

Abstract
Liver injuries are repaired by fibrosis and regeneration. The cause of fibrosis and diminished regeneration, especially in liver cirrhosis, is still unknown. Epithelial-mesenchymal transition (EMT) has been found to be associated with liver fibrosis. The possibility that EMT could contribute to hepatic fibrogenesis reinforced the concept that activated hepatic stellate cells are not the only key players in the hepatic fibrogenic process and that other cell types, either hepatic or bone marrow-derived cells could contribute to this process. Following an initial enthusiasm for the discovery of this novel pathway in fibrogenesis, more recent research has started to cast serious doubts upon the real relevance of this phenomenon in human fibrogenetic disorders. The debate on the authenticity of EMT or on its contribution to the fibrogenic process has become very animated. The overall result is a general confusion on the meaning and on the definition of several key aspects. The aim of this article is to describe how EMT participates to hepatic fibrosis and discuss the evidence of supporting this possibility in order to reach reasonable and useful conclusions.
Keywords: Epithelial-mesenchymal transition, Liver, Fibrosis, Transforming growth factor-beta1, Biological markers
Core tip: The cause of fibrosis and diminished regeneration, especially in liver cirrhosis, is still unknown. Epithelial-mesenchymal transition (EMT) has been found to be associated with liver fibrosis. The possibility that EMT could contribute to hepatic fibrogenesis reinforced the concept that activated hepatic stellate cells are not the only key players in the hepatic fibrogenic process. The aim of this article is to describe how EMT participates to hepatic fibrosis and discuss the evidence of supporting this possibility in order to reach reasonable and useful conclusions.
INTRODUCTION
Chronic liver damage can be triggered by different mechanisms (e.g., viral hepatitis, metabolic liver diseases, or chronic alcohol consumption)[1] and are accompanied by changes in several key biochemical pathways involved in hepatic tissue homeostasis. One of the most important alterations is hepatic fibrosis, which is characterized by deposition of extracellular matrix (ECM) components around the sinusoidal layer in the space of Disse, together with molecular reorganization of the matrix components resulting in an altered composition[2].
Fibrosis is comparable with a wound-healing response being out of control. Repair mechanisms aim at the replacement of injured cells. However, contrary to the pure regeneration of tissue in fibroplasia, connective tissue substitutes normal parenchyma[3]. The ultimate result is organ failure. In the liver, the final common pathway is cirrhosis, characterized by accumulation of ECM. In human beings liver fibrosis is associated with dysregulated growth of hepatocytes and results in the formation of regenerative nodules, dysplastic nodules, and hepatocellular carcinomas[4]. At present, no therapeutic concepts have been developed to treat and reverse fibrosis[5].
Striking increases in our understanding of the pathogenesis of liver fibrosis include the identification of the main cellular effectors, key cytokines regulating the EMT process, and determinants of ECM turnover[3].
FIBROGENESIS OF HEPATIC STELLATE CELLS, MYOFIBROBLASTS AND HEPATOCYTES IN LIVER CIRRHOSIS
Recent work regarding liver fibrosis centers on the myofibroblast as a pivotal cell type due to its contractile nature and synthesis repertoire[6]. The sources of myofibroblasts are still matters of discussion. Undisputedly, a “myofibroblast” phenotype is observed with hepatic stellate cell (HSC) after exposure to profibrogenic cytokines[3].
Liver myofibroblasts stand for a wide repertoire of functions that emphasize the dynamic nature of the wound-healing response, including synthesis of fibrillar collagens, contractile and migratory activities, secretion of chemotactic and vasoactive factors, and the secretion of matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs)[3]. The origin of myofibroblasts in the injured liver is now under scrutiny, although evidence hints at HSCs as a major source[3]. Myofibroblasts can be derived from local mesenchymal cells recruited from the bone marrow or could derive from other cellular sources by EMT, a physiologic process in embryogenesis and of relevance for cancerous cell transformation[7] (Figure 1A).
Figure 1.
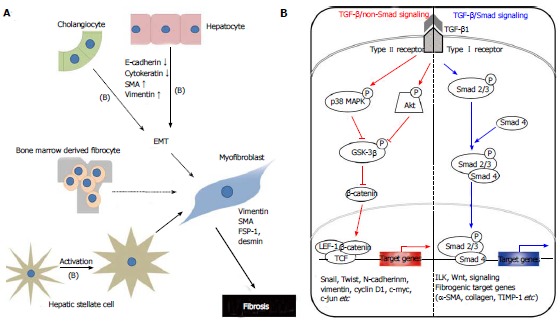
Mechanisms of hepatic fibrogenesis. A: The proposed sources of hepatic myofibroblasts: Resident cells (hepatic stellate cells and portal fibroblasts); bone marrow-derived mesenchymal cells, and EMT from hepatocytes and cholangiocytes. Different insults initiate inflammation and then cause hepatocyte stellate cells activation and hepatocyte and biliary cell damage, necrosis and EMT. Continuous insults will shift those EMT-like cells to complete EMT cells and finally myofibroblasts, the main producer of extracellular matrix, which may be one of the main causes of an early loss of regenerative capacity. A similar process also occurs in biliary cells. Some cytokines play an important role to affect the adjacent cells and promote EMTs, such as TGF-β1 (B); B: Schematic presentation of the major intracellular signal transduction pathways of TGF-β1 in liver fibrosis. TGF-β1 is a chief inducer of the EMT process, and p-Smad2/3, p38 MAPK and ILK function as mediators of the intracellular signaling pathway. TGF-β1 signals via heteromeric transmembrane complexes of type I and type II receptors that are endowed with intrinsic serine/threonine kinase activity (ALK activin receptor-like kinase). Upon type-II-mediated phosphorylation of the type I receptor, the activated type I receptor initiates intracellular signalling by phosphorylating receptor regulated-Smad2 and Smad3. Activated Smads form heteromeric complexes with Smad4 and these complexes accumulate in the nucleus where they mediate transcriptional responses. p-Smad2/3: Phosphorylated-Smad2/3; MAPK: Mitogen-activated protein kinase; GSK-3β: Glycogen synthase kinase-3β; ILK: Integrin-linked kinase; TCF/LEF-1 complex: T cell factor/lymphoid enhancer-binding factor-1 complex; EMT: Epithelial-mesenchymal transition; TGF: Transforming growth factor.
Studies with animals and human tissue indicate that bone marrow stem cells infiltrate the liver and contribute to the myofibroblast population after damage. This may occur directly or through an intermediary cell, such as quiescent HSCs or CD45+ fibrocytes[8,9]. Several studies have indicated that bone marrow-derived mesenchymal stem cells could be a source of multi-lineage cells for various organs. They have the capacity to differentiate into hepatocytes, biliary epithelial cells, sinusoidal endothelial cells and even Kupffer cells in the presence of a suitable hepatic microenvironment[10,11]. There is growing evidence to suggest that bone marrow-derived stem cells are recruited during both progression and regression of liver fibrosis[8,12-16].
Most studies in the past decade have focused on HSCs and analyzed characteristic features like plasticity and transdifferentiation to myofibroblasts, a phenotype that can be readily recapitulated in tissue culture[3]. Current concepts envision activated HSCs as a crucial profibrogenic source, while the majority of hepatocytes are believed to undergo necrosis or apoptosis, thereby providing space for proliferating cells. Besides the resident hepatic cells, infiltrating neutrophils, macrophages, T and B cells, and eosinophils participate in the inflammatory response and may perpetuate the damage, whereby activated macrophages and neutrophils clean up tissue debris, dead cells, and invading organisms[17].
HSCs are located in the space of Disse of hepatic sinusoids between hepatocytes and sinusoidal endothelial cells[1] as liver-specific pericytes interposed by sparse connective tissue and closely adhering to sinusoidal endothelial cells[18]. They also directly face hepatocytes and maintain a quiescent phenotype with the main function to store vitamin A[19]. During liver injury, HSCs lose their vitamin A content[20] and undergo activation triggered by exposure to cytokines and growth factors, such as platelet-derived growth factor (PDGF) and transforming growth factor (TGF)-β1, which derive from activated Kupffer cells and damaged hepatocytes. During activation, HSCs transform into a myofibroblast-like phenotype[21-23] losing their typical star-shape[20], characterized by the expression of α-smooth muscle actin (SMA)[24], by the production of ECM and matrix degrading enzymes, such as MMPs[21-23] and TIMPs[25,26]. Activated HSCs are thought to migrate from the sinusoids into necrotic areas and produce a variety of ECM[19].
Some authors[27] have proposed that HSC could be transitional cells derived from epithelial cells that have undergone partial EMT[28] or even a particular type of oval cell/hepatocyte precursor[29]. This hypothesis was based, at least in part, on the finding of an adult subpopulation of primary rat HSC expressing the progenitor cell marker CD133 and differentiating into either myofibroblasts or hepatocytes when cultured under different in vitro conditions[30].
Another source of myofibroblast may be portal fibroblasts that are described in fibrotic diseases with a portal component (e.g., viral hepatitis and autoimmune conditions)[31]. In coherence with experimental data on hepatocytes, TGF-β1 is required for myofibroblast transdifferentiation of this cell type[32]. The portal connective tissue in healthy liver is surrounded by quiescent portal fibroblasts, which constitute a second population of liver cells implicated in portal fibrosis[33]. Derived from small portal vessels, they express markers distinct from HSC (e.g., elastin)[32]. Proliferation of biliary cells is often accompanied by proliferation of portal fibroblasts, which form onion-like configurations around biliary structures and acquire a myofibroblast phenotype, and are thus implied in the early deposition of ECM in portal zones[34]. It is generally believed that substantial signaling from biliary epithelial cells leads to portal fibroblast activation, although the key factors remain to be identified[20]. In complex studies employing the “lineage tracing” methodological approach have documented, in different animal models of liver fibrogenesis, that some hepatocytes or cholangiocytes acquire “mesenchymal markers” implicated in cell motility and survival, but are not involved in active fibrillar ECM deposition and, therefore, cannot be considered pro-fibrogenic cells[35].
Dooley et al[3] have provided in vitro and in vivo evidence that profibrogenic TGF-β1 functions during liver damage are directed toward hepatocytes. A TGF-β1-induced gene expression profiling of hepatocytes indicates a minor role of apoptosis and induction of fibrogenesis- and EMT-related genes. While the definite occurrence of EMT in this cell type in vivo needs further investigation (e.g., by double-transgenic animals expressing fluorescent proteins under the control of hepatocyte- and myofibroblast-specific promoters), their results suggest that hepatocytes and TGF-β1 signaling in this cell type play a prominent role for fibrogenesis. In patients with chronic hepatitis B virus (HBV) infection, the activation of the TGF-β1 pathway was also shown by the accumulation of phosphorylated Smad2 in hepatocyte nuclei. Furthermore, the induction of Snail, a transcription factor known to repress E-cadherin expression, and the co-expression of type I collagen and transferrin in HBV livers, indicated that hepatocyte EMT was a feature of human liver fibrosis[36]. Therefore, the hepatocytes may be a contributor to hepatic fibrosis, especially when they are chronically injured[4].
TGF-β/SMAD AND NON-SMAD SIGNALLING PATHWAY IN LIVER FIBROSIS
TGF-β1 is recognized as a major profibrogenic cytokine and is a potent inducer of HSC proliferation and collagen production[37]. Furthermore, TGF-β1 expression is also associated with morphologic alterations like EMT in fetal[38] and adult hepatocytes[39], and changes in survival signaling pathways[40].
TGF-β1 binds to TGF-β1 receptor type II (TbR-II), and it recruits the TGF-β1 receptor type I (TbR-I)[41]. TbR-I subsequently phosphorylates Smad2 and Smad3, which form hetero-oligomers with Smad4. They translocate from the cytoplasm to the nucleus, where they regulate transcription of target genes[42] (Figure 1B). R-Smad signaling is limited by the inhibitory effects of inhibitory Smad6 and 7[3].
Dysregulated TGF-β1 signaling is implicated in multiple developmental disorders and various human diseases, including cancer and autoimmune illnesses[43]. Its over-expression is linked to liver fibrosis in diverse animal models[44] and in human patients with chronic liver diseases[45]. TGF-β1 crucially regulates ECM deposition by controlling the expression of ECM network components such as fibrillar collagens and fibronectin, ECM-degrading protease inhibitors, such as plasminogen activator inhibitor (PAI)-1 and TIMPs. Its activity is strongly induced during chronic liver damage with links between TGF-β1 and connective tissue growth factor in the HSC activation process[46], which in turn acquire myofibroblastic features and produce ECM proteins.
Epithelial cell transdifferentiation comprises alterations in cellular morphology characterized by changes in cell polarity and loss of adhesion protein expression[3]. TGF-β1 can initiate and maintain this process in a variety of biological systems and pathophysiological contexts by activating major signaling pathways and transcriptional regulators integrated in extensive cellular networks[47]. In MDCKII cells, claudin-1, claudin-2, occludin and E-cadherin disappear within 72 h of exposure to TGF-β1. It is suggested that this expression loss occurs through a Smad-independent mechanism, involving mitogen-activated protein kinase kinase and phosphatidylinositol 3-kinase pathways with expression of Snail. On the other hand, a complete loss of E-cadherin and transition to the mesenchymal phenotype additionally requires Smad signaling, which results in formation of β-catenin/lymphoid enhancer factor-1 complexes that induce EMT[48]. Participation of TGF-β1 in the regulation of Notch signaling has been reported previously at the onset of EMT in epithelial cells from mammary gland, kidney tubules, and epidermis[49]. A set of the previously mentioned genes and others described to be involved in EMT, including Snail and Notch2, were identified as TGF-β1 target genes in hepatocytes. Some studies[39,50] have suggested on hepatocyte plasticity showing up-regulation of α1(I) collagen mRNA expression and type I collagen deposition in mouse hepatocytes and α (alpha) mouse liver 12 cells as a result of Smad2/3/4-dependent induction of Snail-1.
EMT IN HEPATIC FIBROGENESIS
EMT is a process that is normally evident in embryonic stages of development and recently has been investigated as a mechanism of cancer cell migration and metastasis[51,52]. A classification of EMT has been recently proposed to distinguish between these different types of EMT[53]. It is characterized by the loss of epithelial characteristics (E-cadherin) and the acquisition of a mesenchymal phenotype (vimentin and fibronectin)[54,55]. According to the functional consequences and biological context, EMT is divided into three subtypes[53,55,56]: Type I EMT occurs during embryogenesis, in which it produces motile cells but does not lead to ECM deposition or intravascular invasion. Type II EMT induces a morphogenetic change during organ fibrosis or wound healing, which is associated with ECM production and muscle-like characteristics. Type III EMT is involved in carcinoma-metastatic transition.
Evidence of EMT in fibrosis was first demonstrated in the kidney. In vitro, adult renal tubular epithelial cells were shown to undergo EMT[57,58]. Thereafter, induction of renal fibrosis in mice by unilateral ureteral obstruction (UUO) showed that epithelial marker expression (i.e., E-cadherin) was lost in tubular epithelial cells, while the mesenchymal marker α-SMA expression was increased[59]. A cell tracing method later established that mice submitted to UUO display EMT-derived fibroblasts that contribute to the fibroblastic population[60].
Because the liver is an organ prone to fibrosis and because the origin of fibroblastic cells in fibrotic liver is still debated, the possibility that liver epithelial cells participate to fibrosis by EMT is appealing. Such hypothesis was strengthened by the observation that HSC lines express E-cadherin, while hepatic epithelial progenitor cells are positive for α-SMA[61]. Kaimori et al[39] then demonstrated that freshly isolated hepatocytes were able to convert to mesenchymal cells in vitro. Hepatocyte EMT, characterized by a decrease in E-cadherin expression and concomitant acquisition of mesenchymal markers (vimentin and type I collagen), was observed when cells were incubated with the profibrogenic cytokine, TGF-β1[39]. Zeisberg and co-workers were the first to report in vivo evidence for hepatocyte EMT[50]. They demonstrated that hepatocyte EMT was observed in CCl4 induced liver fibrosis by developing transgenic mice specifically expressing a molecular tag in hepatocytes (i.e., β-galactosidase). In these mice challenged with CCl4, 45% of the cells expressing the mesenchymal marker fibroblast-specific protein-1 (FSP-1) were also positive for β-galactosidase expression. Furthermore, the inhibition of the TGF-β1 pathway limited the extent of liver fibrosis in the CCl4-injected mice[50]. In vitro TGF-β1 treatment induced higher vimentin expression in cirrhotic liver-derived hepatocytes than in normal liver-derived hepatocytes[4]. Taken together, these results suggest that hepatocyte EMT is triggered by TGF-β1 and contributes to liver fibrosis. Some studies have proposed that EMT leads to myofibroblast accumulation through a two-stage process. In the first stage, epithelial cells adopt a mesenchymal phenotype, whereas in the second stage these mesenchymal cells further transition to myofibroblasts as part of what has been termed an epithelial-to-myofibroblast transition (EMyT)[62-64].
Substantial experimental evidence supports the occurrence of EMT in embryonic development and tumor metastasis, processes in which the motility phenotype of the transitioned cells is essential[56,65,66]. For tissue fibrosis, however, there are conflicting data on whether or not EMT occurs[67]. Many studies of EMT in fibrosis have failed to define EMT rigorously or to differentiate between the transition to a mesenchymal (EMT) vs a myofibroblast (EMyT) phenotype. Type I collagen expression is the most direct measure of fibrogenesis, and the literature suggests that α-SMA-positive cells are the primary effectors of fibrogenesis[55,62,68,69]. Nevertheless, surrogate fibroblast markers have often been used to identify EMT, most notably FSP-1, despite some data suggesting that it is nonspecific[68,70,71].
Mendez et al[72] examined the expression of four different mesenchymal markers, including FSP-1, vimentin, α-SMA, and procollagen I. Their lack of colocalization with yellow fluorescent protein (YFP) in the setting of fibrosis supports the conclusion that in these models EMT does not contribute to fibrosis. The complete absence of its colocalization with YFP in their study suggests that liver epithelial cells do not transition to either mesenchymal cells or myofibroblasts in the mouse models examined.
The hypothesis of hepatocyte EMT contributing to liver fibrosis has been also challenged by a cell lineage strategy in mice[67]. A lineage tracing study in which β-galactosidase was expressed under the control of the hepatocyte marker albumin in transgenic mice expressing a collagen marker provided strong evidence against hepatocyte EMT in the CCl4 model of fibrosis[73]. Triple transgenic mice with permanent cell labeling were produced to track hepatocyte-derived cells and type I collagen-expressing cells. Hepatocytes isolated from these triple transgenic mice were able to undergo EMT in culture when incubated with TGF-β1. However, in mice challenged by CCl4, no cells exhibited a double labeling specific for both hepatocytes and collagen expressing cells[73]. These observations also suggest that hepatocytes may not undergo EMT in vivo, while the observed transition in vitro might be an experimental artifact.
EMT IN CHOLANGIOCYTES
The assumption that liver epithelial cells undergo EMT in liver fibrosis cannot however be ruled out for biliary epithelial cells. Indeed, biliary epithelial cell EMT could represent a cellular mechanism supporting histological observations[70]. For instance, primary biliary cirrhosis (PBC), a prototypical biliary-type liver disease, is characterized by both the loss of biliary epithelial cells and the concomitant development of periportal fibrosis.
The bile duct basement membranes undergo degradation in fibrogenic liver diseases and that cholangiocytes, the other major hepatic epithelial cell type, assume fibroblast-like, non-cuboidal shapes. Therefore, it became obvious that the next step was to investigate whether or not biliary cells could undergo EMT in chronic liver disease[27]. It is well established that proliferating cholangiocytes within the so-called ‘‘ductular reaction’’ (i.e., ‘‘reactive cholangiocytes’’), detectable in all types of chronic liver disease, express a variety of pro-fibrogenic growth factors and cytokines and are likely to contribute to fibrosis and inflammation by promoting activation, proliferation, and collagen synthesis in the surrounding pro-fibrogenic cells[74-80]. Nevertheless, the possibility of a direct contribution of cholangiocytes to fibrosis via EMT was suggested by Omenetti and his colleagues[81] showing in vitro a complete EMT in an immature cholangiocyte cell line treated with activated HSC conditioned medium.
Biliary epithelial cell EMT was confirmed by another study analyzing liver of patients with PBC, primary sclerosing cholangitis or alcoholic liver disease. Irrespective of the underlying etiology, biliary epithelial cells from ducts associated with the ductular reaction were positive for FSP-1 and vimentin[82]. In biliary atresia, a disease defined by a destructive inflammatory obliterative cholangiopathy with portal tract fibrosis and ductular proliferation[83], biliary epithelial cells were shown to express FSP-1 and vimentin, while hepatocytes were not. Moreover, the authors of this study show that the expression of mesenchymal markers in biliary epithelial cells is observed in all liver disease with a ductular proliferation component[84]. The common bile duct ligation (BDL) is an experimental liver fibrosis model that induces strong ductular reaction. In mice submitted to BDL, biliary epithelial cells undergo EMT as shown by α-SMA and type I collagen expression[85].
Evidence of cholangiocyte EMT was recently challenged with the lineage-tracing methodology previously used for the investigation of hepatocyte EMT[27]. Along these lines, Scholten and the colleagues[86] employed the Cre-Lox technology for lineage tracing and studied several mouse strains expressing Cre under cholangiocyte-, HSC-, or FSP-1-specific promoters in two established models of liver fibrosis, i.e., chronic CCl4 intoxication and common BDL. In this case the fundamental experiment was tracing the fate of cells expressing K19, a bile ductular cell-specific marker, after permanent genetic Cre-mediated labeling of cholangiocytes. The key result of this study was that, although myofibroblast markers were often found in the close proximity of the K19+ progeny of cholangiocytes, the two signals never overlapped in either CCl4 or BDL fibrosis. Based on these and other observations reported in the paper, the authors concluded that cholangiocyte EMT does not occur in their experimental models.
It may be that in human livers EMT occurs in cirrhosis, a state not well modeled in rodents, and may require a florid ductular reaction, which is also poorly mimicked by rodent models. Alternatively, this discrepancy may reflect the limitations of immunohistochemistry-based lineage-tracing methodology[67]. Future work should focus on better understanding the direct contribution of dysfunctional epithelial cells to liver fibrosis, as well as determining the mechanistic relationships between fibrogenesis and the progenitor cell activation characteristic of the ductular reaction. This will ultimately require the development of animal models of biliary fibrosis that better reflect human disease[67].
SPECIFIC CELLULAR MARKERS IN EMT
The most widely used marker identifying myofibroblasts is the cytoskeletal protein α-SMA that is a part of the contractile machinery and is involved in cell motility[27]. In adult normal tissue, α-SMA expression is mostly restricted to vascular smooth muscle cells, but in most chronic inflammatory and fibrogenic disease states, it is often found in myofibroblasts of different derivation, and this expression is interpreted as an active involvement of these cells in fibrogenesis (i.e., ‘‘activated myofibroblast’’). Accordingly, α-SMA cannot be a good lineage marker since its expression is activated by disease states and, in addition, does not denote function. Regardless of this, there is supportive evidence that epithelial cells express intermediate filaments such as α-SMA and vimentin following tissue injury[4,87].
A multitude of studies have shown that epithelial cells, including hepatocytes, when cultured in vitro retain epithelial features including polarity and specific protein expression (i.e., albumin for hepatocytes), but when chronically stimulated with TGF-β1 or serum factors acquire a pattern of gene expression that is somehow typical of myofibroblasts in vivo and in the mesenchyme during development[39,88-91]. These genes are often represented by Slug, Twist, Snail, α-SMA, vimentin, desmin, FSP-1, and discoidin domain receptor tyrosine kinase 2. Some of these markers have been used to identify epithelial cells that are in the midst of undergoing an EMT associated with chronic inflammation. These cells continue to exhibit epithelial-specific morphology and molecular markers, such as cytokeratin and E-cadherin, but often show the concomitant expression of the FSP-1 and α-SMA. These aspects have been proposed to represent the intermediate stages of EMT, when epithelial markers continue to be expressed but new mesenchymal markers have already been acquired, and, overall, these observations have led to the notion of the so-called ‘‘partial EMT’’[53].
Previous work has demonstrated in a model of fetal hepatocytes that TGF-β1 treatment induces EMT-like morphologic changes in 50%-60% of the hepatocyte population, whereas the remaining hepatocytes undergo apoptosis[38,40]. This means that EMT can be elicited by several oncogenic pathways (Src, Ras, integrin, Wnt/β-catenin and Notch)[27,92]. In particular, Ras-MAPK has been shown to activate two related transcription factors known as Snail and Slug[93]. Both of these proteins are transcriptional repressors of E-cadherin and their expression induces EMT.
Chronic liver damage leads to fibrotic degeneration of parenchyma, characterized by the formation of fibrotic septa. Except for HSCs, portal myofibroblasts can produce collagen in the liver[94]. Although both cell types show similar expression patterns of intercellular adhesion molecule-1, desmin, vimentin, collagen type IV, fibronectin, and α-SMA, several differences between them have also been observed[94]. For instance, cultured portal myofibroblasts are positive for fibulin-2 and interleukin-6 mRNA, whereas CD95L, α2-macroglobulin, P100, and reelin are exclusively expressed by activated HSCs[94-100]. In addition, neural cell adhesion molecule, synaptophysin, neurotrophin, neural growth factor, αB-crystallin, and tyrosine kinases are markers that distinguish HSCs from portal myofibroblasts[99,101]. Experiments using these markers have shown that myofibroblastic cells in fibrotic septa strongly resemble portal myofibroblasts, that they may originate and migrate from the portal tract, and that they are different from sinusoidal HSCs[19].
Uyama et al[19] found that fascin is present in intralobular sinusoidal areas, but not in the periportal areas or fibrotic septa of the human liver. In addition, the localization of fascin in the sinusoidal area is similar to that of vimentin. They concluded that fascin was localized in human HSCs, but not in (myo)fibroblasts of the periportal area or fibrotic septa.
Evidence favoring biliary EMT comes largely from immunohistochemical studies of fibrotic human and rodent livers that identified cholangiocytes coexpressing epithelial markers (especially the cholangiocyte marker K19) and mesenchymal markers (i.e., FSP-1, vimentin, and HSP47)[64]. Table 1 shows the summary of useful biomarkers for identifying EMT.
Table 1.
Useful biomarkers for identifying epithelial-mesenchymal transition
| Biomarkers | Myofibroblast | Hepatic stellate cell | Hepatocyte | Cholangiocyte |
| α-SMA1 | + | + | - | - |
| Vimentin1 | + | + | - | - |
| Desmin1 | + | + | - | - |
| ICAM-1 | + | + | - | - |
| Collagen type IV | + | + | - | - |
| Fibronectin | + | + | - | - |
| Fibulin-2 | + | - | - | - |
| IL-6 mRNA | + | - | - | - |
| NCAM | + | - | - | - |
| Synaptophysin | + | - | - | - |
| Neurotrophin | + | - | - | - |
| Neural growth factor | + | - | - | - |
| αB-crystalline | + | - | - | - |
| Tyrosine kinase | + | - | - | - |
| FSP-11 | + | - | - | |
| HSP471 | + | - | - | |
| CD95L | - | + | - | - |
| α2-macroglobulin | - | + | - | - |
| P100 | - | + | - | - |
| Reelin | - | + | - | - |
| Fascin | - | + | - | - |
| E-cadherin | - | - | + | + |
| Cytokeratin | - | - | + | + |
| K19 | - | - | - | + |
| Albumin | - | - | + | - |
| Slug1 | + | ? | - | - |
| Twist1 | + | ? | - | - |
| Snail1 | + | ? | - | - |
Previously proven markers associated with epithelial-mesenchymal transition. α-SMA: α-smooth muscle actin; ICAM-1: Intercellular adhesion molecule-1; IL-6: Interleukin-6; NCAM: Neural cell adhesion molecule; FSP-1: Fibroblast-specific protein-1.
CONCLUSION
The pathogenesis of liver fibrosis is now better understood than ever before. It is increasingly recognized that the fibrogenic cells in the liver are heterogenous in both their formation and their behaviour.
EMT is an established process in embryo development and plays an important role in liver fibrosis. Discussions now arise on the involvement of EMT in organ fibrosis. The possibility that EMT could contribute to hepatic fibrogenesis in chronic liver diseases reinforced the concept that activated HSCs are not the only key players in the hepatic fibrogenic process and that other cell types, either hepatic or extrahepatic (bone marrow-derived cells and circulating fibrocytes) could contribute to this process. The presence of cells expressing both epithelial and mesenchymal markers suggests that EMT is a feature of liver fibrosis, however the ability of these cells to produce ECM in vivo has not yet been documented.
Therefore, the knowledge relative to the interpretation of what is defined as EMT in chronic fibrogenic disorders of the liver represents a scientific treasure that has prompted discussion, animated debates and has ultimately provided further maturity in this field of research. Definitely, there is now need for a more insightful analysis of the real pathophysiological meaning of these observations beyond their morphologic and biological features. Nevertheless, the future holds great promise for EMT as a viable therapeutic target.
EMT research in the next few years promises to be exciting, as new mouse models and molecular probes are identified to address the identities of the EMT-inducing microenvironmental signals, the nature of the cellular response of such signals and signaling machinery within epithelial cells.
Future research will surely be required on uncovering the origin of all fibrogenic cells within the liver and the molecular similarities and differences among the EMT programs.
Footnotes
P- Reviewers: Anand BS, Kravos M S- Editor: Song XX L- Editor: A E- Editor: Wu HL
Supported by The National Research Foundation of Korea Grant funded by the Korean Government, No. 2012R1A1A401015639
References
- 1.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marsillach J, Ferré N, Camps J, Rull A, Beltran R, Joven J. Changes in the expression of genes related to apoptosis and fibrosis pathways in CCl4-treated rats. Mol Cell Biochem. 2008;308:101–109. doi: 10.1007/s11010-007-9617-0. [DOI] [PubMed] [Google Scholar]
- 3.Dooley S, Hamzavi J, Ciuclan L, Godoy P, Ilkavets I, Ehnert S, Ueberham E, Gebhardt R, Kanzler S, Geier A, et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology. 2008;135:642–659. doi: 10.1053/j.gastro.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 4.Nitta T, Kim JS, Mohuczy D, Behrns KE. Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways. Hepatology. 2008;48:909–919. doi: 10.1002/hep.22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giebeler A, Boekschoten MV, Klein C, Borowiak M, Birchmeier C, Gassler N, Wasmuth HE, Müller M, Trautwein C, Streetz KL. c-Met confers protection against chronic liver tissue damage and fibrosis progression after bile duct ligation in mice. Gastroenterology. 2009;137:297–308, 308.e1-4. doi: 10.1053/j.gastro.2009.01.068. [DOI] [PubMed] [Google Scholar]
- 6.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russo FP, Alison MR, Bigger BW, Amofah E, Florou A, Amin F, Bou-Gharios G, Jeffery R, Iredale JP, Forbes SJ. The bone marrow functionally contributes to liver fibrosis. Gastroenterology. 2006;130:1807–1821. doi: 10.1053/j.gastro.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 9.Forbes SJ, Russo FP, Rey V, Burra P, Rugge M, Wright NA, Alison MR. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology. 2004;126:955–963. doi: 10.1053/j.gastro.2004.02.025. [DOI] [PubMed] [Google Scholar]
- 10.Gao Z, McAlister VC, Williams GM. Repopulation of liver endothelium by bone-marrow-derived cells. Lancet. 2001;357:932–933. doi: 10.1016/s0140-6736(00)04217-3. [DOI] [PubMed] [Google Scholar]
- 11.Fujii H, Hirose T, Oe S, Yasuchika K, Azuma H, Fujikawa T, Nagao M, Yamaoka Y. Contribution of bone marrow cells to liver regeneration after partial hepatectomy in mice. J Hepatol. 2002;36:653–659. doi: 10.1016/s0168-8278(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 12.Chang YJ, Liu JW, Lin PC, Sun LY, Peng CW, Luo GH, Chen TM, Lee RP, Lin SZ, Harn HJ, et al. Mesenchymal stem cells facilitate recovery from chemically induced liver damage and decrease liver fibrosis. Life Sci. 2009;85:517–525. doi: 10.1016/j.lfs.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 13.Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, Brenner DA. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45:429–438. doi: 10.1016/j.jhep.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Roderfeld M, Rath T, Voswinckel R, Dierkes C, Dietrich H, Zahner D, Graf J, Roeb E. Bone marrow transplantation demonstrates medullar origin of CD34+ fibrocytes and ameliorates hepatic fibrosis in Abcb4-/- mice. Hepatology. 2010;51:267–276. doi: 10.1002/hep.23274. [DOI] [PubMed] [Google Scholar]
- 15.Higashiyama R, Inagaki Y, Hong YY, Kushida M, Nakao S, Niioka M, Watanabe T, Okano H, Matsuzaki Y, Shiota G, et al. Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology. 2007;45:213–222. doi: 10.1002/hep.21477. [DOI] [PubMed] [Google Scholar]
- 16.Bird TG, Lorenzini S, Forbes SJ. Activation of stem cells in hepatic diseases. Cell Tissue Res. 2008;331:283–300. doi: 10.1007/s00441-007-0542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wake K. Perisinusoidal stellate cells (fat-storing cells, interstitial cells, lipocytes), their related structure in and around the liver sinusoids, and vitamin A-storing cells in extrahepatic organs. Int Rev Cytol. 1980;66:303–353. doi: 10.1016/s0074-7696(08)61977-4. [DOI] [PubMed] [Google Scholar]
- 19.Uyama N, Iimuro Y, Kawada N, Reynaert H, Suzumura K, Hirano T, Kuroda N, Fujimoto J. Fascin, a novel marker of human hepatic stellate cells, may regulate their proliferation, migration, and collagen gene expression through the FAK-PI3K-Akt pathway. Lab Invest. 2012;92:57–71. doi: 10.1038/labinvest.2011.150. [DOI] [PubMed] [Google Scholar]
- 20.Mormone E, George J, Nieto N. Molecular pathogenesis of hepatic fibrosis and current therapeutic approaches. Chem Biol Interact. 2011;193:225–231. doi: 10.1016/j.cbi.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedman SL. Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. N Engl J Med. 1993;328:1828–1835. doi: 10.1056/NEJM199306243282508. [DOI] [PubMed] [Google Scholar]
- 22.Pinzani M. Novel insights into the biology and physiology of the Ito cell. Pharmacol Ther. 1995;66:387–412. doi: 10.1016/0163-7258(94)00072-b. [DOI] [PubMed] [Google Scholar]
- 23.Gressner AM. Cytokines and cellular crosstalk involved in the activation of fat-storing cells. J Hepatol. 1995;22:28–36. [PubMed] [Google Scholar]
- 24.Ramadori G, Veit T, Schwögler S, Dienes HP, Knittel T, Rieder H, Meyer zum Büschenfelde KH. Expression of the gene of the alpha-smooth muscle-actin isoform in rat liver and in rat fat-storing (ITO) cells. Virchows Arch B Cell Pathol Incl Mol Pathol. 1990;59:349–357. doi: 10.1007/BF02899424. [DOI] [PubMed] [Google Scholar]
- 25.Iredale JP, Benyon RC, Arthur MJ, Ferris WF, Alcolado R, Winwood PJ, Clark N, Murphy G. Tissue inhibitor of metalloproteinase-1 messenger RNA expression is enhanced relative to interstitial collagenase messenger RNA in experimental liver injury and fibrosis. Hepatology. 1996;24:176–184. doi: 10.1002/hep.510240129. [DOI] [PubMed] [Google Scholar]
- 26.Théret N, Musso O, L’Helgoualc’h A, Clément B. Activation of matrix metalloproteinase-2 from hepatic stellate cells requires interactions with hepatocytes. Am J Pathol. 1997;150:51–58. [PMC free article] [PubMed] [Google Scholar]
- 27.Pinzani M. Epithelial-mesenchymal transition in chronic liver disease: fibrogenesis or escape from death? J Hepatol. 2011;55:459–465. doi: 10.1016/j.jhep.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Choi SS, Diehl AM. Epithelial-to-mesenchymal transitions in the liver. Hepatology. 2009;50:2007–2013. doi: 10.1002/hep.23196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang L, Jung Y, Omenetti A, Witek RP, Choi S, Vandongen HM, Huang J, Alpini GD, Diehl AM. Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells. 2008;26:2104–2113. doi: 10.1634/stemcells.2008-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kordes C, Sawitza I, Müller-Marbach A, Ale-Agha N, Keitel V, Klonowski-Stumpe H, Häussinger D. CD133+ hepatic stellate cells are progenitor cells. Biochem Biophys Res Commun. 2007;352:410–417. doi: 10.1016/j.bbrc.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 31.Kinnman N, Housset C. Peribiliary myofibroblasts in biliary type liver fibrosis. Front Biosci. 2002;7:d496–d503. doi: 10.2741/A790. [DOI] [PubMed] [Google Scholar]
- 32.Li Z, Dranoff JA, Chan EP, Uemura M, Sévigny J, Wells RG. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology. 2007;46:1246–1256. doi: 10.1002/hep.21792. [DOI] [PubMed] [Google Scholar]
- 33.Tuchweber B, Desmoulière A, Bochaton-Piallat ML, Rubbia-Brandt L, Gabbiani G. Proliferation and phenotypic modulation of portal fibroblasts in the early stages of cholestatic fibrosis in the rat. Lab Invest. 1996;74:265–278. [PubMed] [Google Scholar]
- 34.Desmoulière A, Darby I, Costa AM, Raccurt M, Tuchweber B, Sommer P, Gabbiani G. Extracellular matrix deposition, lysyl oxidase expression, and myofibroblastic differentiation during the initial stages of cholestatic fibrosis in the rat. Lab Invest. 1997;76:765–778. [PubMed] [Google Scholar]
- 35.Firrincieli D, Boissan M, Chignard N. Epithelial-mesenchymal transition in the liver. Gastroenterol Clin Biol. 2010;34:523–528. doi: 10.1016/j.gcb.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 36.Carmona-Cuenca I, Herrera B, Ventura JJ, Roncero C, Fernández M, Fabregat I. EGF blocks NADPH oxidase activation by TGF-beta in fetal rat hepatocytes, impairing oxidative stress, and cell death. J Cell Physiol. 2006;207:322–330. doi: 10.1002/jcp.20568. [DOI] [PubMed] [Google Scholar]
- 37.Eghbali-Fatourechi G, Sieck GC, Prakash YS, Maercklein P, Gores GJ, Fitzpatrick LA. Type I procollagen production and cell proliferation is mediated by transforming growth factor-beta in a model of hepatic fibrosis. Endocrinology. 1996;137:1894–1903. doi: 10.1210/endo.137.5.8612529. [DOI] [PubMed] [Google Scholar]
- 38.Valdés F, Alvarez AM, Locascio A, Vega S, Herrera B, Fernández M, Benito M, Nieto MA, Fabregat I. The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes. Mol Cancer Res. 2002;1:68–78. [PubMed] [Google Scholar]
- 39.Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. 2007;282:22089–22101. doi: 10.1074/jbc.M700998200. [DOI] [PubMed] [Google Scholar]
- 40.Del Castillo G, Murillo MM, Alvarez-Barrientos A, Bertran E, Fernández M, Sánchez A, Fabregat I. Autocrine production of TGF-beta confers resistance to apoptosis after an epithelial-mesenchymal transition process in hepatocytes: Role of EGF receptor ligands. Exp Cell Res. 2006;312:2860–2871. doi: 10.1016/j.yexcr.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 41.Kitao A, Sato Y, Sawada-Kitamura S, Harada K, Sasaki M, Morikawa H, Shiomi S, Honda M, Matsui O, Nakanuma Y. Endothelial to mesenchymal transition via transforming growth factor-beta1/Smad activation is associated with portal venous stenosis in idiopathic portal hypertension. Am J Pathol. 2009;175:616–626. doi: 10.2353/ajpath.2009.081061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inagaki Y, Okazaki I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56:284–292. doi: 10.1136/gut.2005.088690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rubtsov YP, Rudensky AY. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat Rev Immunol. 2007;7:443–453. doi: 10.1038/nri2095. [DOI] [PubMed] [Google Scholar]
- 44.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:d793–d807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 45.Annoni G, Weiner FR, Zern MA. Increased transforming growth factor-beta 1 gene expression in human liver disease. J Hepatol. 1992;14:259–264. doi: 10.1016/0168-8278(92)90168-o. [DOI] [PubMed] [Google Scholar]
- 46.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 47.Zavadil J, Böttinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 48.Medici D, Hay ED, Goodenough DA. Cooperation between snail and LEF-1 transcription factors is essential for TGF-beta1-induced epithelial-mesenchymal transition. Mol Biol Cell. 2006;17:1871–1879. doi: 10.1091/mbc.E05-08-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zavadil J, Cermak L, Soto-Nieves N, Böttinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23:1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 51.Thompson EW, Newgreen DF, Tarin D. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res. 2005;65:5991–595; discussion 5995. doi: 10.1158/0008-5472.CAN-05-0616. [DOI] [PubMed] [Google Scholar]
- 52.Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005;65:5996–6000; discussion 6000-1. doi: 10.1158/0008-5472.CAN-05-0699. [DOI] [PubMed] [Google Scholar]
- 53.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15:1–12. doi: 10.1097/01.asn.0000106015.29070.e7. [DOI] [PubMed] [Google Scholar]
- 55.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okada H, Danoff TM, Kalluri R, Neilson EG. Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol. 1997;273:F563–F574. doi: 10.1152/ajprenal.1997.273.4.F563. [DOI] [PubMed] [Google Scholar]
- 58.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol. 2001;159:1465–1475. doi: 10.1016/S0002-9440(10)62533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sicklick JK, Choi SS, Bustamante M, McCall SJ, Pérez EH, Huang J, Li YX, Rojkind M, Diehl AM. Evidence for epithelial-mesenchymal transitions in adult liver cells. Am J Physiol Gastrointest Liver Physiol. 2006;291:G575–G583. doi: 10.1152/ajpgi.00102.2006. [DOI] [PubMed] [Google Scholar]
- 62.Masszi A, Speight P, Charbonney E, Lodyga M, Nakano H, Szászi K, Kapus A. Fate-determining mechanisms in epithelial-myofibroblast transition: major inhibitory role for Smad3. J Cell Biol. 2010;188:383–399. doi: 10.1083/jcb.200906155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fan L, Sebe A, Péterfi Z, Masszi A, Thirone AC, Rotstein OD, Nakano H, McCulloch CA, Szászi K, Mucsi I, et al. Cell contact-dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phospho-myosin pathway. Mol Biol Cell. 2007;18:1083–1097. doi: 10.1091/mbc.E06-07-0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Masszi A, Fan L, Rosivall L, McCulloch CA, Rotstein OD, Mucsi I, Kapus A. Integrity of cell-cell contacts is a critical regulator of TGF-beta 1-induced epithelial-to-myofibroblast transition: role for beta-catenin. Am J Pathol. 2004;165:1955–1967. doi: 10.1016/s0002-9440(10)63247-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hay ED, Zuk A. Transformations between epithelium and mesenchyme: normal, pathological, and experimentally induced. Am J Kidney Dis. 1995;26:678–690. doi: 10.1016/0272-6386(95)90610-x. [DOI] [PubMed] [Google Scholar]
- 66.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chu AS, Diaz R, Hui JJ, Yanger K, Zong Y, Alpini G, Stanger BZ, Wells RG. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology. 2011;53:1685–1695. doi: 10.1002/hep.24206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamaoka K, Nouchi T, Marumo F, Sato C. Alpha-smooth-muscle actin expression in normal and fibrotic human livers. Dig Dis Sci. 1993;38:1473–1479. doi: 10.1007/BF01308606. [DOI] [PubMed] [Google Scholar]
- 70.Wells RG. The epithelial-to-mesenchymal transition in liver fibrosis: here today, gone tomorrow? Hepatology. 2010;51:737–740. doi: 10.1002/hep.23529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Le Hir M, Hegyi I, Cueni-Loffing D, Loffing J, Kaissling B. Characterization of renal interstitial fibroblast-specific protein 1/S100A4-positive cells in healthy and inflamed rodent kidneys. Histochem Cell Biol. 2005;123:335–346. doi: 10.1007/s00418-005-0788-z. [DOI] [PubMed] [Google Scholar]
- 72.Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24:1838–1851. doi: 10.1096/fj.09-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Taura K, Miura K, Iwaisako K, Osterreicher CH, Kodama Y, Penz-Osterreicher M, Brenner DA. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology. 2010;51:1027–1036. doi: 10.1002/hep.23368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Milani S, Herbst H, Schuppan D, Stein H, Surrenti C. Transforming growth factors beta 1 and beta 2 are differentially expressed in fibrotic liver disease. Am J Pathol. 1991;139:1221–1229. [PMC free article] [PubMed] [Google Scholar]
- 75.Pinzani M, Milani S, Herbst H, DeFranco R, Grappone C, Gentilini A, Caligiuri A, Pellegrini G, Ngo DV, Romanelli RG, et al. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am J Pathol. 1996;148:785–800. [PMC free article] [PubMed] [Google Scholar]
- 76.Grappone C, Pinzani M, Parola M, Pellegrini G, Caligiuri A, DeFranco R, Marra F, Herbst H, Alpini G, Milani S. Expression of platelet-derived growth factor in newly formed cholangiocytes during experimental biliary fibrosis in rats. J Hepatol. 1999;31:100–109. doi: 10.1016/s0168-8278(99)80169-x. [DOI] [PubMed] [Google Scholar]
- 77.Kinnman N, Hultcrantz R, Barbu V, Rey C, Wendum D, Poupon R, Housset C. PDGF-mediated chemoattraction of hepatic stellate cells by bile duct segments in cholestatic liver injury. Lab Invest. 2000;80:697–707. doi: 10.1038/labinvest.3780073. [DOI] [PubMed] [Google Scholar]
- 78.Pinzani M, Milani S, De Franco R, Grappone C, Caligiuri A, Gentilini A, Tosti-Guerra C, Maggi M, Failli P, Ruocco C, et al. Endothelin 1 is overexpressed in human cirrhotic liver and exerts multiple effects on activated hepatic stellate cells. Gastroenterology. 1996;110:534–548. doi: 10.1053/gast.1996.v110.pm8566602. [DOI] [PubMed] [Google Scholar]
- 79.Caligiuri A, Glaser S, Rodgers RE, Phinizy JL, Robertson W, Papa E, Pinzani M, Alpini G. Endothelin-1 inhibits secretin-stimulated ductal secretion by interacting with ETA receptors on large cholangiocytes. Am J Physiol. 1998;275:G835–G846. doi: 10.1152/ajpgi.1998.275.4.G835. [DOI] [PubMed] [Google Scholar]
- 80.Marra F, DeFranco R, Grappone C, Milani S, Pastacaldi S, Pinzani M, Romanelli RG, Laffi G, Gentilini P. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: correlation with monocyte infiltration. Am J Pathol. 1998;152:423–430. [PMC free article] [PubMed] [Google Scholar]
- 81.Omenetti A, Porrello A, Jung Y, Yang L, Popov Y, Choi SS, Witek RP, Alpini G, Venter J, Vandongen HM, et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest. 2008;118:3331–3342. doi: 10.1172/JCI35875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rygiel KA, Robertson H, Marshall HL, Pekalski M, Zhao L, Booth TA, Jones DE, Burt AD, Kirby JA. Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab Invest. 2008;88:112–123. doi: 10.1038/labinvest.3700704. [DOI] [PubMed] [Google Scholar]
- 83.Hartley JL, Davenport M, Kelly DA. Biliary atresia. Lancet. 2009;374:1704–1713. doi: 10.1016/S0140-6736(09)60946-6. [DOI] [PubMed] [Google Scholar]
- 84.Díaz R, Kim JW, Hui JJ, Li Z, Swain GP, Fong KS, Csiszar K, Russo PA, Rand EB, Furth EE, et al. Evidence for the epithelial to mesenchymal transition in biliary atresia fibrosis. Hum Pathol. 2008;39:102–115. doi: 10.1016/j.humpath.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 85.Xia JL, Dai C, Michalopoulos GK, Liu Y. Hepatocyte growth factor attenuates liver fibrosis induced by bile duct ligation. Am J Pathol. 2006;168:1500–1512. doi: 10.2353/ajpath.2006.050747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scholten D, Osterreicher CH, Scholten A, Iwaisako K, Gu G, Brenner DA, Kisseleva T. Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology. 2010;139:987–998. doi: 10.1053/j.gastro.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ng YY, Fan JM, Mu W, Nikolic-Paterson DJ, Yang WC, Huang TP, Atkins RC, Lan HY. Glomerular epithelial-myofibroblast transdifferentiation in the evolution of glomerular crescent formation. Nephrol Dial Transplant. 1999;14:2860–2872. doi: 10.1093/ndt/14.12.2860. [DOI] [PubMed] [Google Scholar]
- 88.Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999;56:1455–1467. doi: 10.1046/j.1523-1755.1999.00656.x. [DOI] [PubMed] [Google Scholar]
- 89.Gershengorn MC, Hardikar AA, Wei C, Geras-Raaka E, Marcus-Samuels B, Raaka BM. Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells. Science. 2004;306:2261–2264. doi: 10.1126/science.1101968. [DOI] [PubMed] [Google Scholar]
- 90.Russ HA, Ravassard P, Kerr-Conte J, Pattou F, Efrat S. Epithelial-mesenchymal transition in cells expanded in vitro from lineage-traced adult human pancreatic beta cells. PLoS One. 2009;4:e6417. doi: 10.1371/journal.pone.0006417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Godoy P, Hengstler JG, Ilkavets I, Meyer C, Bachmann A, Müller A, Tuschl G, Mueller SO, Dooley S. Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor beta-induced apoptosis. Hepatology. 2009;49:2031–2043. doi: 10.1002/hep.22880. [DOI] [PubMed] [Google Scholar]
- 92.Guarino M, Rubino B, Ballabio G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology. 2007;39:305–318. doi: 10.1080/00313020701329914. [DOI] [PubMed] [Google Scholar]
- 93.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 94.Ramadori G, Saile B. Portal tract fibrogenesis in the liver. Lab Invest. 2004;84:153–159. doi: 10.1038/labinvest.3700030. [DOI] [PubMed] [Google Scholar]
- 95.Dubuisson L, Lepreux S, Bioulac-Sage P, Balabaud C, Costa AM, Rosenbaum J, Desmoulière A. Expression and cellular localization of fibrillin-1 in normal and pathological human liver. J Hepatol. 2001;34:514–522. doi: 10.1016/s0168-8278(00)00048-9. [DOI] [PubMed] [Google Scholar]
- 96.Saile B, Matthes N, Neubauer K, Eisenbach C, El-Armouche H, Dudas J, Ramadori G. Rat liver myofibroblasts and hepatic stellate cells differ in CD95-mediated apoptosis and response to TNF-alpha. Am J Physiol Gastrointest Liver Physiol. 2002;283:G435–G444. doi: 10.1152/ajpgi.00441.2001. [DOI] [PubMed] [Google Scholar]
- 97.Knittel T, Kobold D, Saile B, Grundmann A, Neubauer K, Piscaglia F, Ramadori G. Rat liver myofibroblasts and hepatic stellate cells: different cell populations of the fibroblast lineage with fibrogenic potential. Gastroenterology. 1999;117:1205–1221. doi: 10.1016/s0016-5085(99)70407-5. [DOI] [PubMed] [Google Scholar]
- 98.Kobold D, Grundmann A, Piscaglia F, Eisenbach C, Neubauer K, Steffgen J, Ramadori G, Knittel T. Expression of reelin in hepatic stellate cells and during hepatic tissue repair: a novel marker for the differentiation of HSC from other liver myofibroblasts. J Hepatol. 2002;36:607–613. doi: 10.1016/s0168-8278(02)00050-8. [DOI] [PubMed] [Google Scholar]
- 99.Cassiman D, Libbrecht L, Desmet V, Denef C, Roskams T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J Hepatol. 2002;36:200–209. doi: 10.1016/s0168-8278(01)00260-4. [DOI] [PubMed] [Google Scholar]
- 100.Dranoff JA, Kruglov EA, Robson SC, Braun N, Zimmermann H, Sévigny J. The ecto-nucleoside triphosphate diphosphohydrolase NTPDase2/CD39L1 is expressed in a novel functional compartment within the liver. Hepatology. 2002;36:1135–1144. doi: 10.1053/jhep.2002.36823. [DOI] [PubMed] [Google Scholar]
- 101.Nakatani K, Seki S, Kawada N, Kobayashi K, Kaneda K. Expression of neural cell adhesion molecule (N-CAM) in perisinusoidal stellate cells of the human liver. Cell Tissue Res. 1996;283:159–165. doi: 10.1007/s004410050524. [DOI] [PubMed] [Google Scholar]