

Abstract
Non-alcoholic fatty liver disease (NAFLD) is emerging as one of the most common liver diseases, leading to the increasing interest for new therapeutic approaches for its treatment. NAFLD primarily depends on a hypercaloric and/or unbalanced diet leading to overweight and obesity. The liver, in fact, plays a central role in lipid metabolism by importing free fatty acids from the blood and synthesizing, storing, oxidizing and exporting lipids. Furthermore, the liver is the target for the thyroid hormones, thyroxine (T4) and 3,3’,5-triiodo-L-thyronine (T3), that stimulate the basal metabolic rate and lead to body weight loss. In the last decade, other iodothyronines have been shown to possess biological relevance and play some thyromimetic activities; in particular, 3,5-diiodo-L-thyronine (T2) gained large interest. The global effect of iodothyronines on liver lipid metabolism results from the balance between direct and indirect actions on the hepatocyte, leading to stimulation of lipid synthesis, oxidation and autophagy. In this review, the results so far obtained on both in vivo and in vitro models of hepatosteatosis are summarized in order to obtain an updated picture of the lipid-lowering effects of iodothyronines on mammalian liver.
Keywords: Iodothyronines, Liver steatosis, Lipid metabolism, Non-alcoholic fatty liver disease, Hepatocytes
Core tip: This review summarizes the recent insights about the mechanisms underlying the lipid lowering action of iodothyronines. In the last decades, extensive studies investigated the possible use of iodothyronines in the treatment of obesity and dysmetabolic syndromes. Since the pharmacological use of thyroid hormones has found severe limitations because of their thyrotoxic effects, the identification of iodothyronines retaining anti-obesity and hypolipemic efficacies, while being devoid of thyrotoxicity, gained great interest. The review discusses the recent studies employing both in vivo and in vitro models of hepatosteatosis, with particular attention to the in vitro studies demonstrating the direct anti-steatotic effect of iodothyronines.
IODOTHYRONINES AND METABOLISM
Thyroid hormones (THs) secreted by the thyroid gland comprise two main iodothyronines: 3,5,3’,5’-tetraiodothyronine (thyroxine or T4) and 3,5,3’-triiodo-L-thyronine (T3) (Figure 1). T4 is the major form secreted by the thyroid and the most abundant TH in circulation, while T3, the active form, is mainly generated by peripheral deiodination of T4. T3 may be further deiodinated to yield different diiodothyronines such as 3,5-diiodo-L-thyronine (T2) (Figure 1). In the past, T3 was assumed to be the only active iodothyronine in vivo, but recent evidence suggested that other iodothyronines, such as 3’,5’,3-l-triiodothyronine (rT3) and T2, may be of biological relevance[1,2].
Figure 1.
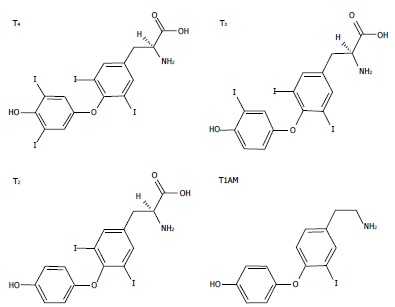
The chemical structures of three biologically active iodothyronines and one derivative: thyroxine, 3,3’,5-triiodothyronine, 3,5-diiodothyronine and 3-iodothyronamine. T4: Thyroxine; T3: 3,3’,5-triiodothyronine; T2: 3,5-diiodothyronine; T1AM: 3-iodothyronamine.
THs influence a large number of physiological processes in vertebrates, including growth, development and differentiation. THs have stimulatory effects on metabolic activity, thus inducing thermogenesis (the so-called calorigenic effect) that represents a major component of the energy expenditure in endotherms. Of particular interest is the effect of THs on lipid metabolism resulting from the balance between stimulation of lipid synthesis and lipid oxidation (Figure 2). The liver represents one of the main target tissues of THs. At the hepatic level, T3 stimulates cholesterol synthesis and its metabolism into bile acids[3] and cholesterol uptake[4], and it induces lipogenic enzymes, including fatty acid synthase (FAS) and acetyl-CoA-carboxylase[5]. In addition to the lipogenic action, T3 also leads to a general reduction in the hepatic triglyceride (TAG) content, likely through stimulation of lipolytic pathways[6]. Moreover, it has been suggested that TH-stimulated lipogenesis/lipolysis “futile” cycle may contribute to the calorigenic effect[7].
Figure 2.
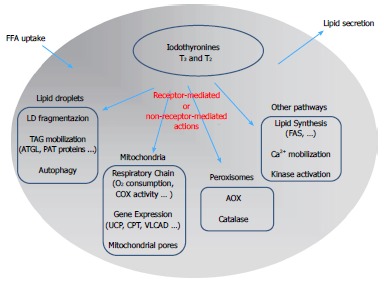
Schematic representation of the mechanisms underlying the control of lipid metabolism by iodothyronines in the hepatic cell: A summary of the possible signaling pathways involved in iodothyronine actions is presented. The classic “receptor-mediated” pathway describes the action of iodothyronines through the thyroid hormone receptors (TR). The “non receptor-mediated” pathway occurs through the interaction of iodothyronines with different cellular targets. FFA: Free fatty acids; LD: Lipid droplet; AOX: Acyl-CoA oxidase; ATGL: Adipose triglyceride lipase; CPT: Carnitine palmitoyl-transferase 1; FAS: fatty acid synthase.
The metabolic effects of iodothyronines have long been investigated because of their potential use as drugs to treat obesity and lipid metabolism disorders[8]. However, due to the simultaneous undesirable side effects, such as the induction of a thyrotoxic state (tachycardia, muscle wasting, bone loss), the employment of T3 or T4 to stimulate body weight loss or treat metabolic syndrome has been limited. At the same time, the development of TH agonists/analogs retaining lipid-lowering and anti-obesity efficacies, while being devoid of thyrotoxic effects, has received great interest as a potential therapeutic advancement. The research of the last years has identified several iodothyronines other than T3 and T4 that display some thyromimetic activities. Among them, T2 assumed a great interest as it mimics several effects of T3 on energy metabolism[9-11] without inducing thyrotoxic effects[12]. A single dose of T2 (25 μg/100 g body wt) stimulated the resting metabolic rate (RMR) of hypothyroid rats and increased the liver oxidative capacity to the same extent as the same dose of T3[13]. Moreover, T2 significantly reduced serum triglyceride and cholesterol levels and increased liver oxygen consumption[10].
With regards to the calorigenic effects of THs, several cellular targets have been proposed but none has received universal acceptance. By virtue of their central role in the energy-transduction pathway, mitochondria are natural candidates to mediate the calorigenic activity of iodothyronines[14]. Single injections of T2 or T3 into hypothyroid rats stimulated RMR[15], in association with an increase in oxygen consumption[16]. It is widely accepted that iodothyronines may exert two kinds of effects on mitochondria: (1) a rapid stimulation of respiration (within minutes/hours); and (2) a long term effect leading to mitochondrial biogenesis and mitochondrial mass increase. The calorigenic activity of THs has long been ascribed to uncoupling of mitochondrial oxidative phosphorylation, but the mode by which they promote mitochondrial proton leak is still unresolved. Harper et al[17] related the T3-induced increase in mitochondrial proton leak to an increased permeability of the phospholipid bilayer due to a change in the lipid composition of the inner mitochondrial membrane. On the other hand, T2, and to a lesser extent T3, was shown to bind the Va subunit of the cytochrome oxidase complex, thus abolishing the allosteric inhibition due to ATP binding and stimulating enzyme activity[18]. Recently, Yehuda-Shnaidman et al[19] reported that mitochondrial uncoupling by T3 was transduced both in vivo (in rats) and in vitro (Jurkat cells) by gating of the mitochondrial permeability transition pore.
Interestingly, T2 administration has been demonstrated to be able to stimulate RMR and to also reduce body weight in humans. In a pilot study, two euthyroid subjects were treated with increasing doses of T2 (from 100 to 900 mcg/d, three times a day for 8 d) and for a further 3 weeks with 3000 mcg/d. A reduction in body weight of -4% was observed without effects at the cardiac level[20].
The pharmacological effects of some derivatives of thyronines called thyronamines have been also investigated. Scanlan et al[21] described the synthesis and biological properties of 3-iodothyronamine (T1AM), a novel thyronamine that was shown to be an endogenous component of biogenic amine extracts from rodents. T1AM has a carbon skeleton identical to that of T4 and theoretically, it could be produced from T4 by enzymatic decarboxylation and deiodination (Figure 1). T1AM treatment rapidly induced a hypometabolic state and hypothermia in rodents, with opposite effects compared with those typical of THs.
MECHANISMS OF ACTION OF IODOTHYRONINES
In the past, it was a common notion that TH actions were mediated by specific nuclear thyroid hormone receptors (TRs) acting as ligand-dependent transcription factors binding the “thyroid hormone response elements” (TREs) on the promoter region of thyroid hormone-responsive genes[22]. In the early 1960s, Tata and co-workers provided the first evidence for a “receptor-mediated” mechanism of T3 action on energy metabolism[23]. In the 1980s, two distinct genes, THRA and THRB, were identified in humans and rodents, each encoding a different TR isoform (TRα and TRβ, respectively). The THRA gene was originally identified in chicken[24], while THRB was cloned from human and rat cDNA libraries[25]. Each isoform shows alternative splice variants (TRα1, TRα2, TRβ1 and TRβ2) with specific and distinct functions and tissue localization.
Although the “receptor-mediated” mechanism accounts for several actions of THs, other effects independent of TRs have been described, suggesting an alternative model for their action. Effects of iodothyronines that are not initiated by binding to TRs are termed ‘non-receptor-mediated’ mechanisms[26] and could involve a multiplicity of signaling pathways, such as phosphorylation of effector proteins[27], binding to surface receptors[28], Ca2+ mobilization[29], alteration of mRNA stability[30], modification of membrane fluidity and permeability[2]. The possibility that TH action was mediated by interactions with membrane surface receptors was confirmed by using cell impermeant agarose-conjugated T3. The results clearly indicated that both free and conjugated hormones led to activation of extracellular signal-regulated kinase (ERK1/2s)[31] and affected Ca2+ homeostasis[32]. Moreover, THs were shown to interact with theαVβ3 integrin receptor triggering the ERK1/2 pathway[33]. Although the “non-receptor mediated” effects are sometimes called “non-genomic”, this term is rather confusing as these pathways may also in turn affect gene transcription [34].
In conclusion, it is now widely accepted that TH effects may result from a synergism between “receptor-mediated” and “non-receptor mediated” mechanisms. Moreover, we can distinguish between early and late effects of THs (also called “short-term” and “long-term” effects), the first being evident within minutes or a few hours, whereas the second occurs over several hours or days[34,35]. However, the latency of a response is not sufficient to discriminate between “receptor-mediated” and “non-receptor” mediated effects.
HEPATIC STEATOSIS: IN VITRO AND IN VIVO MODELS
With the rapidly growing prevalence of obesity throughout the Western countries, morbidity and mortality related to its complications are on the rise. Severe obesity is generally associated with TAG accumulation in non-adipose tissues like liver, muscle and pancreas and leads to a high risk of co-morbidities, including nonalcoholic fatty liver disease (NAFLD), cardiovascular disease and diabetes (for a review see[36]). NAFLD is a pathological condition associated with over-accumulation of TAGs in the liver and represents the most common of all hepatic disorders and the most frequent cause of chronic liver disease[37,38]. The earliest stage of NAFLD is hepatic steatosis characterized by the deposition of cytoplasmic TAGs as macro- and/or micro-vesicular lipid droplets in more than 5% of hepatocytes. Simple steatosis may progress to nonalcoholic steatohepatitis (NASH), cirrhosis and finally hepatocellular carcinoma[39]. NAFLD is now considered the hepatic manifestation of the metabolic syndrome and has insulin resistance as its hallmark. NAFLD is a syndrome with multifactorial etiology for which there is no effective treatment, although weight loss may halt disease progression and revert histological changes[36].
In hepatocytes, steatosis results from an imbalance between lipid availability (deriving from circulating lipid uptake or de novo lipid synthesis) and lipid disposal (through FFA oxidation or TAG secretion)[40]. Typically, the main cause of steatosis is an overflow of free fatty acids (FFAs) into the liver that may eventually trigger lipoperoxidative stress and hepatic injury[39,41]. In the liver, FFAs are stored as TAGs through their esterification with glycerol or, alternatively, catabolized by oxidation to generate adenosine triphosphate (ATP). Excess TAGs are accumulated inside lipid droplets (LDs) that regulate storage and traffic of lipids (for a review see[42]). Typically, LDs are composed of a core of neutral lipids surrounded by phospholipids and proteins of the PAT protein family (acronym referring to the first members identified)[43]. The main PAT proteins are the adipocyte differentiation-related protein (ADRP, also called PLIN2), the oxidative tissue-enriched PAT protein (OXPAT or PLIN5) and the tail-interacting protein (TIP47 or PLIN3)[44]. ADRP expression is increased in rat models of NAFLD and in isolated hepatocytes[45]. PAT proteins are under the control of peroxisome proliferator-activated receptors (PPARs), a subfamily of lipid-activated transcription factors[46] consisting of three members, PPARα, PPARγ and PPARδ, with distinct functional roles[47,48]. In the liver, PPARα enhances lipid catabolism and mobilization[49], PPARδ induces glycolysis/lipogenesis and PPARγ promotes lipid synthesis and LD formation[50]. In summary, PPARα and PPARδ mainly act in energy burning, whereas PPARγ regulates energy storage, although an overlapping in their function has been described[40-51]. Moreover, PAT proteins regulate action of hepatic lipases that mobilize TAGs stored in LDs towards oxidation or secretion[52], in particular, the adipose triglyceride lipase (ATGL) performs the first step in TAG hydrolysis.
In vivo models
Steatosis and steatohepatitis can be modeled in rodents by two main dietary protocols: a methionine and choline deficient (MCD) diet or a high-fat diet (HFD). Different dietary approaches produce different disease severities and work by specific mechanisms[53]. In rodents, a MCD diet quickly induces (2-4 wk) hepatic steatosis (mainly macrovesicular) that may progress to inflammation and fibrosis. MCD diet-induced NASH is reversible by switching to a diet with methionine and choline. Rodents fed MCD diets lose weight (due to the lower caloric intake) and do not show insulin resistance. By contrast, HFD increases body weight, body fat and induces insulin resistance in rodent models. In general, HFD feeding induces only mild steatosis (mainly microvesicular) and does not produce liver fibrosis. The term “HFD” encompasses a wide variety of diet formulas but in all of them about 30%-75% of total calories is derived from saturated fatty acids. This diet closely resembles the pathological and molecular alterations found in humans with NAFLD[53]. It can be emphasized that fatty liver is typically characterized by altered lipid metabolism, increased oxidative stress and abnormal pattern of cytokine production.
In vitro models
Hepatic steatosis in humans is typically associated with excess accumulation of oleic acid, a monounsaturated omega-9 fatty acid which represents the end product of de novo fatty acid synthesis. A number of studies using both primary cell cultures[54] and immortalized cell lines[55,56] proposed reliable cell models of hepatosteatosis in which the steatosis severity might be modulated and the TAG content was exactly quantifiable. These in vitro models represent a simple experimental system to investigate the mechanisms underlying the steatosis progression and the hepatocyte alterations by excluding the interference from the matrix and other non-hepatocytic cells. Over the past decade, several cellular models of hepatosteatosis have employed palmitate (C16:0) and oleate (C18:1) as exogenous fatty acids since these are common dietary long-chain FFAs and the most abundant FFAs in liver in both normal subjects and patients with NAFLD[57]. The human hepatoma cell line (HepG2) incubated with a mixture of oleate/palmitate (2:1 ratio) was used to study the cellular mechanisms involved in FFA-mediated lipotoxicity[55,58]. The same FFA mixture was used to induce steatosis in primary human hepatocytes[58]. In order to assess the different toxicity of saturated and unsaturated FFAs, primary mice hepatocytes and HepG2 cells were treated with various concentrations (0.05-0.5 mmol/L) of long chain FFAs with different degrees of saturation; exposure to monounsaturated fatty acids resulted in lipid accumulation without changes in hepatocyte viability; in contrast, saturated fatty acids significantly decreased cell viability[59]. The effect of increasing concentrations of oleate alone (0.1-2.0 mmol/L) was also evaluated in order to clarify the pathophysiological changes associated with NAFLD[60].
LIPID-LOWERING EFFECTS OF IODOTHYRONINES ON IN VIVO MODELS OF HEPATOSTEATOSIS
In 1994, a first study reported that a daily intraperitoneal (ip) injection of T3 (from 0 to 25 μg/100 g b.w.) to ob/ob mice decreased body weight and body fat and increased oxygen consumption and oxidative metabolism[61]. About ten years later, Goglia and coworkers described similar effects for T2[10]. They showed that a daily ip injection of T2 (25 μg/100 g b.w.) to rats simultaneously receiving HFD reduced both adiposity (about -50%) and body weight gain (about -13%) when compared with rats receiving HFD alone. Moreover, T2 administration resulted in an almost complete disappearance of fat accumulation in the liver, a reduction in serum TAG and cholesterol levels (-52% and -18%, respectively), and a stimulation (about +42%) of FFA oxidation rate without inducing thyrotoxicity [10]. The effects of T2 on liver metabolism seemed to involve mitochondria, even although peroxisomes are the main site for fat oxidation. In fact, long chain FFAs enter mitochondria through the activity of carnitine palmitoyl-transferase 1 (CPT1) that was stimulated by HFD and further increased by T2 [10].
Interestingly, dietary administration of T3 was also able to both prevent and reverse hepatic steatosis in rats[62]. In fact, concurrent dietary administration of T3 and MCD diet resulted in prevention of fatty liver and decrease in lipid peroxidation in rats fed a MCD diet for 10 weeks and then co-fed T3 for 1 week. Similar effects were observed using the potent TR selective agonist GC-1 [62].
The hepatic effects of T2 administration to HFD rats were investigated in more detail by Grasselli et al[63,64]. HFD feeding resulted in hepatic lipid accumulation under the form of numerous LDs, a condition resembling the microvesicular steatosis typical of NAFLD. Fat accumulation was associated with increased transcription of PPARα, a regulator for a number of genes involved in FFA catabolism, and of ATGL, a lipase mobilizing fat from LDs, together with a stimulation of anti-oxidant agents such as catalase and metallothioneins, in line with the increased production of reactive oxygen species (ROS) from mitochondria and peroxisomes as a consequence of fat accumulation[65]. In the liver of HFD rats, concomitant T2 administration was able to prevent lipid accumulation, but also oxidative stress conditions associated with the diet[63]. Moreover, T2 prevented the HFD-induced up-regulation of both PPARα and ATGL and stimulated - (AOX) expression, indicating a stimulation of peroxisomal FFA oxidation[64].
In addition to the above described reports demonstrating the ability of T2 to prevent liver steatosis when administered simultaneously to HFD, other studies demonstrated that T2 was also able to reverse hepatic steatosis after its induction through long term HFD and these effects were associated with a stimulation of mitochondrial uncoupling and a reduction in mitochondrial oxidative stress[66].
A recent paper investigated the changes in the rat liver proteome induced by T2 treatment. The proteomic approach allowed identification of which proteins were differentially expressed in the liver of HFD rats as a function of T2 treatment[67]. Upon T2 administration, the rat liver proteome resembled that typical of a non-steatotic condition. In particular, high-fat feeding led to changes in the expressions of enzymes involved in a multiplicity of pathways (i.e., lipid metabolism, antioxidant defense, respiratory chain, oxidative metabolism). Mitochondria, in particular, appeared as the major target for the metabolic/energy adaptations induced by lipid overload in the liver and showed the more marked changes in terms of proteome as a response to T2 treatment. In mitochondria from HFD rats, enhanced activities of complexes I and V and reduced activities of complexes II and IV were detected, even although the protein levels for all the complexes were increased. T2-treatment stimulated complexes I and II and normalized complex IV activity. On this basis, the authors suggest that the T2-induced enhancement of oxidative capacity may actually be based on a stimulation of the individual respiratory chain complexes (I, II and IV)[67].
LIPID LOWERING EFFECTS OF IODOTHYRONINES ON IN VITRO MODELS OF HEPATOSTEATOSIS
The above described in vivo studies could not distinguish between the direct antisteatosic effects of THs on the liver and their secondary effects due to upstream changes in endocrine or metabolic pathways. The employment of isolated hepatocytes allowed overcoming these problems.
Grasselli et al[51] assessed in vitro the direct effects of T2 and T3 (10-7-10-5 mol/L doses for 24 h) using primary cultures of rat hepatocytes overloaded of lipids (“steatotic” hepatocytes) by exposure to the classical oleate/palmitate (2:1 ratio) mixture. The use of supraphysiological doses of iodothyronines depends on both their rapid metabolism in vitro and on their binding to the high concentration (1%) of albumin present in the culture medium. In accordance with reports showing altered expression of PPARs in murine models developing fatty livers[47], isolated “steatotic” hepatocytes exhibited increased expression of both PPAR-γ and PPAR-δ, as well as of ADRP, a PPAR-regulated PAT protein. As in liver of HFD rats[64], also in isolated “steatotic” hepatocytes an increased activity of AOX, the enzyme catalyzing peroxisomal β-oxidation, as well as of SOD and catalase, two antioxidant enzymes protecting cells from the higher ROS production associated with FFA catabolism, was described. A reduction in the number and average sizes of LDs was observed after treatment with T2 or T3, suggesting that iodothyronines lead to dispersion/fragmentation of LDs, thus making the stored TAGs more accessible to enzymes acting on catabolism/secretion of FFAs. Moreover, both T2 and T3 were able to reduce the FFA-induced up-regulation of PPARγ and PPARδ, the stimulation of AOX, SOD and catalase activities. These results clearly indicate the lipid-lowering effect of iodothyronines mainly depends on a direct action on the hepatic cell[51].
The use of primary rat hepatocytes allowed verification that the lipid-lowering effect of iodothyronines was a direct action on the hepatocyte but the involvement of thyroid hormone receptors in mediating this action remained to be elucidated. To this end, the same experiments were repeated using the FaO rat hepatoma cell line defective for functional TRs. FaO cells were exposed to the classical oleate/palmitate (2:1) mixture and then treated with T2 or T3 for 24 h (10-7-10-5 mol/L doses). In FaO cells, TAG accumulation was associated with an increase in number and size of LDs and in PPARγ mRNA expression. The addition of T2 or T3 to “steatotic” cells reduced both the TAG content and the number and size of LDs and down-regulated expression of PPARα and PPARγ. Moreover, iodothyronines stimulated the fuel-induced O2 consumption. Since iodothyronines prevented the ADP-induced transient stimulation of O2 consumption, this indicated a mitochondrial uncoupling action. In conclusion, this study demonstrated that the lipid-lowering actions of both T2 and T3 on the hepatocyte occur via “non-receptor-mediated” mechanisms and involve a short-term action by stimulation of mitochondrial O2 consumption[68].
IODOTHYRONINES AND AUTOPHAGY OF LIPID DROPLETS
Despite the advances in the understanding of the effects of THs on cellular metabolism, little is known about the mechanisms by which THs regulate energy consumption within the cell. This is particularly true for the events involved in the delivery of FFAs to mitochondria, a necessary step in converting stored intracellular triglyceride fuel into ATP.
Autophagy is a stress-induced catabolic process involving lysosome fusion that is conserved in almost all eukaryotes. Autophagy of lipid droplets, termed “lipophagy”, has been shown to be a major pathway of lipid mobilization in hepatocytes[69] and its inhibition has been linked to development of fatty liver and insulin resistance[70]. The regulation of autophagy also appears to be important in the context of metabolic diseases, such as obesity. In a recent paper, Sinha et al[71] showed that T3 induced both lipophagy in cultured liver cell lines and hepatic autophagy in the mouse liver. The authors observed that the T3-stimulated autophagy of LDs depends on the presence of functional TRs and occurred before any stimulation of hepatic lipases or oxidation enzyme[71]. Moreover, in animals with impaired autophagy, the effect of THs on FFA oxidation was abolished. Therefore, they propose that T3 may increase the delivery of FFAs to mitochondria for β-oxidation through induction of autophagy of LDs. In this light, T3 or its analogs, through their proautophagic action, may be useful in the treatment or prevention of NAFLD and its associated complications.
CONCLUSION
In the last decades, extensive studies investigated the possible use of iodothyronines as pharmacological tools in the treatment of obesity, hyperlipidemia and dysmetabolic syndromes. The possible pharmacological use of the thyroid hormones T3 or T4 to stimulate body weight loss has found severe limitations because of the thyrotoxic effects associated with their long-term administration. For this reason, the identification of TH agonists/analogs retaining anti-obesity and hypolipemic efficacies, while being devoid of thyrotoxic effects, would represent a potential therapeutic advance.
Recent in vivo and in vitro studies have accumulated evidence on the lipid-lowering action of iodothyronines in the liver (Figure 2). The first studies showed that systemic administration of iodothyronines to rats receiving HFD resulted in a significant reduction in body weight gain and in the serum levels of triglycerides and cholesterol. At the organ level, the effects on the liver were very interesting, where iodothyronines could lower the excess lipid accumulation associated with HFD. These studies prosecuted by investigating the mechanisms of iodothyronine action. The development of in vitro models of hepatosteatosis using both primary cultures of rat hepatocytes and rat hepatoma cell lines allowed demonstration that the lipid lowering effects of iodothyronines depend on a direct interaction with the hepatic cell and is not mediated by thyroid hormone receptors. In conclusion, all the data summarized in this review clearly indicates that T2 is able to reduce the lipid content of “steatotic hepatocytes”, thus supporting the possible utilization of T2 as a pharmacological tool in the treatment of dysmetabolic syndromes, such as NAFLD, and also in the light of its lack of thyrotoxic effects.
Although a preliminary study on humans has been published, clinical trials are needed to translate these effects to the treatment of human obesity. If reproduced in humans, these results may offer an interesting perspective on the possible pharmacological approaches to the above mentioned lifestyle-related dysfunctions.
ACKNOWLEDGMENTS
The author wishes to thank Elena Grasselli, Fernando Goglia, Adriana Voci, Laura Canesi and Gabriella Gallo for valuable discussion and their contributions are cited in the article.
Footnotes
P- Reviewers: Goglia F, Hutz RJ, Kim JB S- Editor: Song XX L- Editor: Roemmele A E- Editor: Wu HL
References
- 1.Goglia F. Biological effects of 3,5-diiodothyronine (T(2)) Biochemistry (Mosc) 2005;70:164–172. doi: 10.1007/s10541-005-0097-0. [DOI] [PubMed] [Google Scholar]
- 2.Hulbert AJ. Thyroid hormones and their effects: a new perspective. Biol Rev Camb Philos Soc. 2000;75:519–631. doi: 10.1017/s146479310000556x. [DOI] [PubMed] [Google Scholar]
- 3.Ness GC, Lopez D. Transcriptional regulation of rat hepatic low-density lipoprotein receptor and cholesterol 7 alpha hydroxylase by thyroid hormone. Arch Biochem Biophys. 1995;323:404–408. doi: 10.1006/abbi.1995.0061. [DOI] [PubMed] [Google Scholar]
- 4.Lopez D, Abisambra Socarrás JF, Bedi M, Ness GC. Activation of the hepatic LDL receptor promoter by thyroid hormone. Biochim Biophys Acta. 2007;1771:1216–1225. doi: 10.1016/j.bbalip.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Cunningham BA, Moncur JT, Huntington JT, Kinlaw WB. “Spot 14” protein: a metabolic integrator in normal and neoplastic cells. Thyroid. 1998;8:815–825. doi: 10.1089/thy.1998.8.815. [DOI] [PubMed] [Google Scholar]
- 6.Jackson-Hayes L, Song S, Lavrentyev EN, Jansen MS, Hillgartner FB, Tian L, Wood PA, Cook GA, Park EA. A thyroid hormone response unit formed between the promoter and first intron of the carnitine palmitoyltransferase-Ialpha gene mediates the liver-specific induction by thyroid hormone. J Biol Chem. 2003;278:7964–7972. doi: 10.1074/jbc.M211062200. [DOI] [PubMed] [Google Scholar]
- 7.Oppenheimer JH, Schwartz HL, Lane JT, Thompson MP. Functional relationship of thyroid hormone-induced lipogenesis, lipolysis, and thermogenesis in the rat. J Clin Invest. 1991;87:125–132. doi: 10.1172/JCI114961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pucci E, Chiovato L, Pinchera A. Thyroid and lipid metabolism. Int J Obes Relat Metab Disord. 2000;24 Suppl 2:S109–S112. doi: 10.1038/sj.ijo.0801292. [DOI] [PubMed] [Google Scholar]
- 9.Horst C, Rokos H, Seitz HJ. Rapid stimulation of hepatic oxygen consumption by 3,5-di-iodo-L-thyronine. Biochem J. 1989;261:945–950. doi: 10.1042/bj2610945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lanni A, Moreno M, Lombardi A, de Lange P, Silvestri E, Ragni M, Farina P, Baccari GC, Fallahi P, Antonelli A, et al. 3,5-diiodo-L-thyronine powerfully reduces adiposity in rats by increasing the burning of fats. FASEB J. 2005;19:1552–1554. doi: 10.1096/fj.05-3977fje. [DOI] [PubMed] [Google Scholar]
- 11.O’Reilly I, Murphy MP. Treatment of hypothyroid rats with T2 (3,5-di-iodo-L-thyronine) rapidly stimulates respiration in subsequently isolated mitochondria. Biochem Soc Trans. 1992;20:59S. doi: 10.1042/bst020059s. [DOI] [PubMed] [Google Scholar]
- 12.Cimmino M, Mion F, Goglia F, Minaire Y, Géloën A. Demonstration of in vivo metabolic effects of 3,5-di-iodothyronine. J Endocrinol. 1996;149:319–325. doi: 10.1677/joe.0.1490319. [DOI] [PubMed] [Google Scholar]
- 13.Lanni A, Moreno M, Cioffi M, Goglia F. Effect of 3,3’-diiodothyronine and 3,5-diiodothyronine on rat liver oxidative capacity. Mol Cell Endocrinol. 1992;86:143–148. doi: 10.1016/0303-7207(92)90138-v. [DOI] [PubMed] [Google Scholar]
- 14.Harper ME, Seifert EL. Thyroid hormone effects on mitochondrial energetics. Thyroid. 2008;18:145–156. doi: 10.1089/thy.2007.0250. [DOI] [PubMed] [Google Scholar]
- 15.Lanni A, Moreno M, Lombardi A, Goglia F. Calorigenic effect of diiodothyronines in the rat. J Physiol. 1996;494(Pt 3):831–837. doi: 10.1113/jphysiol.1996.sp021536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kvetny J. 3,5-T2 stimulates oxygen consumption, but not glucose uptake in human mononuclear blood cells. Horm Metab Res. 1992;24:322–325. doi: 10.1055/s-2007-1003323. [DOI] [PubMed] [Google Scholar]
- 17.Harper ME, Brand MD. The quantitative contributions of mitochondrial proton leak and ATP turnover reactions to the changed respiration rates of hepatocytes from rats of different thyroid status. J Biol Chem. 1993;268:14850–14860. [PubMed] [Google Scholar]
- 18.Arnold S, Goglia F, Kadenbach B. 3,5-Diiodothyronine binds to subunit Va of cytochrome-c oxidase and abolishes the allosteric inhibition of respiration by ATP. Eur J Biochem. 1998;252:325–330. doi: 10.1046/j.1432-1327.1998.2520325.x. [DOI] [PubMed] [Google Scholar]
- 19.Yehuda-Shnaidman E, Kalderon B, Azazmeh N, Bar-Tana J. Gating of the mitochondrial permeability transition pore by thyroid hormone. FASEB J. 2010;24:93–104. doi: 10.1096/fj.09-133538. [DOI] [PubMed] [Google Scholar]
- 20.Antonelli A, Fallahi P, Ferrari SM, Di Domenicantonio A, Moreno M, Lanni A, Goglia F. 3,5-diiodo-L-thyronine increases resting metabolic rate and reduces body weight without undesirable side effects. J Biol Regul Homeost Agents. 2011;25:655–660. [PubMed] [Google Scholar]
- 21.Scanlan TS, Suchland KL, Hart ME, Chiellini G, Huang Y, Kruzich PJ, Frascarelli S, Crossley DA, Bunzow JR, Ronca-Testoni S, et al. 3-Iodothyronamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat Med. 2004;10:638–642. doi: 10.1038/nm1051. [DOI] [PubMed] [Google Scholar]
- 22.Bassett JH, Harvey CB, Williams GR. Mechanisms of thyroid hormone receptor-specific nuclear and extra nuclear actions. Mol Cell Endocrinol. 2003;213:1–11. doi: 10.1016/j.mce.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 23.Tata JR, ERNSTER L, LINDBERG O, ARRHENIUS E, PEDERSEN S, HEDMAN R. The action of thyroid hormones at the cell level. Biochem J. 1963;86:408–428. doi: 10.1042/bj0860408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sap J, Muñoz A, Damm K, Goldberg Y, Ghysdael J, Leutz A, Beug H, Vennström B. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature. 1986;324:635–640. doi: 10.1038/324635a0. [DOI] [PubMed] [Google Scholar]
- 25.Weinberger C, Thompson CC, Ong ES, Lebo R, Gruol DJ, Evans RM. The c-erb-A gene encodes a thyroid hormone receptor. Nature. 1986;324:641–646. doi: 10.1038/324641a0. [DOI] [PubMed] [Google Scholar]
- 26.Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–170. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shih A, Zhang S, Cao HJ, Tang HY, Davis FB, Davis PJ, Lin HY. Disparate effects of thyroid hormone on actions of epidermal growth factor and transforming growth factor-alpha are mediated by 3’,5’-cyclic adenosine 5’-monophosphate-dependent protein kinase II. Endocrinology. 2004;145:1708–1717. doi: 10.1210/en.2003-0742. [DOI] [PubMed] [Google Scholar]
- 28.Davis PJ, Davis FB, Cody V. Membrane receptors mediating thyroid hormone action. Trends Endocrinol Metab. 2005;16:429–435. doi: 10.1016/j.tem.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 29.Del Viscovo A, Secondo A, Esposito A, Goglia F, Moreno M, Canzoniero LM. Intracellular and plasma membrane-initiated pathways involved in the [Ca2+]i elevations induced by iodothyronines (T3 and T2) in pituitary GH3 cells. Am J Physiol Endocrinol Metab. 2012;302:E1419–E1430. doi: 10.1152/ajpendo.00389.2011. [DOI] [PubMed] [Google Scholar]
- 30.Serrano-Nascimento C, Calil-Silveira J, Nunes MT. Posttranscriptional regulation of sodium-iodide symporter mRNA expression in the rat thyroid gland by acute iodide administration. Am J Physiol Cell Physiol. 2010;298:C893–C899. doi: 10.1152/ajpcell.00224.2009. [DOI] [PubMed] [Google Scholar]
- 31.Lin HY, Davis FB, Gordinier JK, Martino LJ, Davis PJ. Thyroid hormone induces activation of mitogen-activated protein kinase in cultured cells. Am J Physiol. 1999;276:C1014–C1024. doi: 10.1152/ajpcell.1999.276.5.C1014. [DOI] [PubMed] [Google Scholar]
- 32.Hummerich H, Soboll S. Rapid stimulation of calcium uptake into rat liver by L-tri-iodothyronine. Biochem J. 1989;258:363–367. doi: 10.1042/bj2580363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. J Biol Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 34.Davis PJ, Davis FB. Nongenomic actions of thyroid hormone on the heart. Thyroid. 2002;12:459–466. doi: 10.1089/105072502760143827. [DOI] [PubMed] [Google Scholar]
- 35.Soboll S. Thyroid hormone action on mitochondrial energy transfer. Biochim Biophys Acta. 1993;1144:1–16. doi: 10.1016/0005-2728(93)90024-a. [DOI] [PubMed] [Google Scholar]
- 36.Mirza MS. Obesity, Visceral Fat, and NAFLD: Querying the Role of Adipokines in the Progression of Nonalcoholic Fatty Liver Disease. ISRN Gastroenterol. 2011;2011:592404. doi: 10.5402/2011/592404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Youssef WI, McCullough AJ. Steatohepatitis in obese individuals. Best Pract Res Clin Gastroenterol. 2002;16:733–747. doi: 10.1053/bega.2002.0334. [DOI] [PubMed] [Google Scholar]
- 38.Adams LA, Angulo P. Recent concepts in non-alcoholic fatty liver disease. Diabet Med. 2005;22:1129–1133. doi: 10.1111/j.1464-5491.2005.01748.x. [DOI] [PubMed] [Google Scholar]
- 39.Bradbury MW. Lipid metabolism and liver inflammation. I. Hepatic fatty acid uptake: possible role in steatosis. Am J Physiol Gastrointest Liver Physiol. 2006;290:G194–G198. doi: 10.1152/ajpgi.00413.2005. [DOI] [PubMed] [Google Scholar]
- 40.Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD) Prog Lipid Res. 2009;48:1–26. doi: 10.1016/j.plipres.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olofsson SO, Boström P, Andersson L, Rutberg M, Perman J, Borén J. Lipid droplets as dynamic organelles connecting storage and efflux of lipids. Biochim Biophys Acta. 2009;1791:448–458. doi: 10.1016/j.bbalip.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Bickel PE, Tansey JT, Welte MA. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta. 2009;1791:419–440. doi: 10.1016/j.bbalip.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimmel AR, Brasaemle DL, McAndrews-Hill M, Sztalryd C, Londos C. Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT-family of intracellular lipid storage droplet proteins. J Lipid Res. 2010;51:468–471. doi: 10.1194/jlr.R000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grasselli E, Voci A, Pesce C, Canesi L, Fugassa E, Gallo G, Vergani L. PAT protein mRNA expression in primary rat hepatocytes: Effects of exposure to fatty acids. Int J Mol Med. 2010;25:505–512. doi: 10.3892/ijmm_00000370. [DOI] [PubMed] [Google Scholar]
- 46.Motomura W, Inoue M, Ohtake T, Takahashi N, Nagamine M, Tanno S, Kohgo Y, Okumura T. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem Biophys Res Commun. 2006;340:1111–1118. doi: 10.1016/j.bbrc.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 47.Everett L, Galli A, Crabb D. The role of hepatic peroxisome proliferator-activated receptors (PPARs) in health and disease. Liver. 2000;20:191–199. doi: 10.1034/j.1600-0676.2000.020003191.x. [DOI] [PubMed] [Google Scholar]
- 48.Patsouris D, Reddy JK, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology. 2006;147:1508–1516. doi: 10.1210/en.2005-1132. [DOI] [PubMed] [Google Scholar]
- 49.Pyper SR, Viswakarma N, Yu S, Reddy JK. PPARalpha: energy combustion, hypolipidemia, inflammation and cancer. Nucl Recept Signal. 2010;8:e002. doi: 10.1621/nrs.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 51.Grasselli E, Voci A, Canesi L, De Matteis R, Goglia F, Cioffi F, Fugassa E, Gallo G, Vergani L. Direct effects of iodothyronines on excess fat storage in rat hepatocytes. J Hepatol. 2011;54:1230–1236. doi: 10.1016/j.jhep.2010.09.027. [DOI] [PubMed] [Google Scholar]
- 52.Zimmermann R, Lass A, Haemmerle G, Zechner R. Fate of fat: the role of adipose triglyceride lipase in lipolysis. Biochim Biophys Acta. 2009;1791:494–500. doi: 10.1016/j.bbalip.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 53.Kanuri G, Bergheim I. In Vitro and in Vivo Models of Non-Alcoholic Fatty Liver Disease (NAFLD) Int J Mol Sci. 2013;14:11963–11980. doi: 10.3390/ijms140611963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vinciguerra M, Sgroi A, Veyrat-Durebex C, Rubbia-Brandt L, Buhler LH, Foti M. Unsaturated fatty acids inhibit the expression of tumor suppressor phosphatase and tensin homolog (PTEN) via microRNA-21 up-regulation in hepatocytes. Hepatology. 2009;49:1176–1184. doi: 10.1002/hep.22737. [DOI] [PubMed] [Google Scholar]
- 55.Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185–194. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 56.Giudetti AM, Leo M, Geelen MJ, Gnoni GV. Short-term stimulation of lipogenesis by 3,5-L-diiodothyronine in cultured rat hepatocytes. Endocrinology. 2005;146:3959–3966. doi: 10.1210/en.2005-0345. [DOI] [PubMed] [Google Scholar]
- 57.Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n - 6/n - 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 2004;106:635–643. doi: 10.1042/CS20030326. [DOI] [PubMed] [Google Scholar]
- 58.Gómez-Lechón MJ, Donato MT, Martínez-Romero A, Jiménez N, Castell JV, O’Connor JE. A human hepatocellular in vitro model to investigate steatosis. Chem Biol Interact. 2007;165:106–116. doi: 10.1016/j.cbi.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Li ZZ, Berk M, McIntyre TM, Feldstein AE. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J Biol Chem. 2009;284:5637–5644. doi: 10.1074/jbc.M807616200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cui W, Chen SL, Hu KQ. Quantification and mechanisms of oleic acid-induced steatosis in HepG2 cells. Am J Transl Res. 2010;2:95–104. [PMC free article] [PubMed] [Google Scholar]
- 61.Oh SS, Kaplan ML. Early treatment of obese (ob/ob) mice with triiodothyronine increases oxygen consumption and temperature and decreases body fat content. Proc Soc Exp Biol Med. 1994;207:260–267. doi: 10.3181/00379727-207-43814. [DOI] [PubMed] [Google Scholar]
- 62.Perra A, Simbula G, Simbula M, Pibiri M, Kowalik MA, Sulas P, Cocco MT, Ledda-Columbano GM, Columbano A. Thyroid hormone (T3) and TRbeta agonist GC-1 inhibit/reverse nonalcoholic fatty liver in rats. FASEB J. 2008;22:2981–2989. doi: 10.1096/fj.08-108464. [DOI] [PubMed] [Google Scholar]
- 63.Grasselli E, Canesi L, Voci A, De Matteis R, Demori I, Fugassa E, Vergani L. Effects of 3,5-diiodo-L-thyronine administration on the liver of high fat diet-fed rats. Exp Biol Med (Maywood) 2008;233:549–557. doi: 10.3181/0710-RM-266. [DOI] [PubMed] [Google Scholar]
- 64.Grasselli E, Voci A, Demori I, Canesi L, De Matteis R, Goglia F, Lanni A, Gallo G, Vergani L. 3,5-Diiodo-L-thyronine modulates the expression of genes of lipid metabolism in a rat model of fatty liver. J Endocrinol. 2012;212:149–158. doi: 10.1530/JOE-11-0288. [DOI] [PubMed] [Google Scholar]
- 65.Reddy JK, Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G852–G858. doi: 10.1152/ajpgi.00521.2005. [DOI] [PubMed] [Google Scholar]
- 66.Mollica MP, Lionetti L, Moreno M, Lombardi A, De Lange P, Antonelli A, Lanni A, Cavaliere G, Barletta A, Goglia F. 3,5-diiodo-l-thyronine, by modulating mitochondrial functions, reverses hepatic fat accumulation in rats fed a high-fat diet. J Hepatol. 2009;51:363–370. doi: 10.1016/j.jhep.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 67.Silvestri E, Cioffi F, Glinni D, Ceccarelli M, Lombardi A, de Lange P, Chambery A, Severino V, Lanni A, Goglia F, et al. Pathways affected by 3,5-diiodo-l-thyronine in liver of high fat-fed rats: evidence from two-dimensional electrophoresis, blue-native PAGE, and mass spectrometry. Mol Biosyst. 2010;6:2256–2271. doi: 10.1039/c0mb00040j. [DOI] [PubMed] [Google Scholar]
- 68.Grasselli E, Voci A, Canesi L, Goglia F, Ravera S, Panfoli I, Gallo G, Vergani L. Non-receptor-mediated actions are responsible for the lipid-lowering effects of iodothyronines in FaO rat hepatoma cells. J Endocrinol. 2011;210:59–69. doi: 10.1530/JOE-11-0074. [DOI] [PubMed] [Google Scholar]
- 69.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, Zhu X, Privalsky ML, Cheng SY, Stevens RD, Summers SA, et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest. 2012;122:2428–2438. doi: 10.1172/JCI60580. [DOI] [PMC free article] [PubMed] [Google Scholar]