, Y. , Ao, X. , Chen, Z. , 2008. Knockdown of Snail, a novel zinc finger transcription factor, via RNA interference increases A549 cell sensitivity to cisplatin via JNK/mitochondrial pathway. Lung Cancer. 62, 8–14. [DOI] [PubMed] [Google Scholar]
- Zhuo, W.L. , Wang, Y. , Zhuo, X.L. , Zhang, Y.S. , Chen, Z.T. , 2008. Short interfering RNA directed against TWIST, a novel zinc finger transcription factor, increases A549 cell sensitivity to cisplatin via MAPK/mitochondrial pathway. Biochem. Biophys. Res. Commun.. 369, 1098–1102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data